
Pyran Rings Containing Polyketides from Penicillium raistrickii

Abstract
:1. Introduction
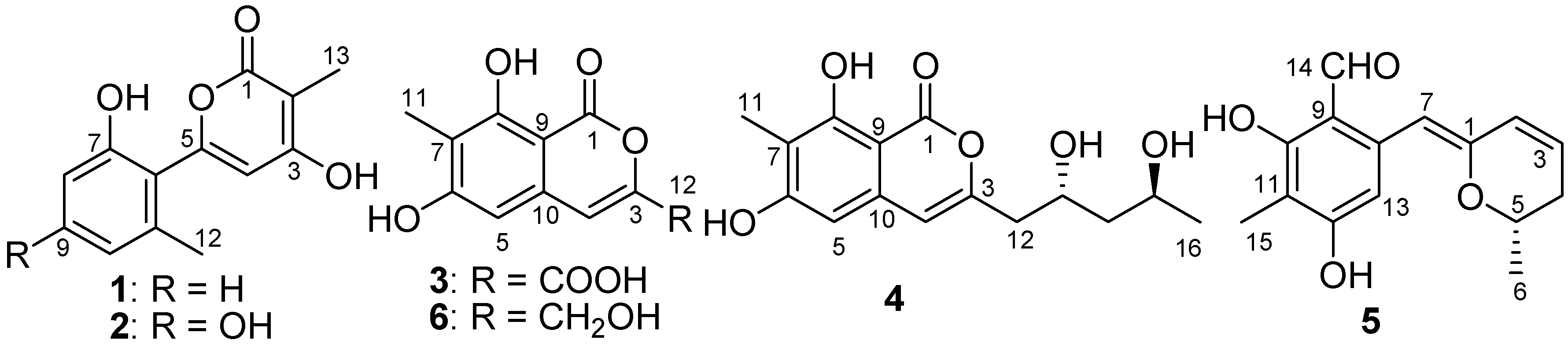
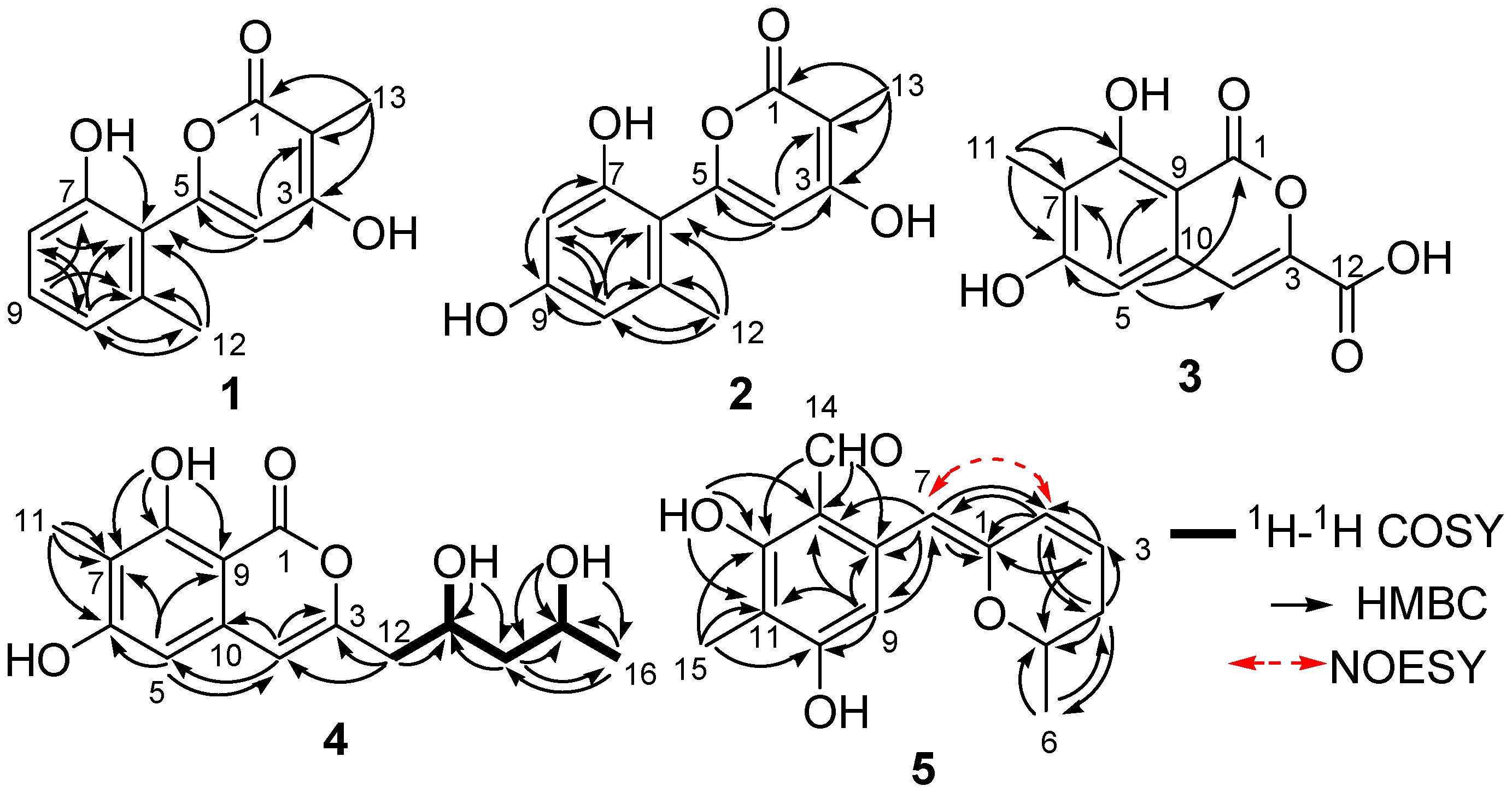
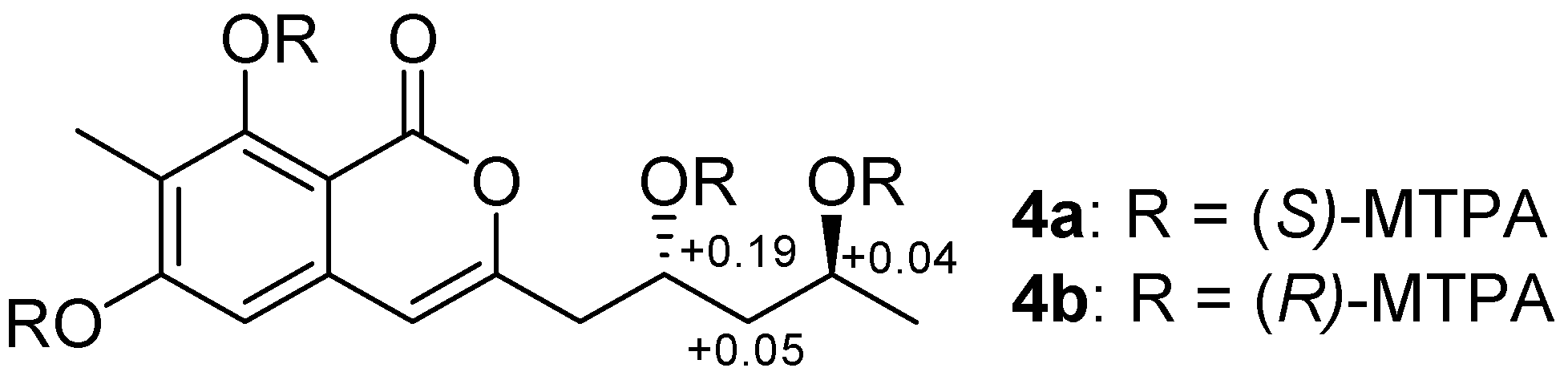
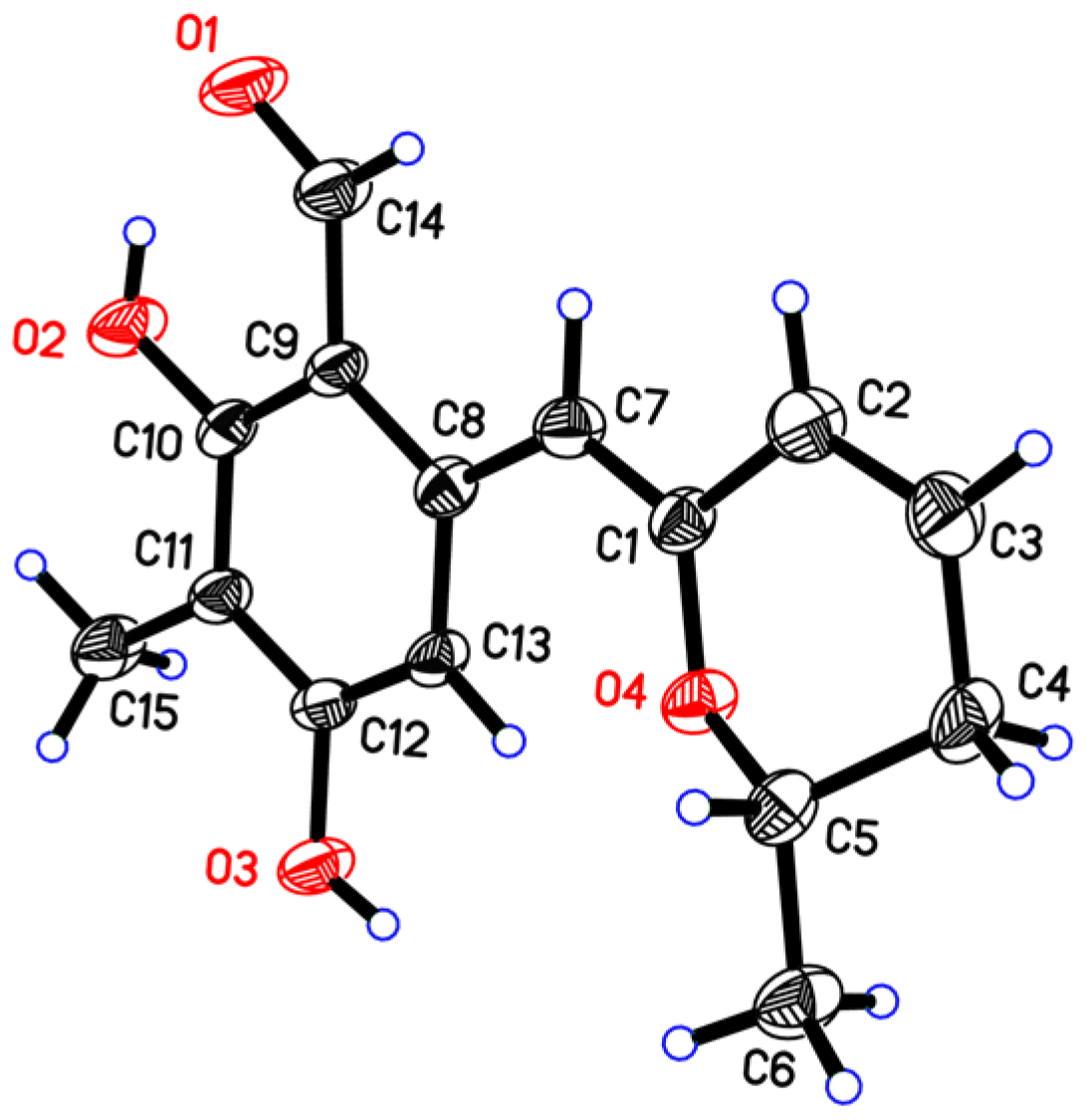
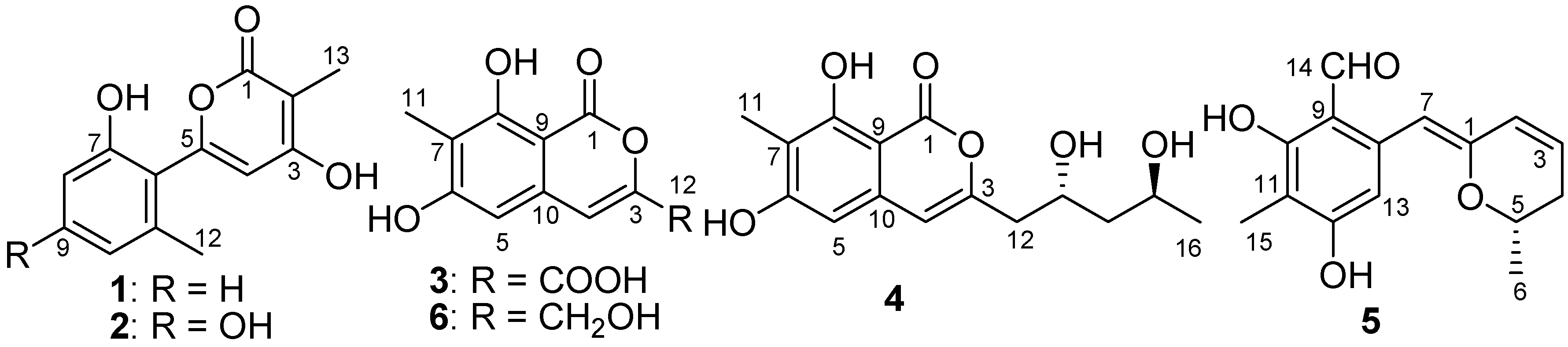
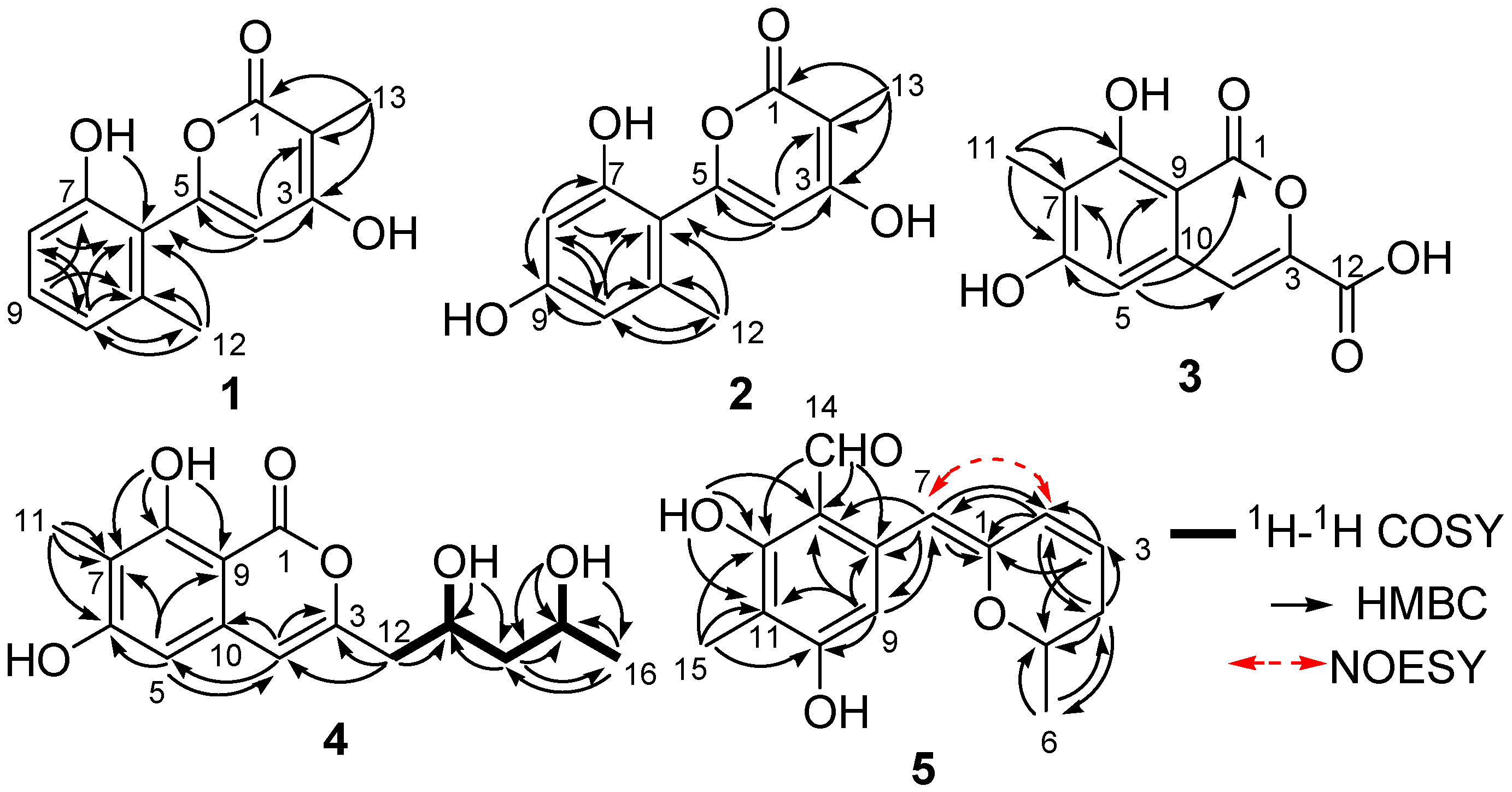
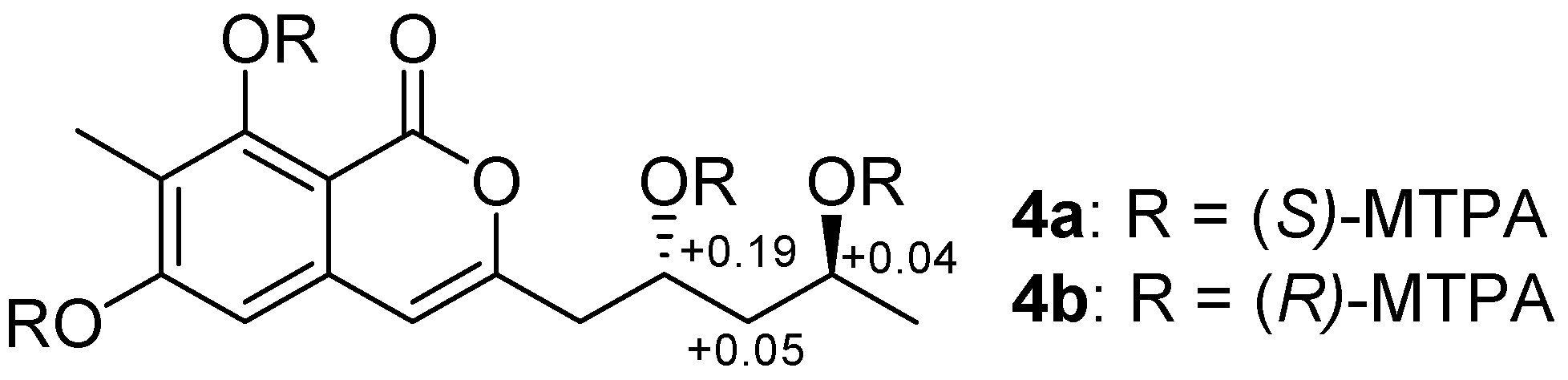
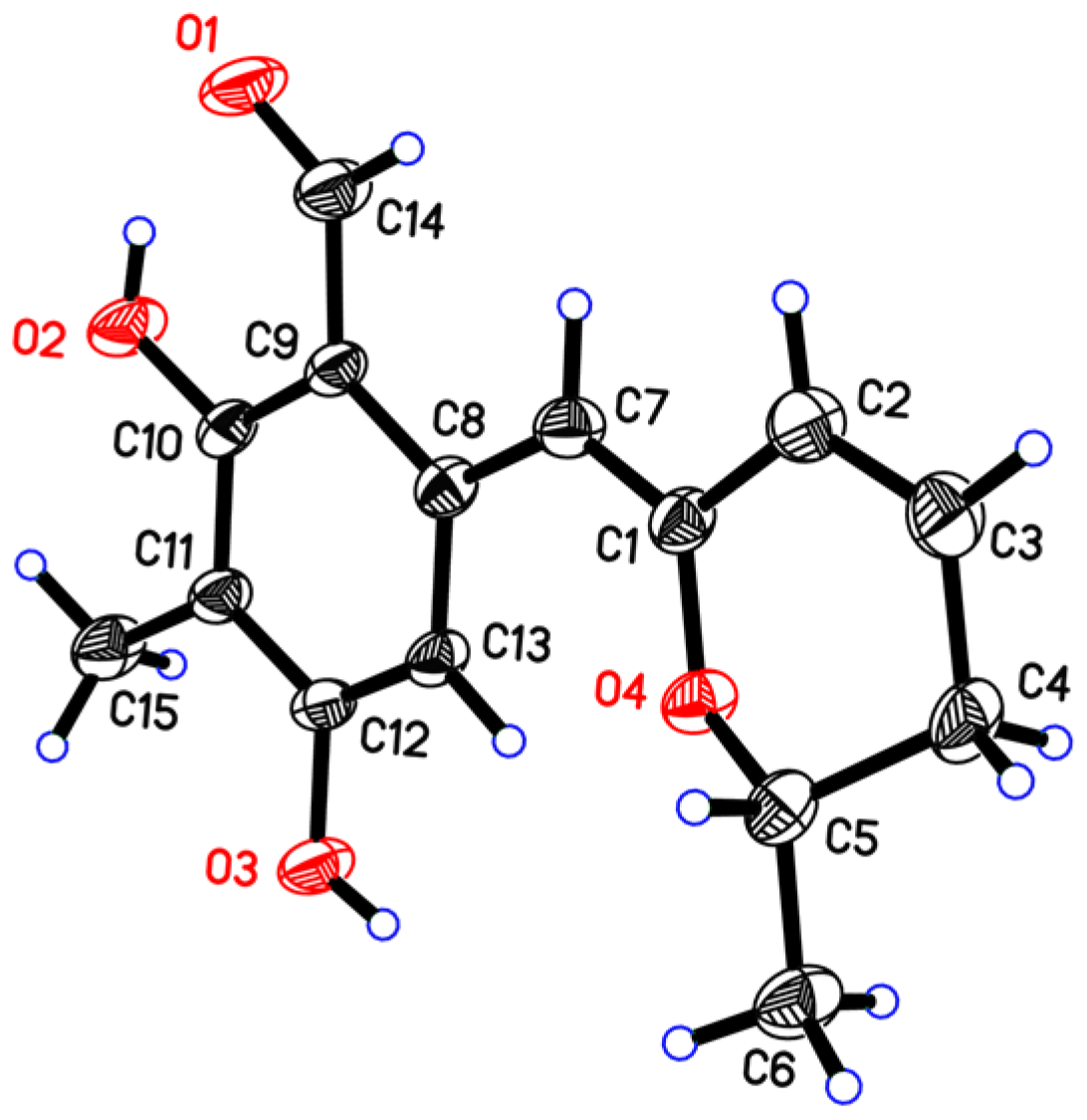
2. Results
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Extraction and Isolation
3.4. X-ray Crystal Data for Penicipyran E (5)
3.5. Preparation of (R)- and (S)-MTPA Esters of Penicipyran D (4)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Barnes, E.C.; Kumar, R.; Davis, R.A. The use of isolated natural products as scaffolds for the generation of chemically diverse screening libraries for drug discovery. Nat. Prod. Rep. 2016, 33, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Gogineni, V.; Schinazi, R.F.; Hamann, M.T. Role of marine natural products in the genesis of antiviral agents. Chem. Rev. 2015, 115, 9655–9706. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ye, D.; Shao, Z.; Cui, C.; Che, Y. A sterol and spiroditerpenoids from a Penicillium sp. isolated from a deep sea sediment sample. Mar. Drugs 2012, 10, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Li, X.M.; Li, X.; Xu, G.M.; Liu, Y.; Wang, B.G. Aspewentins D–H, 20-nor-isopimarane derivatives from the deep sea sediment-derived fungus Aspergillus wentii SD-310. J. Nat. Prod. 2016, 79, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.; Liu, P.; Qu, H.; Wang, Y.; Chen, D.; Wang, H.; Li, J.; Zhu, W. Α-pyrones and diketopiperazine derivatives from the marine-derived actinomycete Nocardiopsis dassonvillei HR10-5. J. Nat. Prod. 2011, 74, 2219–2223. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yan, L.; Ma, L.; Huang, Y.; Pan, X.; Liu, W.; Lv, Z. Diphenyl derivatives from coastal saline soil fungus Aspergillus iizukae. Arch. Pharm. Res. 2015, 38, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Liu, W.; Huang, Y.; Xian, G. Two acid sorbicillin analogues from saline lands-derived fungus Trichoderma sp. J. Antibiot. 2011, 64, 645–647. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.Z.; Ma, L.Y.; Liu, D.S.; Huang, Y.L.; Wang, C.H.; Shi, S.S.; Pan, X.H.; Song, X.D.; Zhu, R.X. Peniciketals A–C, New spiroketals from saline soil derived Penicillium raistrichii. Org. Lett. 2014, 16, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.Y.; Liu, W.Z.; Shen, L.; Huang, Y.L.; Rong, X.G.; Xu, Y.Y.; Gao, X.D. Spiroketals, isocoumarin, and indoleformic acid derivatives from saline soil derived fungus Penicillium raistrickii. Tetrahedron 2012, 68, 2276–2282. [Google Scholar] [CrossRef]
- Liu, W.Z.; Ma, L.Y.; Liu, D.S.; Huang, Y.L.; Wang, C.H. Pranes from Penicillium raistrickii. In Proceedings of the Chinese Chemical Society 10th National Symposium on Natural Organic Chemistry, Guangzhou, China, 21–23 November 2014; P-79. p. 180.
- Zhang, J.; Jiang, Y.; Cao, Y.; Liu, J.; Zheng, D.; Chen, X.; Han, L.; Jiang, C.; Huang, X. Violapyrones A–G, α-pyrone derivatives from Streptomyces violascens isolated from Hylobates hoolock feces. J. Nat. Prod. 2013, 76, 2126–2130. [Google Scholar] [CrossRef] [PubMed]
- Le, D.H.; Takenaka, Y.; Hamada, N.; Miyawaki, H.; Tanahashi, T.A. Fourteen-membered macrolide and isocoumarin derivatives from the cultured lichen mycobionts of Graphis vestitoides. Chem. Pharm. Bull. 2013, 61, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Hemphill, C.F.P.; Daletos, G.; Liu, Z.; Lin, W.; Proksch, P. Polyketides from the mangrove-derived fungal endophyte Pestalotiopsis clavispora. Tetrahedron Lett. 2016, 57, 2078–2083. [Google Scholar] [CrossRef]
- Lai, S.; Shizuri, Y.; Furukawa, H.; Yamamura, S.; Kawai, H.; Furukawa, H. Three new phenolic metabolites from Penicillium species. Heterocycles 1991, 32, 297–305. [Google Scholar]
- Seco, J.M.; Martino, M.; Quinoa, E.; Riguera, R. Absolute configuration of 1,n-diols by NMR: The importance of the combined anisotropic effects in bis-arylmethoxyacetates. Org. Lett. 2000, 2, 3261–3264. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Liu, Y.; Nie, Y.; Liu, Z.; Chen, S.; Zhang, Z.; Lu, Y.; He, L.; Huang, X.; She, Z. Polyketides from the mangrove-derived endophytic fungus Nectria sp. HN001 and their α-glucosidase inhibitory activity. Mar. Drugs 2016, 14, 86. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Chen, S.; Niu, S.; Guo, L.; Yin, J.; Che, Y. Exserolides A–F, new isocoumarin derivatives from the plant endophytic fungus Exserohilum sp. Fitoterapia 2014, 96, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Li, X.M.; Meng, L.; Li, C.S.; Gao, S.S.; Shang, Z.; Proksch, P.; Huang, C.G.; Wang, B.G. Nigerapyrones A–H, α-pyrone derivatives from the marine mangrove-derived endophytic fungus Aspergillus niger MA-132. J. Nat. Prod. 2011, 74, 1787–1791. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Zhu, T.; Du, L.; Zhao, B.; Li, D.; Gu, Q. Sterigmatocystins from the deep-sea-derived fungus Aspergillus versicolor. J. Antibiot. 2011, 64, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Lin, W.L.; Yang, P.; Mao, S.C. A novel citrinin derivative from the marine-source fungus Penicillium citrinum. Yao Xue Xue Bao 2015, 50, 203–206. [Google Scholar] [PubMed]
- Stout, E.P.; Hasemeyer, A.P.; Lane, A.L.; Davenport, T.M.; Engel, S.; Hay, M.E.; Fairchild, C.R.; Prudhomme, J.; le Roch, K.; Aalbersberg, W.; et al. Antibacterial neurymenolides from the Fijian red alga Neurymenia fraxinifolia. Org. Lett. 2009, 11, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Franchin, M.; Rosalen, P.L.; da Cunha, M.G.; Silva, R.L.; Colón, D.F.; Bassi, G.S.; de Alencar, S.M.; Ikegaki, M.; Alves-Filho, J.C.; Cunha, F.Q.; et al. Cinnamoyloxy-mammeisin isolated from geopropolis attenuates inflammatory process by inhibiting cytokine production: involvement of MAPK, AP-1, and NF-κB. J. Nat. Prod. 2016, 79, 1828–1833. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.Y.; Kim, Y.C. Neuroprotective coumarins from the root of Angelica gigas: Structure-activity relationships. Arch. Pharm. Res. 2007, 30, 1368–1373. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Hiep, N.T.; Kim, D.W.; Hong, S.; Guo, Y.; Hwang, B.Y.; Lee, H.J.; Mar, W.; Lee, D. Chemical constituents isolated from the root bark of Cudrania tricuspidata and their potential neuroprotective effects. J. Nat. Prod. 2016, 79, 1938–1951. [Google Scholar] [CrossRef] [PubMed]
- Mackey, K.; Pardo, L.M.; Prendergast, A.M.; Nolan, M.T.; Bateman, L.M.; McGlacken, G.P. Cyclization of 4-phenoxy-2-coumarins and 2-pyrones via a double C-H activation. Org. Lett. 2016, 18, 2540–2543. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 a | 2 b | 3 a | |||
|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 165.3, C | 166.2, C | 166.3, C | |||
| 2 | 97.5, C | 98.3, C | ||||
| 3 | 164.5, C | 164.7, C | 149.2, C | |||
| 4 | 103.6, CH | 6.17, s | 104.1, CH | 6.28, s | 108.4, CH | 7.21, s |
| 5 | 155.7, C | 156.6, C | 103.4, CH | 6.69, s | ||
| 6 | 120.2, C | 112.5, C | 163.8, C | |||
| 7 | 155.4, C | 156.7, C | 111.0, C | |||
| 8 | 113.3, CH | 6.76, d (8.2) | 100.4, CH | 6.34, s | 159.9, C | |
| 9 | 130.5, CH | 7.16, dd (8.2, 7.5) | 159.3, C | 98.7, C | ||
| 10 | 120.6, CH | 6.73, d (7.5) | 109.0, CH | 6.30, s | 135.7, C | |
| 11 | 137.8, C | 139.6, C | 8.1, CH3 | 2.04, s | ||
| 12 | 19.4, CH3 | 2.15, s | 19.4, CH3 | 2.17, s | 162.1, C | |
| 13 | 8.5, CH3 | 1.83, s | 7.8, CH3 | 1.92, s | ||
| OH-3 | 11.21, s | |||||
| OH-7 | 9.73, s | |||||
| OH-8 | 11.39, br s | |||||
| Position | 4 a | 5 b | ||
|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 166.3, C | 152.5, C | ||
| 2 | 124.8, CH | 6.24, dd (9.7, 2.5) | ||
| 3 | 154.6, C | 127.9, CH | 6.09, ddd (9.7, 6.5, 2.6) | |
| 4 | 105.3, CH | 6.47, s | 31.4, CH2 | 2.34, ddd (18.0, 6.1, 3.5); 2.16, ddt (18.0, 10.3, 2.5) |
| 5 | 101.5, CH | 6.44, s | 71.6, CH | 4.11, m |
| 6 | 163.5, C | 20.3, CH3 | 1.34, d (6.2) | |
| 7 | 109.6, C | 100.6, CH | 5.86, s | |
| 8 | 159.9, C | 139.3, C | ||
| 9 | 97.8, C | 111.1, C | ||
| 10 | 136.4, C | 163.5, C | ||
| 11 | 7.9, CH3 | 2.01, s | 108.8, C | |
| 12 | 41.7, CH2 | 2.47, m | 162.4, C | |
| 13 | 64.8, CH | 4.02, m | 108.6, CH | 7.08, s |
| 14 | 46.4, CH2 | 1.40, m | 193.8, CH | 10.08, s |
| 15 | 62.7, CH | 3.83, m | 6.5, CH3 | 2.04, s |
| 16 | 24.5, CH3 | 0.90, d (6.1) | ||
| OH-6/10 | 10.84, s | 12.87, s | ||
| OH-8/12 | 11.33, s | 9.32, s | ||
| OH-13 | 4.72, d (5.6) | |||
| OH-15 | 4.38, d (4.9) | |||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, L.-Y.; Liu, D.-S.; Li, D.-G.; Huang, Y.-L.; Kang, H.-H.; Wang, C.-H.; Liu, W.-Z. Pyran Rings Containing Polyketides from Penicillium raistrickii. Mar. Drugs 2017, 15, 2. https://doi.org/10.3390/md15010002
Ma L-Y, Liu D-S, Li D-G, Huang Y-L, Kang H-H, Wang C-H, Liu W-Z. Pyran Rings Containing Polyketides from Penicillium raistrickii. Marine Drugs. 2017; 15(1):2. https://doi.org/10.3390/md15010002
Chicago/Turabian StyleMa, Li-Ying, De-Sheng Liu, De-Guo Li, Yu-Ling Huang, Hui-Hui Kang, Chun-Hua Wang, and Wei-Zhong Liu. 2017. "Pyran Rings Containing Polyketides from Penicillium raistrickii" Marine Drugs 15, no. 1: 2. https://doi.org/10.3390/md15010002
APA StyleMa, L.-Y., Liu, D.-S., Li, D.-G., Huang, Y.-L., Kang, H.-H., Wang, C.-H., & Liu, W.-Z. (2017). Pyran Rings Containing Polyketides from Penicillium raistrickii. Marine Drugs, 15(1), 2. https://doi.org/10.3390/md15010002