1. Introduction
Natural products are an important resource for drug discovery. Isolation, identification, and syntheses of these products or their derivatives, as well as their subsequent biological studies are of interest. In recent years, marine natural products have attracted much interest from scientists. A vast range of chemically diverse biologically active compounds that have antibacterial and anticancer activities have been discovered [
1,
2,
3,
4]. The increasingly serious problem of bacterial resistance toward antibiotics has made the search for new antimicrobial agents from natural sources urgent. Consequently, novel and effective antimicrobial compounds have attracted much interest of scientists since its isolation. In 2011, the antimicrobial compounds ieodomycins A–D were first isolated by Shin and coworkers from the EtOAc extract of a marine strain of
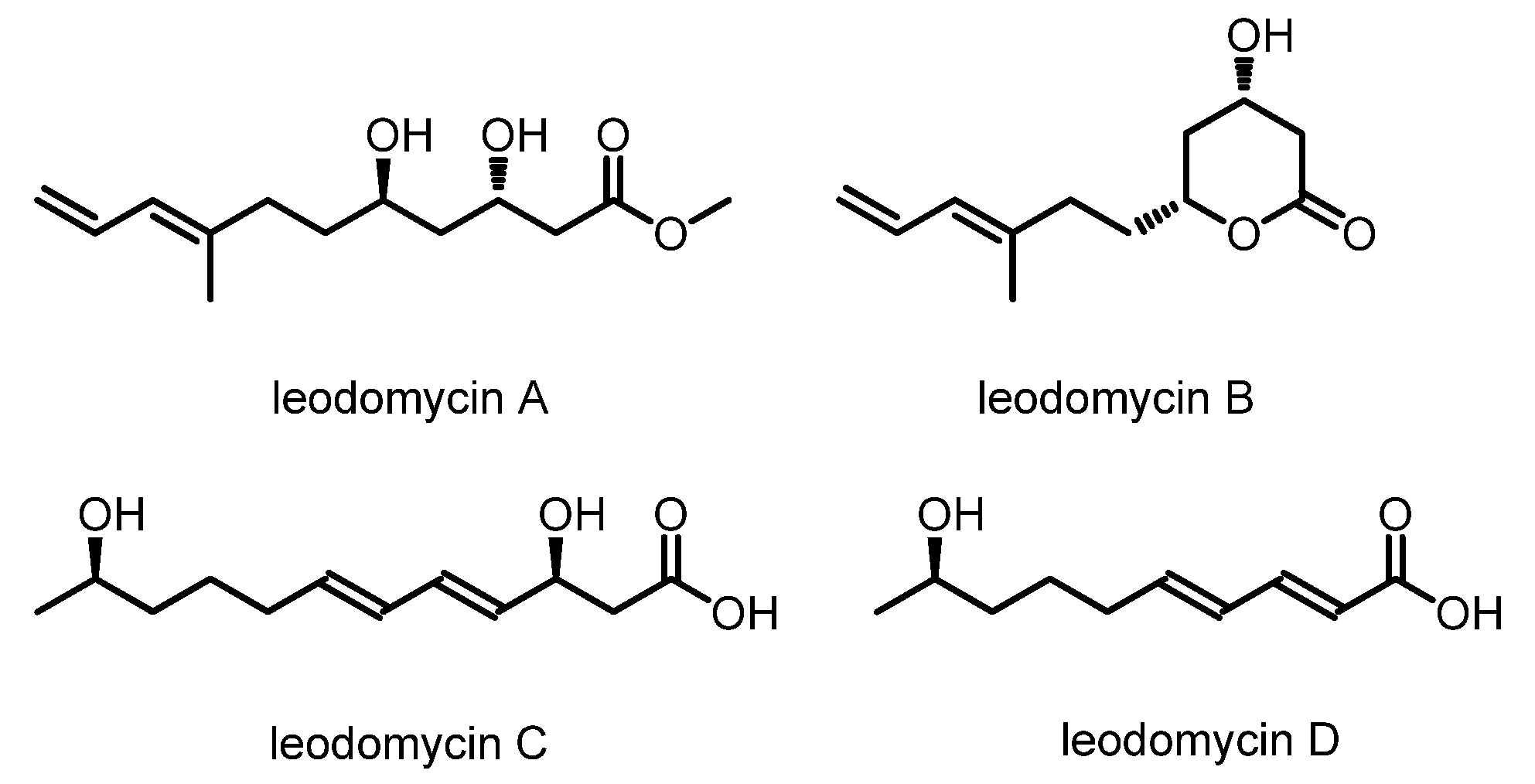
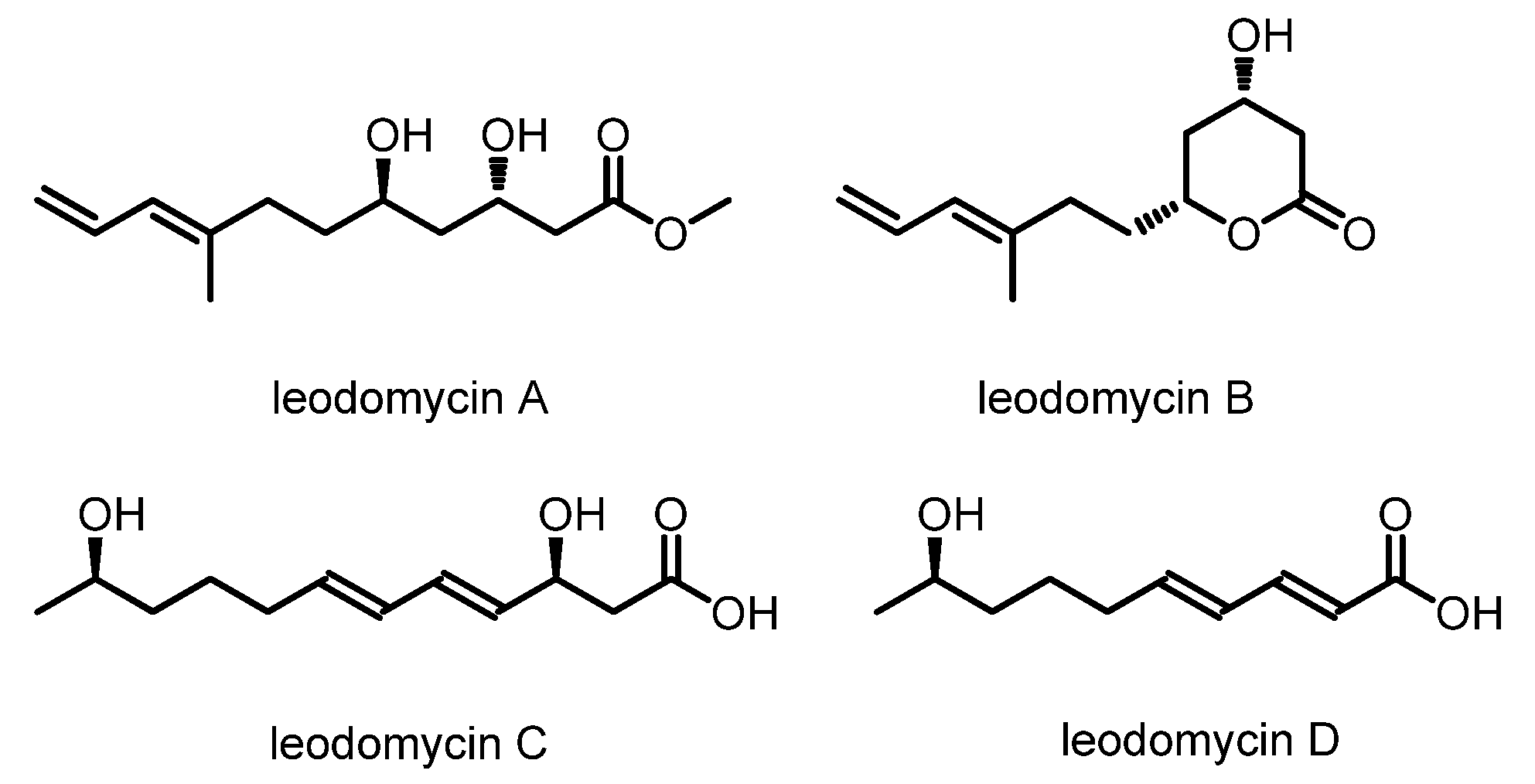
Bacillus species. Through NMR and HRMS analysis and modified Mosher’s method, the planar structures and the absolute configurations were determined, as shown in
Figure 1. Subsequent in vitro antimicrobial experiments showed that all of these compounds possess a broad spectrum of antimicrobial activities. They are active against
Bacillus subtilis and
Escherichia coli, with minimum inhibitory concentrations (MICs) of 32–64 μg/mL [
5].
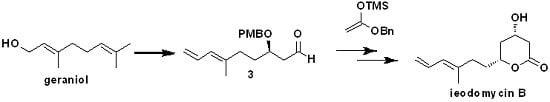
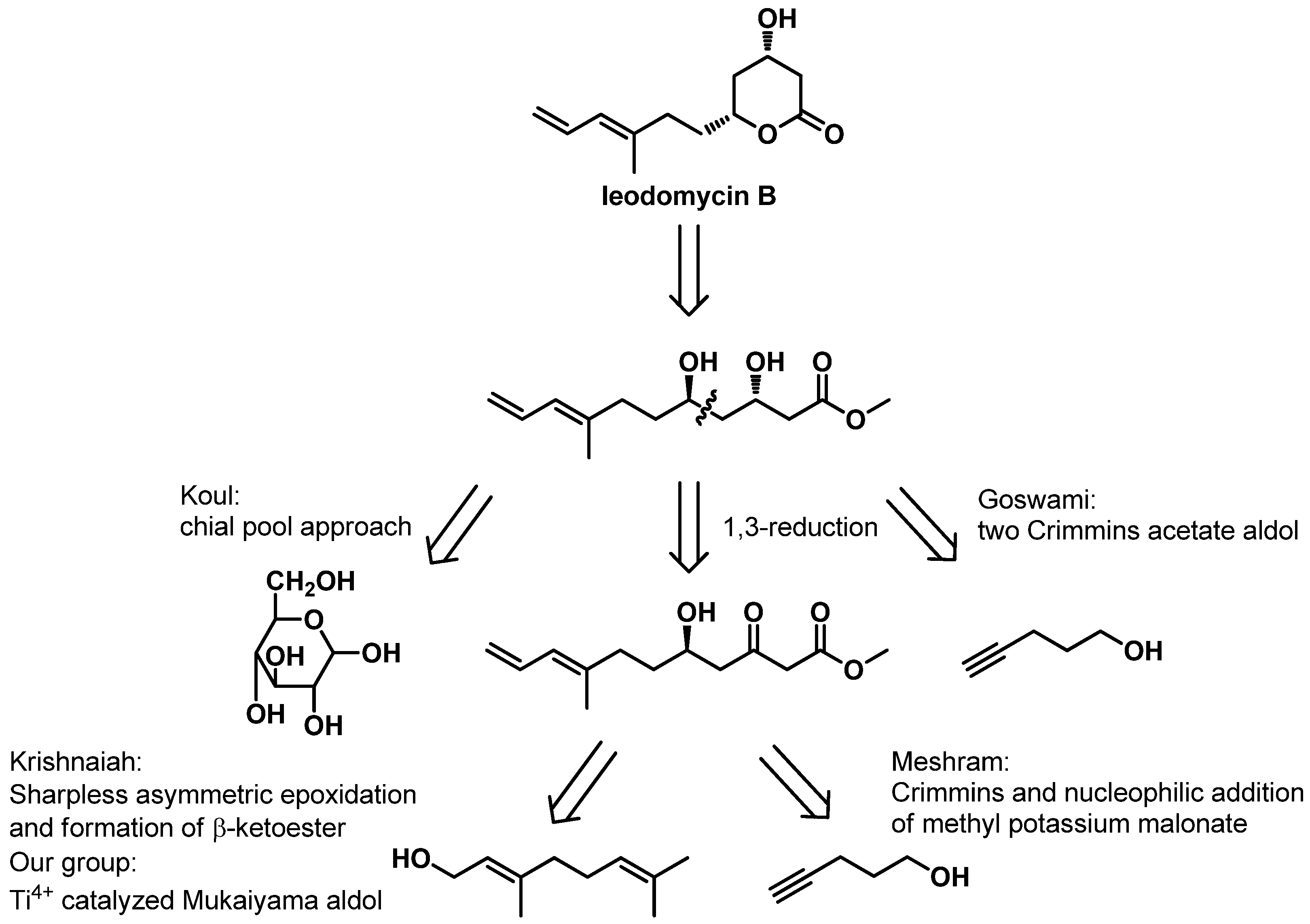
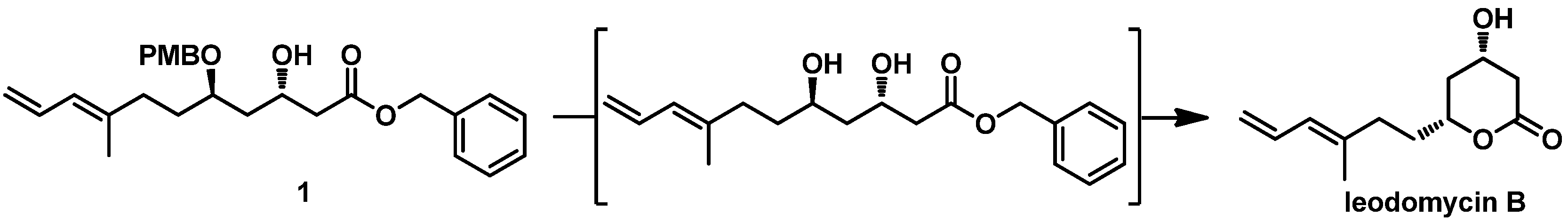
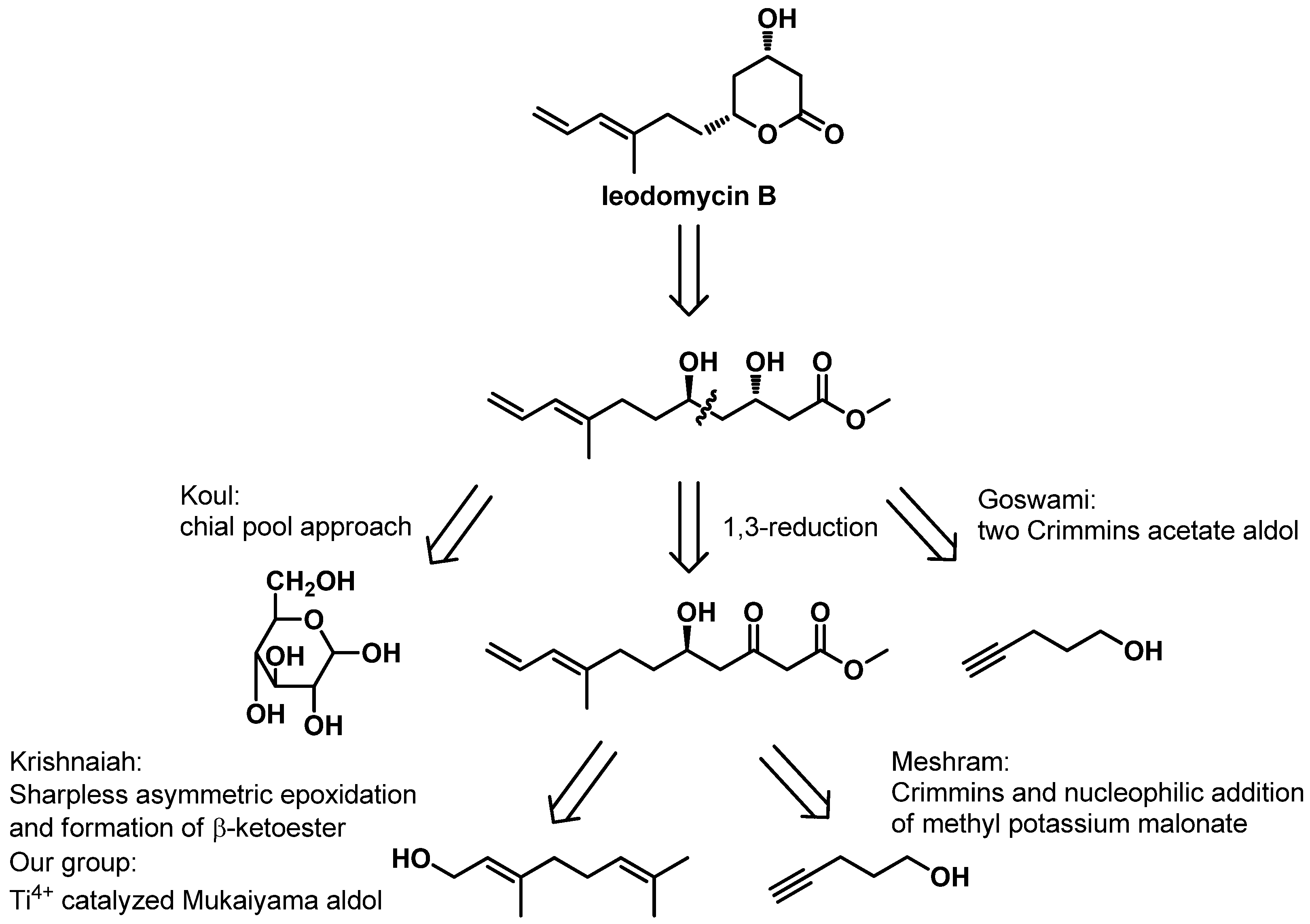
Ieodomycin B has a potential therapeutic application as an antimicrobial agent, and it has an extremely low isolation yield (3.4 mg of ieodomycin B was obtained from 100 L of culture broth) and the greatest complexity among all ieodomycins. Consequently, it has attracted attention from chemical researchers. Several approaches to the synthesis of antimicrobial fatty acids have been developed. As depicted in
Scheme 1, four groups besides our own have achieved different total syntheses of ieodomycin B in the same year. Koul first reported the total synthesis of ieodomycin B using the chiral-pool approach starting from
d-glucose, which consists of more than 15 steps [
6]. Almost simultaneously, Krishnaiah published another stereoselective total synthetic route for ieodomycin B starting from geraniol, which also requires 15 steps [
7]. In Krishnaiah’s route, a protocol that includes Sharpless asymmetric epoxidation-epoxide opening and subsequent 1,3-reduction is used to construct the chiral hydroxyl groups at the C-3 and C-5 positions. Meshram described a novel protection-free, stereoselective, eight-step synthesis of ieodomycin B from commercially available 4-pentyne-1-ol [
8]. This route involves a Crimmins-modified Evans aldol reaction and a nucleophilic addition of the potassium salt of monomethyl malonate, forming the δ-hydroxyl β-keto ester. It then ends in a 1,3-reduction, affording the desired chiral centers. Goswami and coworkers achieved ieodomycin B in nine linear steps starting from the same alcohol. In their route, they used the Crimmins-modified Evans aldol reaction twice to construct the two chiral hydroxyls [
9]. Recently, our group reported a short total synthesis of ieodomycin B involving seven steps [
10]. In this approach, we used Ti
4+-catalyzed asymmetric Mukaiyama aldol reaction and the subsequent 1,3-induced reduction to construct the chiral centers. However, its enantioselectivity is only 88% ee. This unsatisfactory result prompted us to develop an alternative route to ieodomycin B.
In continuation of our studies on developing a new approach toward the synthesis of lactones and on obtaining the natural product at a higher optical purity, we focused on the total synthesis of ieodomycin B. Although the chemical structure of ieodomycin B appears to be simple, a novel and effective protocol to its total synthesis is important. It is well-known that chelation-controlled Mukaiyama aldol addition using alkoxy groups such as
p-methoxybenzyl (PMB) ether or benzyl (Bn) ether is a very important methodology for the highly diastereoselective construction of the
syn/
anti-diol unit (for an example, see [
11]). Using this methodology, we developed in the present study a total synthesis of ieodomycin B using a Crimmins-modified Evans aldol reaction. This reaction is followed by a Mukaiyama aldol reaction induced by PMB ether to selectively construct C-5 and C-3 chiral centers.
2. Results
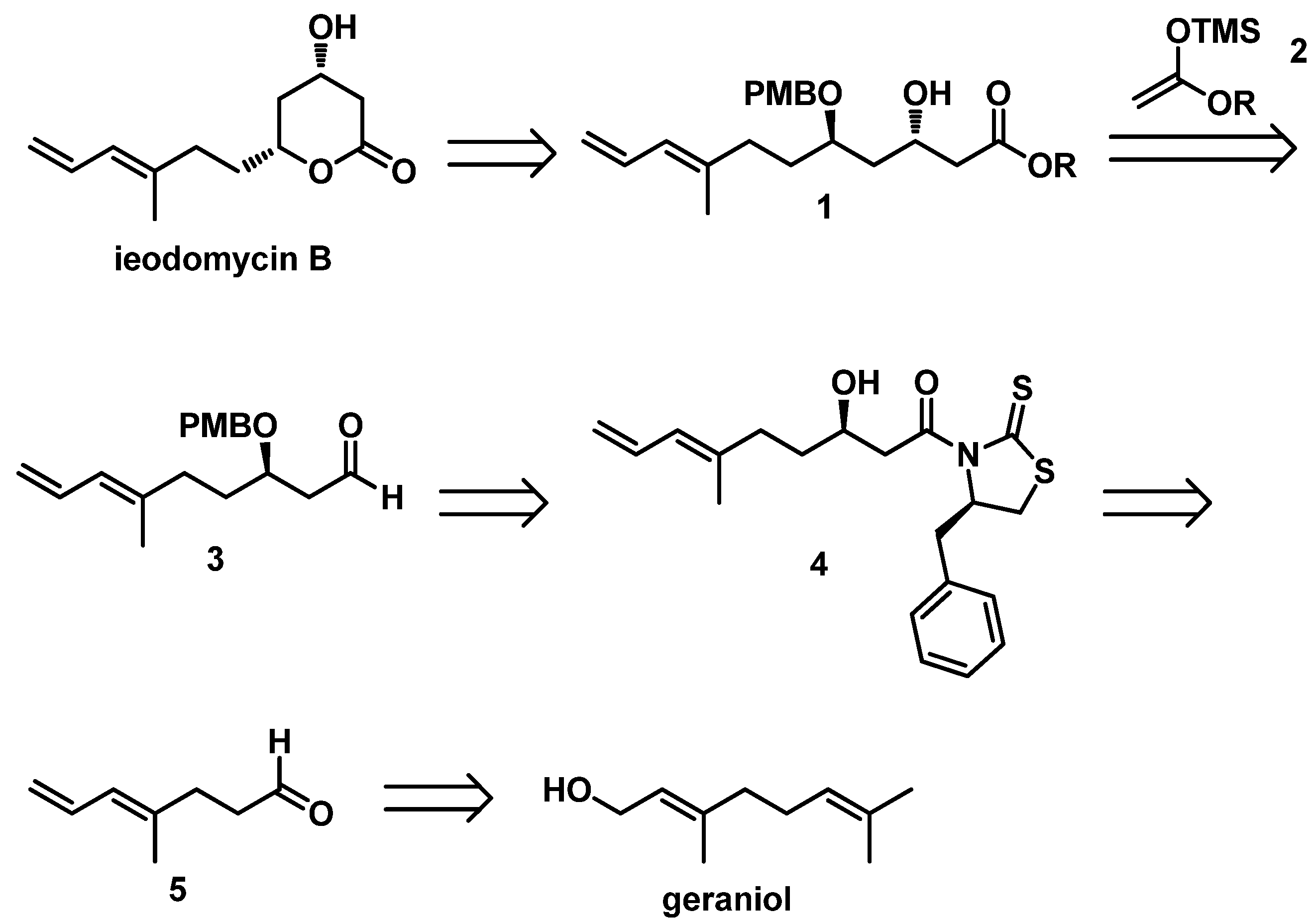
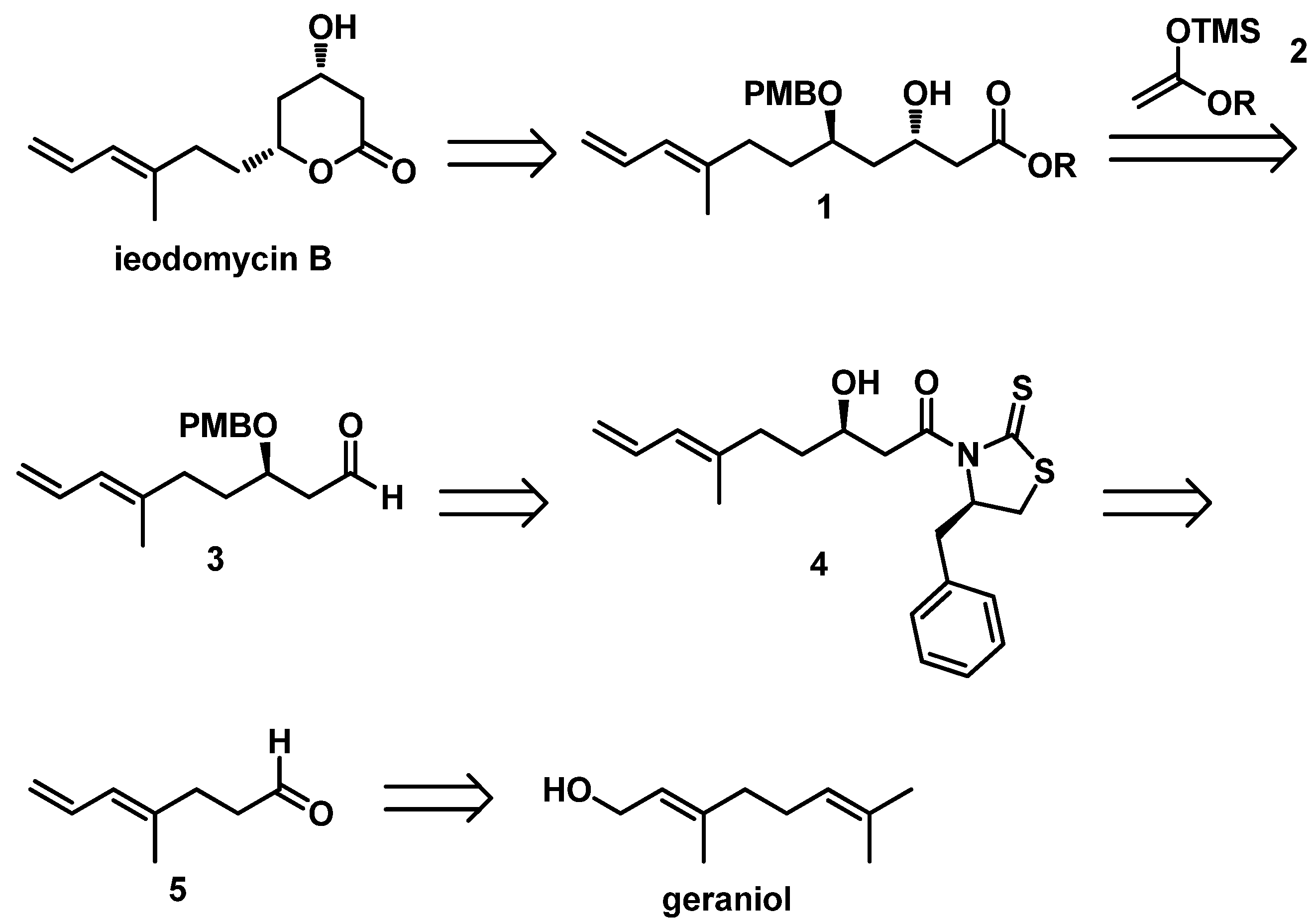
On the basis of the above considerations, we utilized the 1,3-asymmetrically inductive Mukaiyama aldol reaction in the total synthesis of ieodomycin B. Therefore, we performed our retrosynthetic analysis based on this strategy, as depicted in
Scheme 2. We envisioned retrosynthetically that the target molecule ieodomycin B could be prepared from the intermediate
1, which is suitable for disconnection from the protected aldehyde
3 and silyl enol ether
2 via the 1,3-inductive Mukaiyama aldol reaction. We noticed that
1 has
trans configuration. Considering that the chelation-controlled step and deprotection are simple, we decided to protect the hydroxyl in
1 using PMB, in accordance with the studies of Munro et al. [
12,
13]. The total synthesis was mainly focused on the formation of the precursor
3. We propose that
3 can be obtained through a few conversion steps from
4, which can be prepared by a Crimmins-modified Evans aldol reaction of aldehyde
5. Compound
5 could be easily prepared from geraniol.
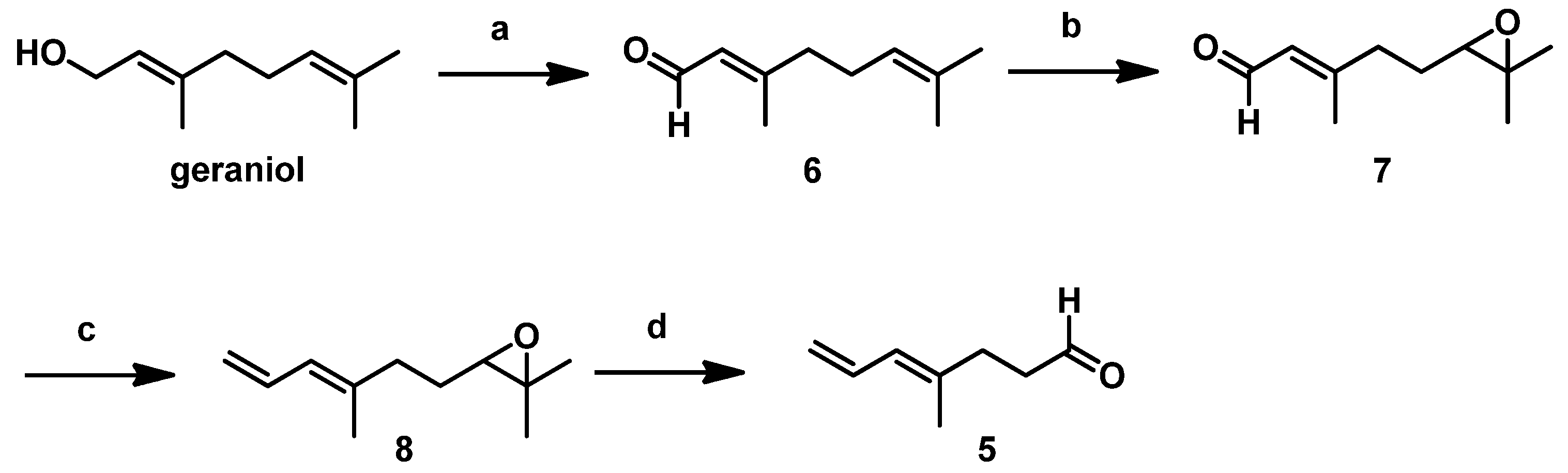
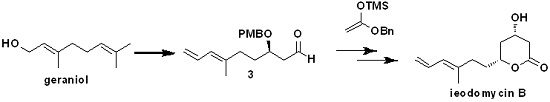
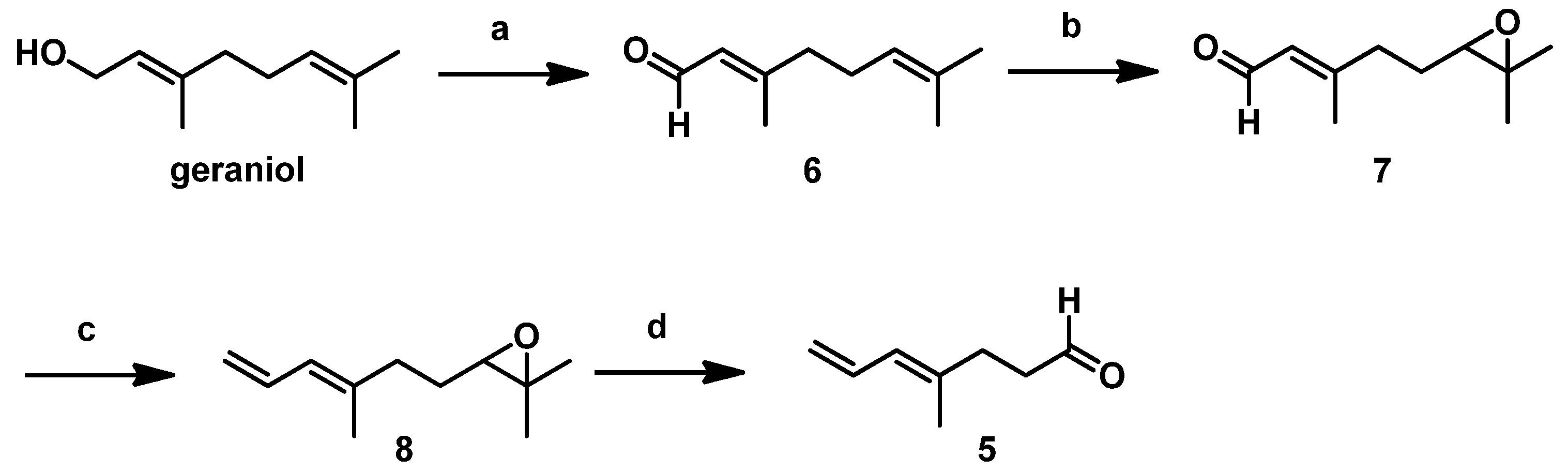
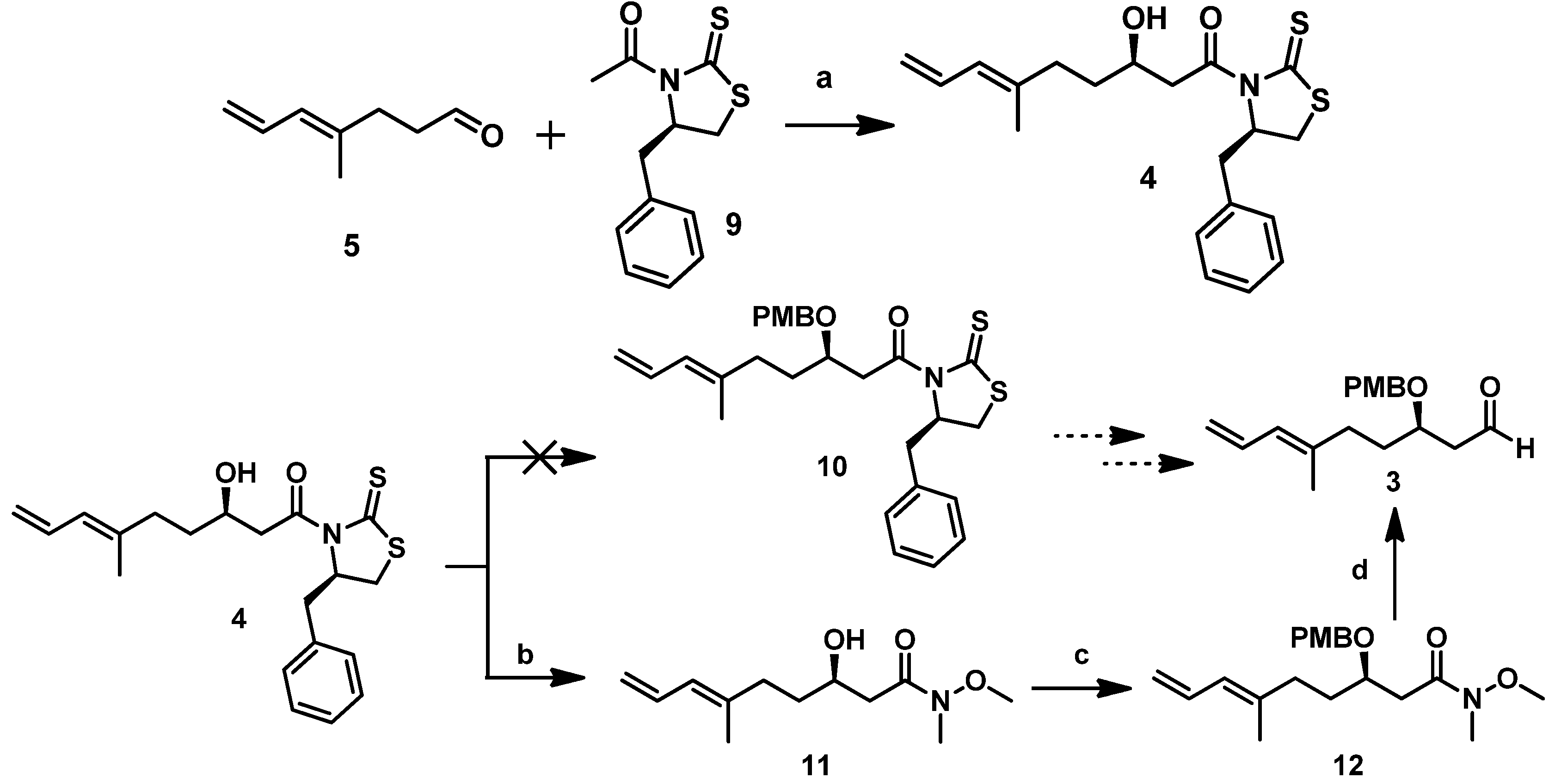
The detailed synthetic route for
5 starting from commercially available geraniol followed procedures described in our previous report [
10]. As shown in
Scheme 3, geraniol underwent Swern oxidation, regioselective epoxidation of the isolated double bond using
m-CPBA, and subsequent Wittig olefination with Ph
3P=CH
2 to the stable epoxide
8. Using HIO
4, we cleaved the resulting epoxide to afford the low-boiling-point
5, which we then immediately subjected to the Crimmins-modified Evans aldol reaction after aborative manipulation.
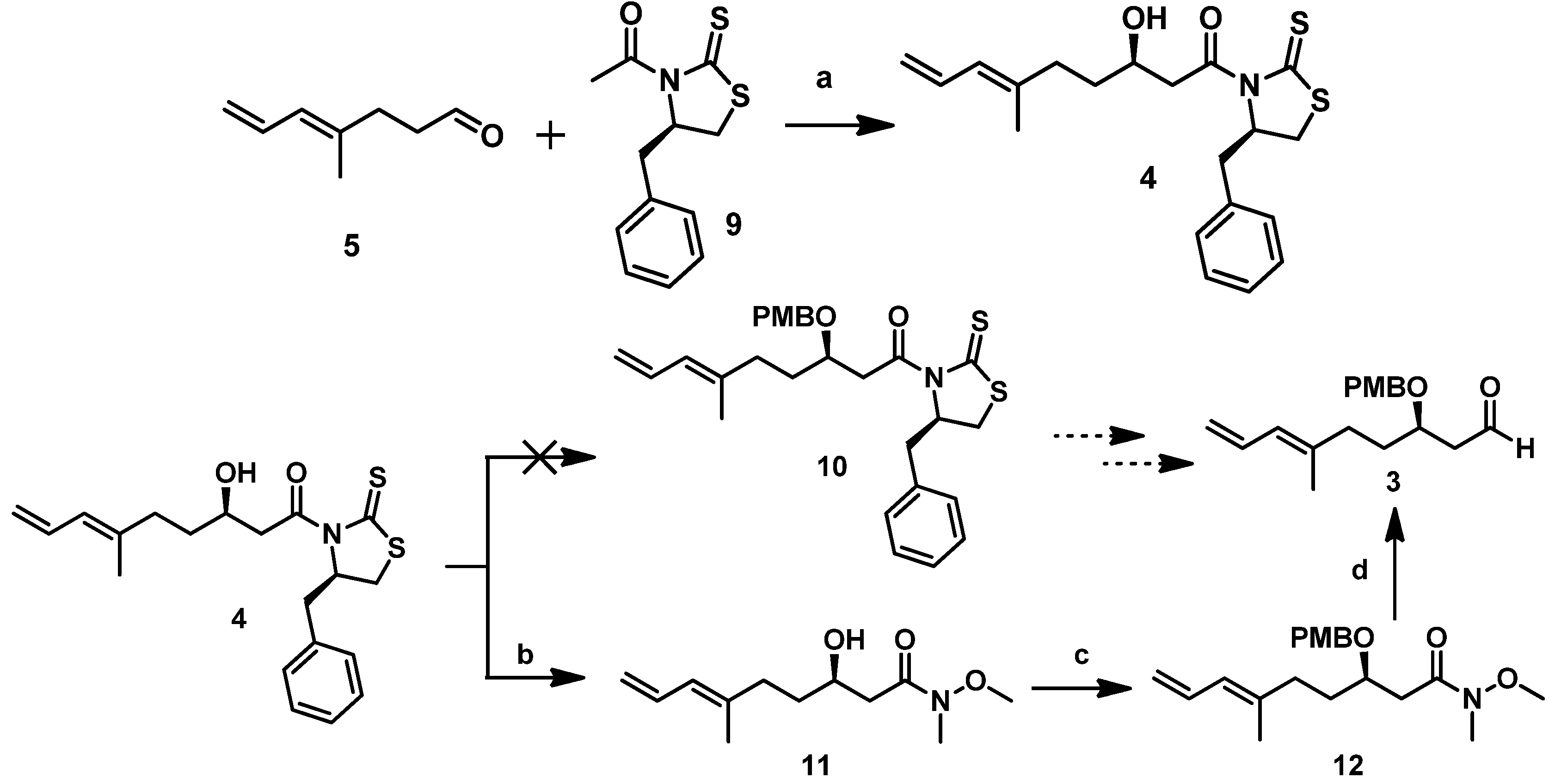
With
5 in hand, secondary alcohol
4 could be obtained by using a Crimmins-modified Evans aldol addition with the acetylthiazolidine thione
9. In the presence of TiCl
4 and (i-Pr)
2NEt (DIPEA), titanium enolate generated from
9 reacted with
5, forming the predominant desired isomer
4 in excellent yield [
8]. As described above, we initially attempted to protect the hydroxyl in
4 using the PMB group under certain conditions, but we could not obtain the desired compound
10. Therefore, we proceeded to convert
4 to a Weinreb amide and then protected it. Direct amidation of
4 with MeONHMe•HCl catalyzed by imidazole yielded Weinreb amide
11. Subsequent treatment of the latter with PMBBr in the presence of NaH afforded the PMB ether
12 in a 67% yield (
Scheme 4). Subsequently,
12 was reduced with DIBAL-H at −78 °C for 1 h to produce the Mukaiyama aldol precursor aldehyde fragment
3 in about a 61% yield.
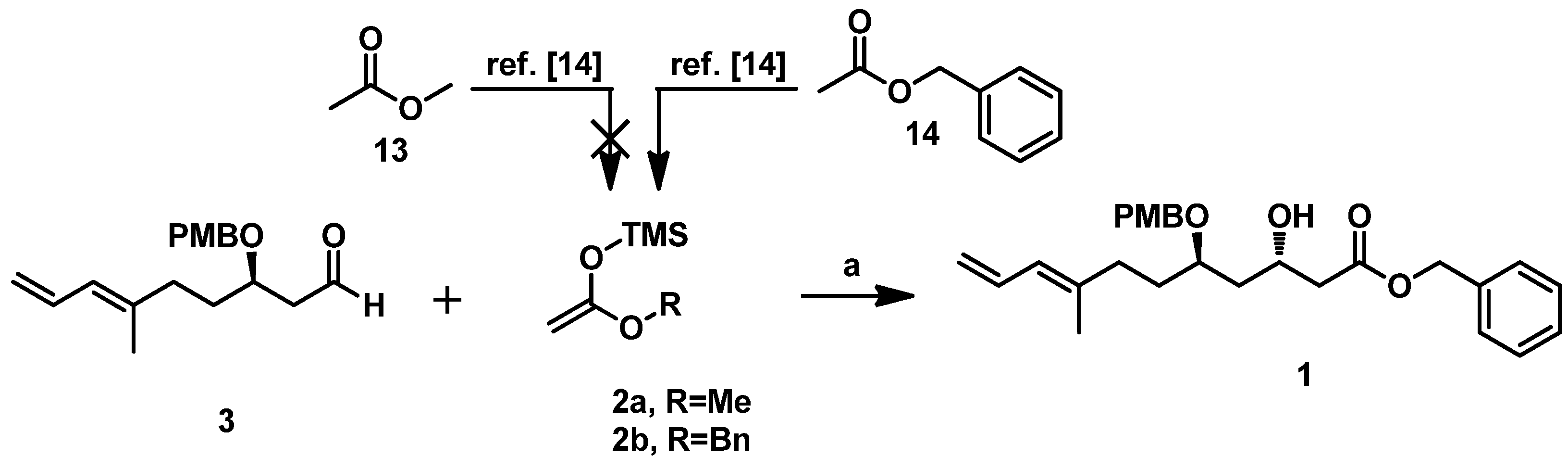
With aldehyde
3 in hand, our next objective was to construct the 1,3-
trans diol unit. We retrosynthetically devised a Mukaiyama aldol reaction induced by a chiral PMB ether in C-5 to selectively construct the C-3 chiral hydroxyl. In our previous study, we found that the mixed titanium species Ti(O
iPr)
2Cl
2 can effectively induce a chelation-controlled Mukaiyama aldol reaction between the β-alkoxy aldehyde and silyl enol ether, affording the 1,3-
trans diol in high yield and with high diastereoselectivity. Meanwhile, common Lewis acids such as BF
3•OEt
2, SnCl
4, TiCl
4, and MgBr
2•OEt
2 afforded 1,3-
trans diol in a low to moderate yield. Thus, we used Ti(O
iPr)
2Cl
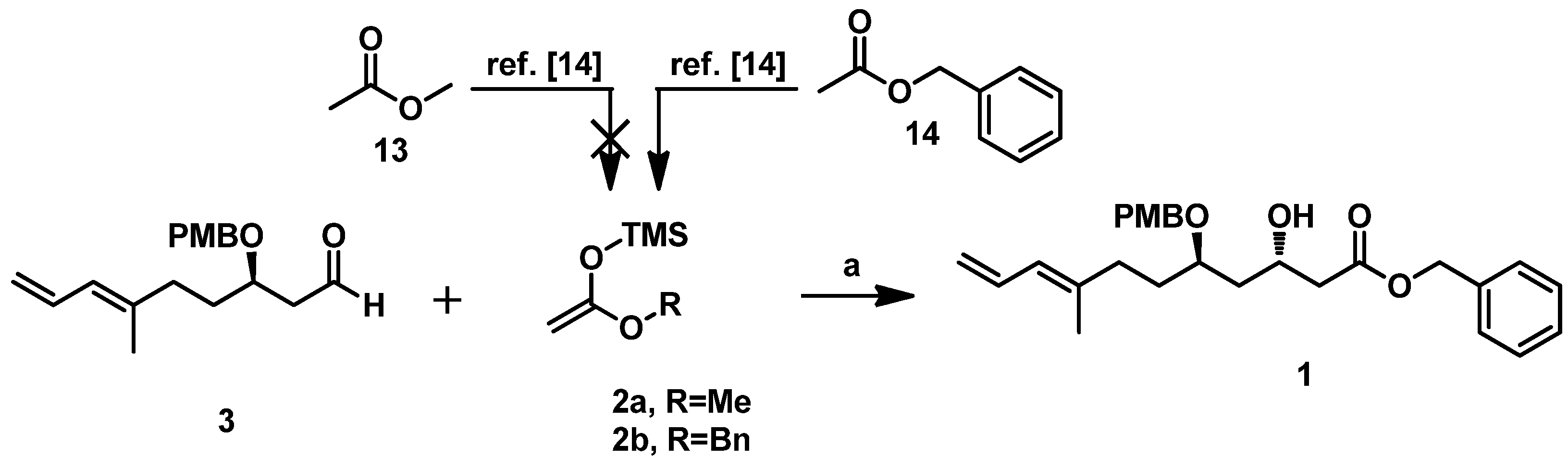
2 in the key Mukaiyama aldol condensation. Considering the possibility of obtaining ieodomycin A and the simplicity of the lactonization, we first attempted to use the ((1-methoxyvinyl)oxy)trimethylsilane
2a as the nucleophilic reagent for reaction with
3, but failed. We could hardly obtain the related silyl enol ether of methyl acetate upon treatment of methyl acetate with lithium diisopropylamide (LDA) and trimethyl chlorosilane (TMSCl). Believing that the ester group effects the reaction, we switched the methyl to the benzyl group and thus obtained the desired silyl enol ether
2b [
14]. Reaction of the unpurified crude product of silyl–enol etherification of
2b with
3 was carried out at −78 °C in the presence of the catalyst Ti(O
iPr)
2Cl
2 (freshly prepared with TiCl
4 and Ti(O
iPr)
4 at 1:1 ratio in the solvent DCM at 0 °C). This step readily produced the desired 1,3-
anti product
1 as the single isomer in about a 72% yield (
Scheme 5).
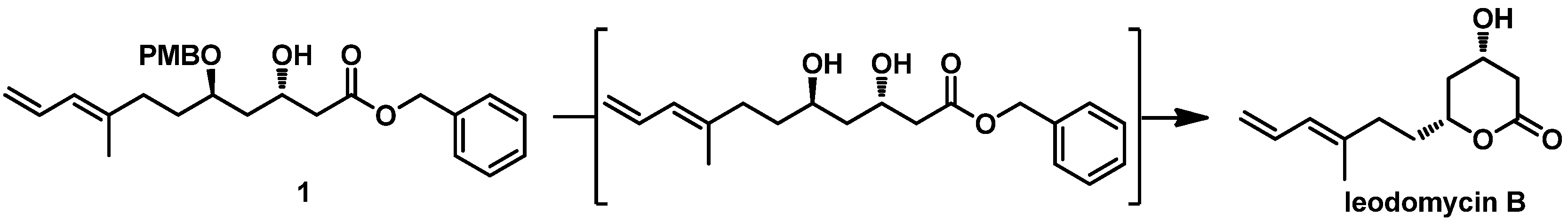
Our final procedure was the removal of the PMB protective group. We originally conducted a stepwise protocol consisting of deprotection and lactonization to obtain ieodomycin B. However, when we used typical reagents such as
p-TsOH, PPTS, DDQ, and CAN, we observed only fuzzy spots in thin-layer chromatography (TLC) along with a decomposition of
1. Finally, we found that brief treatment with trifluoroacetic acid (TFA) in DCM at 0 °C afforded ieodomycin B in a 50% yield (
Figure 2 and
Table 1). Extending the treatment, however, resulted in decomposition and low yield.
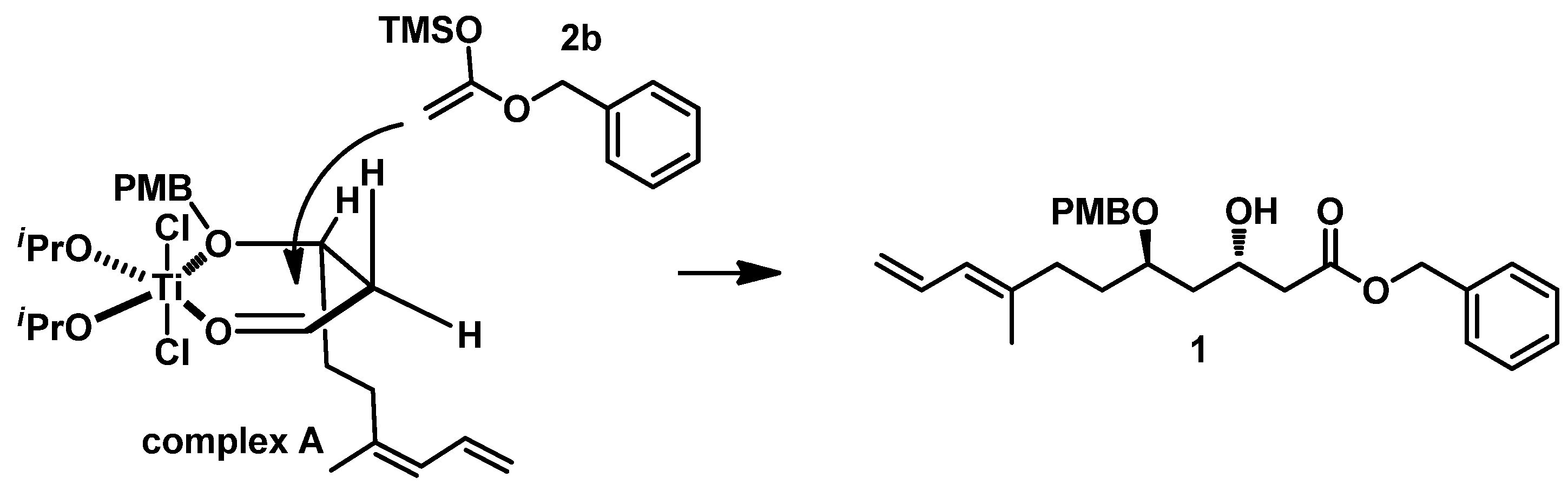
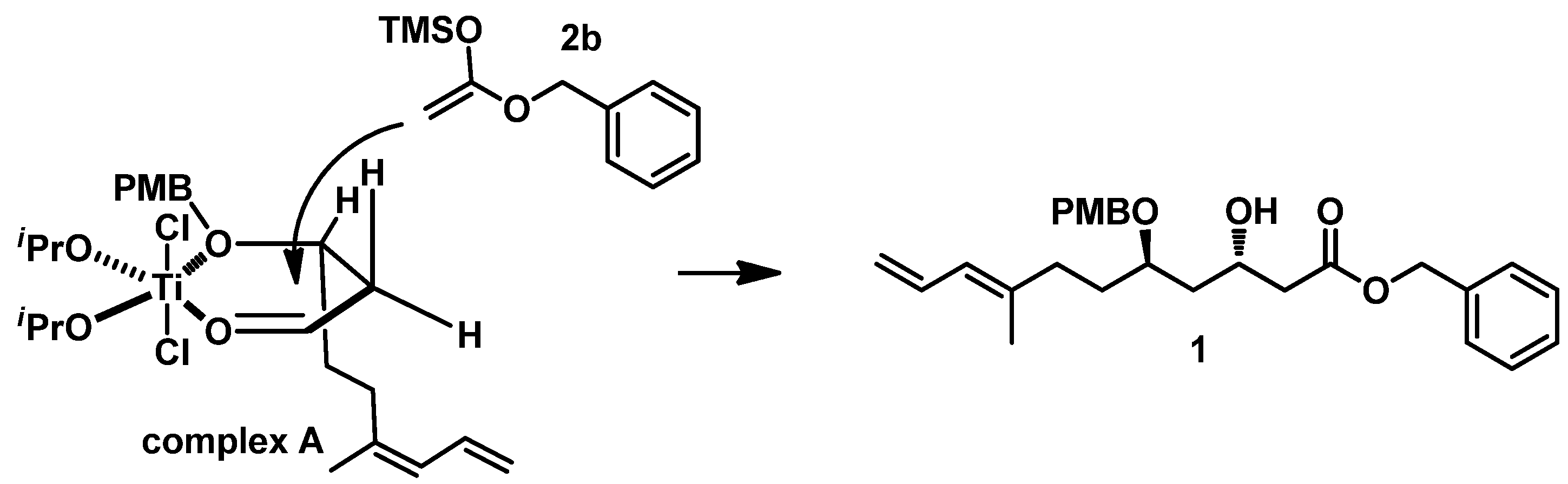
On the basis of Munro’s work [
12], we propose that the highly diastereoselective formation of 1,3-
trans diol arises from the following mechanism (described in
Figure 3): Titanium first forms a chelated complex A with the aldehyde carbonyl in
3 and the ether oxygen of hydroxyl protected by PMB, and the diene group in its preferred conformation occupies a pseudoaxial position. Subsequently, the silyl enol ether ((1-(benzyloxy)vinyl)oxy)trimethylsilane
2b attacks from the less-hindered side, which is opposite to the diene group, giving the
anti-diol product
1.
3. Experimental Section
3.1. General
All reactions were carried out under N2 atmosphere with dry solvents unless otherwise noted and monitored via thin-layer chromatography (TLC) carried out on 0.25 mm silica gel plates (60F-254). Silica gel (200–300 mesh) for flash column chromatography was supplied by Qingdao Marine chemical factory in Qingdao, China. Anhydrous toluene and tetrahydrofuran (THF) was distilled from sodium-benzophenone. Dichloromethane (DCM) and N,N-dimethyl formamide (DMF) was distilled from CaH2. Yield refers to chromatographic and spectroscopic 1H and 13C NMR, unless otherwise stated. NMR spectra were recorded on a Bruker AV 400 NMR spectrometer (Bruker, Fällanden, Switzerland) (1H: 400 MHz, 13C: 100 MHz). High-resolution mass spectra were obtained from an Applied Biosystems Q-Star Elite MALDI-TOF mass spectrometer (Applied Biosystems, Carlsbad, CA, USA). Optical rotations were measured on a Rudolph Autopol IV automatic polarimeter (Rudolph, Hackettstown, NJ, USA) in CHCl3 at 25 °C.
3.2. (R,E)-1-((R)-4-Benzyl-2-thioxothiazolidin-3-yl)-3-hydroxy-6-methylnona-6,8-dien-1-one (4)
To a solution of thiazolidinethione 9 (4.8 g, 19.3 mmol, 1.2 equiv) in anhydrous DCM (60 mL) TiCl4 (3.2 mL, 29.0 mmol, 1.8 equiv) was added dropwise at −40 °C, and the resultant yellowish slurry was stirred for 5 min at the same temperature. Then, DIPEA (4.1 mL, 29.0 mmol, 1.8 equiv) was added slowly. The deep reddish solution was stirred for another 1 h at −40 °C before being cooled to −78 °C, and aldehyde 5 (2.0 g, 16.1 mmol, 1.0 equiv) in DCM (24 mL) was added via a cannula. Stirring of the mixture continued at −78 °C for 20 min and was quenched by saturated aqueous NH4Cl (20 mL). It was then extracted with DCM (3 × 80 mL), washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. Purification via silica gel column chromatography (petroleum ether/EtOAc = 20:1) yielded compound 4 (3.3 g, 55%) as a light yellow oil.
1H NMR (400 MHz, CDCl3), δ 7.28–7.27 (m, 2H), 7.23–7.19 (m, 3H), 6.51 (dt, J = 16.8 Hz, 12.0 Hz, 1H), 5.84 (d, J = 10.4 Hz, 1H), 5.38–5.32 (m, 1H), 5.05 (d, J = 16.8, 1H), 4.93 (d, J = 10.4 Hz, 1H), 4.11–4.06 (m, 1H), 3.60 (dd, J = 17.6 Hz, 2.4 Hz, 1H), 3.28 (dd, J = 11.5 Hz, 7.4 Hz, 1H), 3.12 (dd, J = 13 Hz, 3.1 Hz, 1H), 3.05 (dd, J = 17.7 Hz, 9.4 Hz, 1H), 2.97 (dd, J = 13 Hz, 10.4 Hz, 1H), 2.84 (d, J = 11.6 Hz, 1H), 2.71 (s, 1H), 2.21–2.12 (m, 1H), 2.12–2.08 (m, 1H), 1.72 (s, 3H), 1.67–1.53 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ 201.4, 173.1, 138.6, 136.3, 133.2, 129.4, 128.9, 127.2, 125.9, 115.0, 68.2, 67.5, 45.8, 36.8, 35.6, 34.3, 32.0, 16.6.
3.3. (R,E)-3-Hydroxy-N-methoxy-N,6-dimethylnona-6,8-dienamide (11)
To a solution of compound 4 (3.0 g, 8.0 mmol, 1.0 equiv) in DCM (45 mL), MeONHMe•HCl (3.12 g, 32.0 mmol, 4.0 equiv) and imidazole (2.72 g, 40.0 mmol, 5.0 equiv) were added, and the resultant mixture was stirred at room temperature overnight. When it was clear via TLC that compound 4 had been consumed, the reaction was quenched with a saturated aqueous NH4Cl solution (20 mL), and the resultant mixture was extracted with DCM (3 × 80 mL). The organic layer was then washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. Purification of the residue by silica gel column chromatography (petroleum ether/EtOAc = 3:1) yielded the desired Weinreb amide 11 (1.23 g, 68%) as a yellow oil.
[α = +42.4 (c = 1.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 6.56 (dt, J = 16.8 Hz, 10.7 Hz, 1H), 5.89 (d, J = 10.8 Hz, 1H), 5.09 (d, J = 16.8, 1H), 4.98 (d, J = 10.0 Hz, 1H), 4.04–3.96 (m, 1H), 3.83–3.80 (m, 1H), 3.68 (s, 3H), 3.19 (s, 3H), 2.69–2.65 (m, 1H), 2.50–2.43 (m, 1H), 2.34–2.20 (m, 1H), 2.20–2.10 (m, 1H), 1.77 (s, 3H), 1.74–1.52 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ 173.8, 139.0, 133.2, 125.7, 114.8, 67.5, 61.2, 38.1, 35.6, 34.6, 31.8, 16.6. HRMS (ESI): m/z calcd. for C12H21NO3Na [M + Na]+ 250.1414, found 250.1416.
3.4. (R,E)-N-Methoxy-3-((4-methoxybenzyl)oxy)-N,6-dimethylnona-6,8-dienamide (12)
NaH (0.21 g, 5.28 mmol, 1.5 equiv, 60% in mineral oil) was added to the solution of compound 11 (0.80 g, 3.52 mmol, 1.0 equiv) in DMF (12 mL) at −15 °C and PMB-Br (1.03 mL, 7.04 mmol, 2.0 equiv, stabilized with 5 wt % K2CO3). The suspension was stirred at −15 °C for 1.5 h until it was observed via TLC that compound 11 was consumed; then, it was poured onto H2O (5 mL) and ether (3 × 40 mL). The layers were separated and the organic layer was then washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated at reduced pressure. The residue was purified via silica gel column chromatography (petroleum ether/EtOAc = 10:1) to yield PMB ether 12 (0.82 g, 67%) as a yellow oil.
[α = −7.5 (c = 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.26 (d, J = 8.0 Hz, 2H), 6.86 (d, J = 8.0 Hz, 2H), 6.48 (dt, J = 16.8 Hz, 10.5 Hz, 1H), 5.85 (d, J = 10.8 Hz, 1H), 5.08 (d, J = 16.8, 1H), 4.98 (d, J = 10.1 Hz, 1H), 4.47 (q, J = 10.8 Hz, 2H), 3.98–3.92 (m, 1H), 3.79 (s, 3H), 3.66 (s, 3H), 3.19 (s, 3H), 2.86 (dd, J = 14.6, 6.2 Hz, 1H), 2.48 (dd, J = 15.0, 5.3 Hz, 1H), 2.25–2.07 (m, 2H), 1.75 (s, 3H), 1.80–1.66 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ 159.1, 139.1, 133.3, 130.8, 129.4, 125.6, 114.7, 113.7, 75.6, 71.6, 61.3, 55.2, 37.2, 35.4, 33.1, 29.7, 16.6. HRMS (ESI): m/z calcd. for C20H29NO4Na [M + Na]+ 370.1989, found 370.1989.
3.5. (R,E)-3-((4-Methoxybenzyl)oxy)-6-methylnona-6,8-dienal (3)
DIBAL-H (1.20 mL, 1.80 mmol, 1.5 M in toluene, 1.3 equiv) was added dropwise to a solution of the Weinreb amide 12 (0.57 g, 1.64 mmol, 1.0 equiv) in freshly distilled DCM (25 mL) at −78 °C. Stirring of the reaction continued at −78 °C for 1 h until it was observed via TLC that compound 12 had been consumed. Then, it was quenched with a saturated aqueous NaCl solution (5 mL). The mixture was extracted with DCM (3 × 20 mL), dried over anhydrous Na2SO4, filtered, and concentrated at reduced pressure. The residue was purified via silica gel column chromatography (petroleum ether/EtOAc = 20:1) to afford aldehyde 3 as a yellow oil (0.29 g, 61%). The aldehyde 3 was unstable so it did not characterize and was used in the next step immediately.
3.6. (3S,5R,E)-Benzyl 3-hydroxy-5-((4-methoxybenzyl)oxy)-8-methylundeca-8,10-dienoate (1)
To a solution of Ti(OiPr)4 (10.1 mL, 33.8 mmol) in toluene (30 mL), TiCl4 (3.37 mL, 30.7 mmol) was added dropwise. The solution was stirred at ambient temperature for 30 min, and the resultant 1 M Ti(OiPr)2Cl2 solution was used in the next step.
The above freshly prepared Ti(O
iPr)
2Cl
2 (2.08 mL, 2.08 mmol, 0.6 equiv) solution was cooled to −78 °C to yield a milky white slurry. This slurry was then treated with a solution of aldehyde
3 (1.0 g, 3.47 mmol, 1 equiv) in toluene (25 mL) via a cannula over the course of 10 min at −78 °C. The resultant pale yellow homogeneous solution was stirred at −78 °C for 15 min before being treated with a solution of the ((1-(benzyloxy)vinyl)oxy)trimethylsilane
2b (4.63 g, 20.8 mmol, 6.0 equiv, prepared following [
14]) in toluene (6 mL) via a cannula. The bright yellow reaction mixture was then stirred at −78 °C for another 40 min before being quenched with a saturated aqueous NaHCO
3 solution (20 mL) when it was observed via TLC that compound
3 was consumed. The mixture was warmed to room temperature and extracted with DCM (3 × 50 mL). The organic extracts were combined, washed with brine, dried over anhydrous Na
2SO
4, filtered, and concentrated in vacuo. The residue was purified via flash chromatography (petroleum ether/EtOAc = 10:1) to yield compound
1 (1.09 g, 72%) as a yellow oil.
[α = +4.1 (c = 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.41–7.35 (m, 5H), 7.28 (d, J = 7.1 Hz, 2H), 6.88 (d, J = 8.3 Hz, 2H), 6.59 (dt, J = 16.9 Hz, 10.6 Hz, 1H), 5.86 (d, J = 10.8 Hz, 1H), 5.17 (s, 2H), 5.12 (d, J = 16.8, 1H), 5.02 (d, J = 10.2 Hz, 1H), 4.54–4.45 (m, 2H), 4.41–4.31 (m, 1H), 3.81 (s, 3H), 3.75–3.70 (m, 1H), 3.39 (brs, 1H), 2.59–2.50 (m, 2H), 2.13–2.09 (m, 2H), 1.78 (s, 3H), 1.83–1.60 (m, 4H); 13C NMR (CDCl3, 100 MHz) δ 172.2, 159.2, 138.9, 135.6, 133.2, 130.3, 129.5, 128.5, 128.2, 128.2, 125.6, 114.9, 113.8, 75.6, 71.0, 66.3, 65.2, 55.2, 41.8, 39.9, 35.2, 31.8, 16.6. HRMS (ESI): m/z calcd. for C27H34NO5Na [M + Na]+ 461.2298, found 461.2302.
3.7. Ieodomycin B
To a solution of compound 1 (0.25 g, 0.57 mmol, 1 equiv) in toluene (15 mL), TFA (0.3 mL, 5.1 mmol, 7.0 equiv) was added dropwise at 0 °C and stirred at 0 °C for 30 min. When it was observed via TLC that compound 1 was consumed, the reaction was quenched with a saturated aqueous NaHCO3 solution (20 mL). The mixture was warmed to room temperature and extracted with DCM (3 × 20 mL). The organic extracts were combined, washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified via flash chromatography (petroleum ether/EtOAc = 1:1) to afford the desired natural product, ieodomycin B (60 mg, 50%), as a light yellow oil.
[α = +22.6 (c = 1, CHCl3 ); 1H NMR (400 MHz, CDCl3): δ 6.53 (ddd, J = 16.8, 10.4, 10.4 Hz, 1H), 5.84 (d, J = 10.4 Hz, 1H), 5.09 (d, J = 16.8 Hz, 1H), 4.98 (d, J = 10.0 Hz, 1H), 4.28–4.02 (m, 2H), 2.85 (dd, J = 16.8 Hz, 3.6 Hz, 1H), 2.44 (dd, J = 16.8 Hz, 7.2 Hz, 1H), 2.32–2.12 (m, 3H), 1.88–1.79 (m, 2H), 1.73 (s, 3H), 1.61–1.51 (m, 1H). 13C NMR (CDCl3, 100 MHz) δ 171.4, 137.5, 132.9, 126.2, 115.4, 76.7, 63.4, 39.3, 37.5, 34.7, 33.4, 16.5. HRMS (ESI): m/z calcd. for C12H18O3Na [M + Na]+ 233.1148, found 233.1149.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}