
Rare Chromones from a Fungal Mutant of the Marine-Derived Penicillium purpurogenum G59

 ,
, 
Abstract
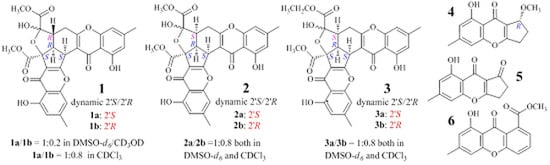
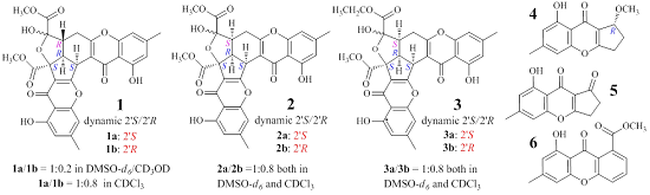
:
1. Introduction
2. Results and Discussion
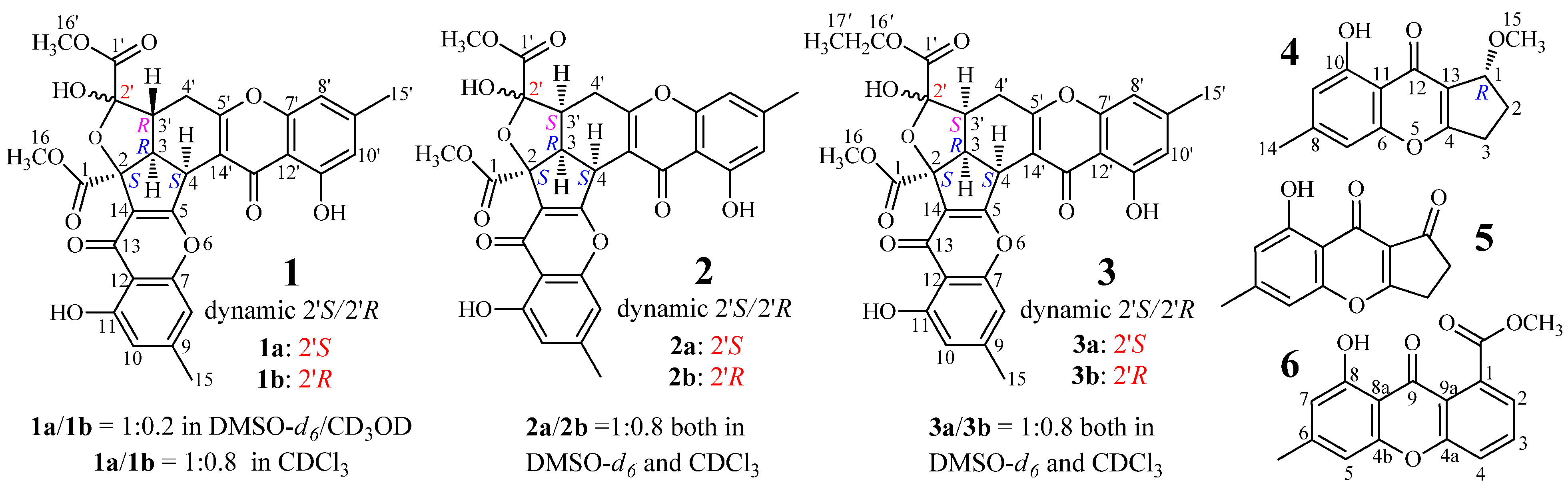
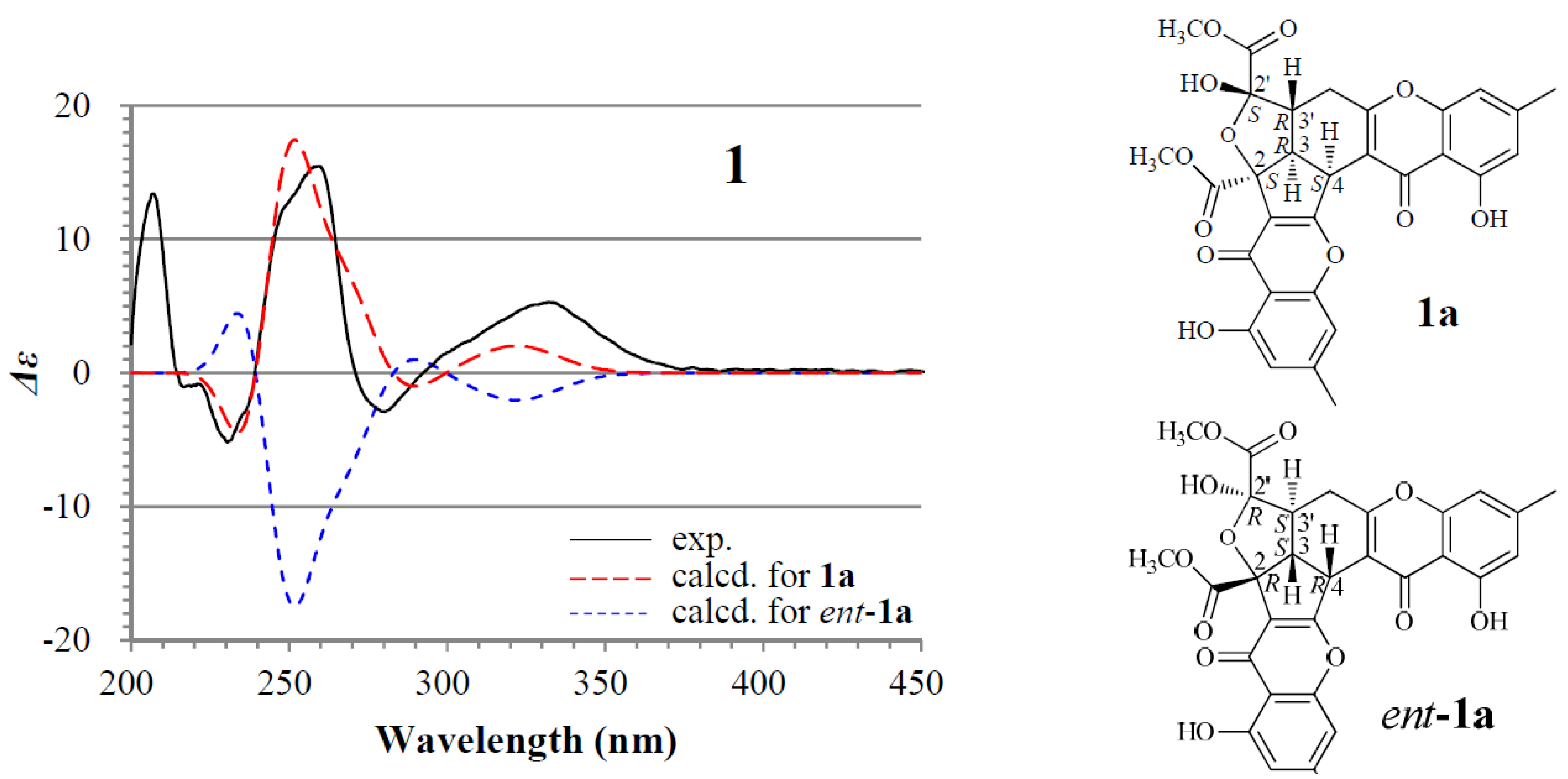
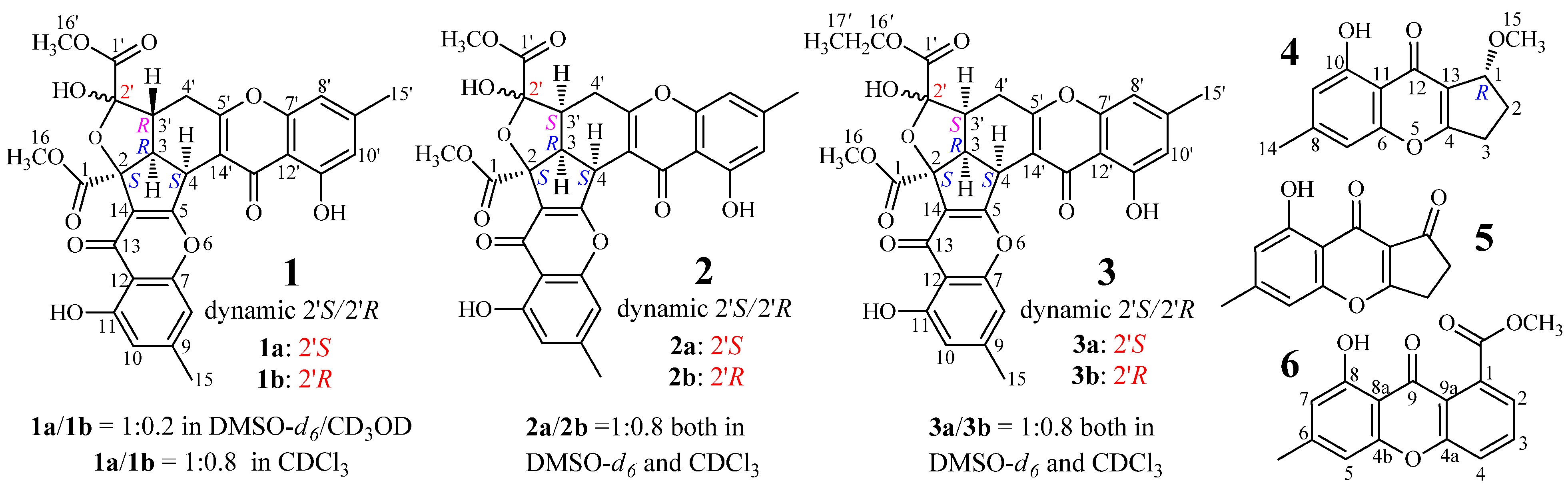
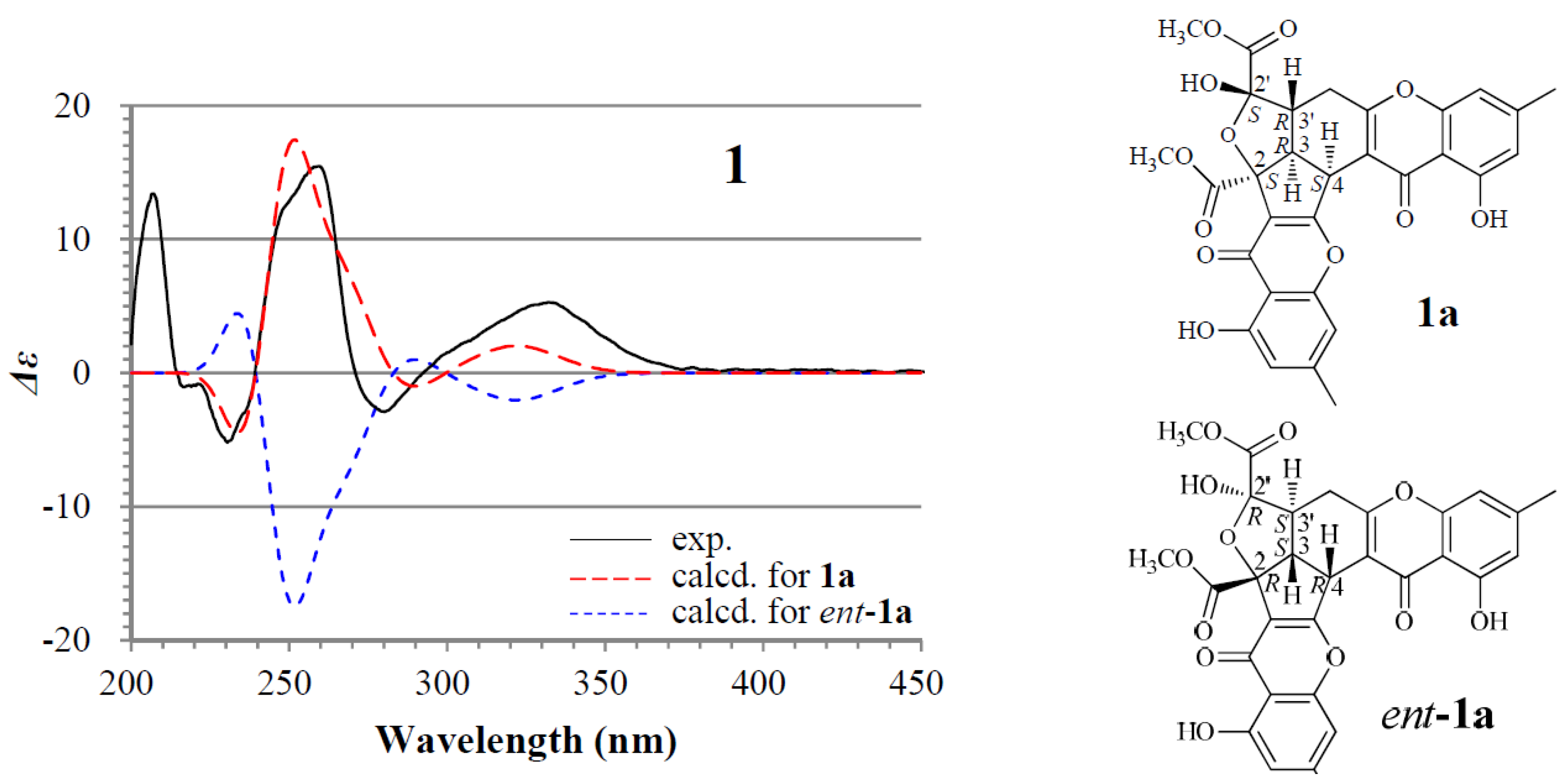
2.1. Identification and Absolute Configuration Determination of Remisporine B (1)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1a b | 2a c | 2b c | 3a c | 3b c | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | — | 170.0 s | — | 170.8, s | — | 171.3 s | — | 170.8 s | — | 171.4 s |
| 2 | — | 86.6 s | — | 88.2 s | — | 89.5 s | — | 88.2 s | — | 89.4 s |
| 3 | 3.29 (dd, 12.2, 6.0) | 47.2 d | 3.78 (dd, 9.2, 9.0) | 47.0 d | 3.88 (dd, 9.0, 8.4) | 46.6 d | 3.81 (dd, 9.4, 8.9) | 47.3 d | 3.87 (dd, 9.0, 8.4) | 46.6 d |
| 4 | 4.60 (d, 6.0) | 39.1 d | 4.98 (d, 9.0) | 36.9 d | 5.02 (d, 9.0) | 36.2 d | 4.99 (d, 8.9) | 36.9 d | 5.01 (d, 9.0) | 36.2 d |
| 5 | — | 171.5 s | — | 169.3 s | — | 168.9 s | — | 168.9 s | — | 168.9 s |
| 7 | — | 156.7 s | — | 156.78 s | — | 156.75 s | — | 156.8 s | — | 156.7 s |
| 8 | 6.86 (s) | 108.3 d | 6.79 (s) | 108.3 d | 6.77 (s) | 108.3 d | 6.81 (s) | 108.3 d | 6.79 (s) | 108.3 d |
| 9 | — | 147.5 s | — | 147.5 s | — | 147.5 s | — | 147.47 s | — | 147.51 s |
| 10 | 6.69 (s) | 112.5 d | 6.64 (s) | 112.3 d | 6.64 (s) | 112.3 d | 6.65 (s) | 112.3 d | 6.65 (s) | 112.3 d |
| 11 | — | 159.8 s | — | 159.87 s | — | 159.85 s | — | 159.9 s | — | 159.8 s |
| 12 | — | 108.5 s | — | 108.4 s | — | 108.4 s | — | 108.4 s | — | 108.4 s |
| 13 | — | 179.7 s | — | 178.87 s | — | 178.89 s | — | 178.8 s | — | 178.9 s |
| 14 | — | 119.3 s | — | 119.2 s | — | 118.7 s | — | 119.3 s | — | 118.7 s |
| 15 | 2.35 (s) | 21.5 q | 2.30 (s) | 21.5 q | 2.28 (s) | 21.4 q | 2.30 (s) | 21.45 q | 2.29 (s) | 21.42 q |
| 16 | 3.71 (s) | 52.7 q | 3.69 (s) | 52.8 q | 3.70 (s) | 52.6 q | 3.68 (s) | 52.6 q | 3.70 (s) | 52.5 q |
| 1′ | — | 168.1 s | — | 169.4 s | — | 167.7 s | — | 169.2, s | — | 167.4 s |
| 2′ | — | 102.8 s | — | 105.7 s | — | 106.2 s | — | 105.7 s | — | 106.1 s |
| 3′ | 2.63 (tdd, 12.2, 4.3, 1.3) | 45.5 d | 3.10 (ddd, 10.1, 9.2, 6.4) | 42.7 d | 2.79 (ddd, 12.5, 8.4, 5.9) | 47.3 d | 3.09 (ddd, 10.3, 9.4, 6.5) | 42.9 d | 2.79 (ddd, 11.3, 8.4, 6.6) | 47.2 d |
| 4′α | 2.91 (dd, 16.8, 12.2) | 28.4 t | 2.70 (dd, 17.0, 6.4) | 26.3 t | 2.48 (dd, 15.9, 5.9) | 26.9 t | 2.69 (dd, 16.9, 6.5) | 26.3 t | 2.48 (dd, 15.9, 6.6) | 27.0 t |
| β | 2.83 (dd, 16.8, 4.3) | 2.64 (dd, 17.0, 10.1) | 2.43 (dd, 15.9, 12.4) | 2.64 (dd, 16.9, 10.3) | 2.44 (dd, 15.9, 11.3) | |||||
| 5′ | — | 169.0 s | — | 168.0 s | — | 167.4 s | — | 168.1 s | — | 167.1 s |
| 7′ | — | 155.5 s | — | 155.5 s | — | 155.6 s | — | 155.5 s | — | 155.6 s |
| 8′ | 6.91 (s) | 107.4 d | 6.90 (s) | 107.6 d | 6.88 (s) | 107.6 d | 6.92 (s) | 107.6 d | 6.91 (s) | 107. 7 d |
| 9′ | — | 147.7 s | — | 147.40 s | — | 147.44 s | — | 147.40 s | — | 147.43 s |
| 10′ | 6.72 (s) | 111.8 d | 6.71 (s) | 112.0 d | 6.71 (s) | 112.0 d | 6.72 (s) | 111.9 d | 6.72 (s) | 111.9 d |
| 11′ | — | 159.4 s | — | 159.52 s | — | 159.55 s | — | 159.5 s | — | 159.5 s |
| 12′ | — | 107.3 s | — | 107.7 s | — | 107.8 s | — | 107.8 s | — | 107.7 s |
| 13′ | — | 181.4 s | — | 179.4 s | — | 179.3 s | — | 179.4 s | — | 179.3 s |
| 14′ | — | 113.1 s | — | 111.9 s | — | 111.7 s | — | 111.9 s | — | 111.8 s |
| 15′ | 2.39 (s) | 21.8 q | 2.38 (s) | 21.77 q | 2.38 (s) | 21.79 q | 2.39 (s) | 21.8 q | 2.38 (s) | 21.8 q |
| 16′ | 3.67 (s) | 52.4 q | 3.74 (s) | 52.28 q | 3.75 (s) | 52.31 q | 4.25–4.18 (m) | 61.3 t | 4.17–4.12 (m) | 61.2 t |
| 17′ | — | — | — | — | — | — | 1.26 (t, 7.1) | 13.8 q | 1.26 (t, 7.1) | 14.0 q |
| 11–OH | 12.20 (s) | — | 12.15 (s) | — | 12.12 (s) | — | 12.16 (s) | — | 12.13 (s) | — |
| 2′–OH | 7.96 (d, 1.3) | — | 7.80 (s) | — | 7.53 (s) | — | 7.74 (s) | — | 7.49 (s) | — |
| 11′–OH | 12.50 (s) | — | 12.49 (s) | — | 12.47 (s) | — | 12.50 (s) | — | 12.48 (s) | — |
| Proton | 1 | Position | 4 | ||
|---|---|---|---|---|---|
| δH (J in Hz) of 1a | δH (J in Hz) of 1b | δH (J in Hz) | δC | ||
| 3 | 3.25 (dd, 12.0, 6.0) | 3.54 (dd, 11.3, 7.0) | 1 | 4.94 (dt, 6.8, 1.5) | 79.5 d |
| 4 | 4.78 (d, 6.0) | 4.98 (d, 7.0) | 2α | 2.14 (dddd, 14.0, 8.6, 2.6, 1.5) | 27.8 t |
| 8 | 6.71 (s) | 6.70 (s) | β | 2.31 (dddd, 14.0, 9.4, 7.4, 6.8) | |
| 10 | 6.63 (s) | 6.63 (s) | 3α | 3.17 (dddd, 18.0, 8.6, 7.4, 1.5) | 30.3 t |
| 15 | 2.38 (s) | 2.37 (s) | β | 2.77 (ddd, 18.0, 9.4, 2.6) | |
| 16 | 3.82 (s) | 3.90 (s) | 4 | — | 174.0 s |
| 3′ | 2.78 (ddd, 12.7, 12.0, 4.0) | 3.93 (td, 11.3, 5.1) | 6 | — | 157.5 s |
| 4′α | 2.94 (dd, 17.0, 12.7) | 2.75 (dd, 17.6, 11.9) | 7 | 6.70 (br s) | 107.8 d |
| β | 2.84 (dd, 17.0, 4.0) | 3.05 (dd, 17.6, 5.1) | 8 | — | 146.8 s |
| 8′ | 6.78 (s) | 6.73 (s) | 9 | 6.63 (br s) | 112.8 d |
| 10′ | 6.71 (s) | 6.70 (s) | 10 | — | 161.2 d |
| 15′ | 2.43 (s) | 2.43 (s) | 11 | — | 109.2 d |
| 16′ | 3.80 (s) | 3.85 (s) | 12 | — | 181.3 d |
| 11–OH | 12.12 (s) | 11.89 (s) | 13 | — | 120.1 d |
| 2′–OH | 4.60 (br s) | 4.49 (s) | 14 | 2.39 (s) | 22.4 q |
| 11′–OH | 12.37 (s) | 12.34 (s) | 15 | 3.49 (s) | 57.5 q |
| — | — | — | 10-OH | 12.55 (s) | — |
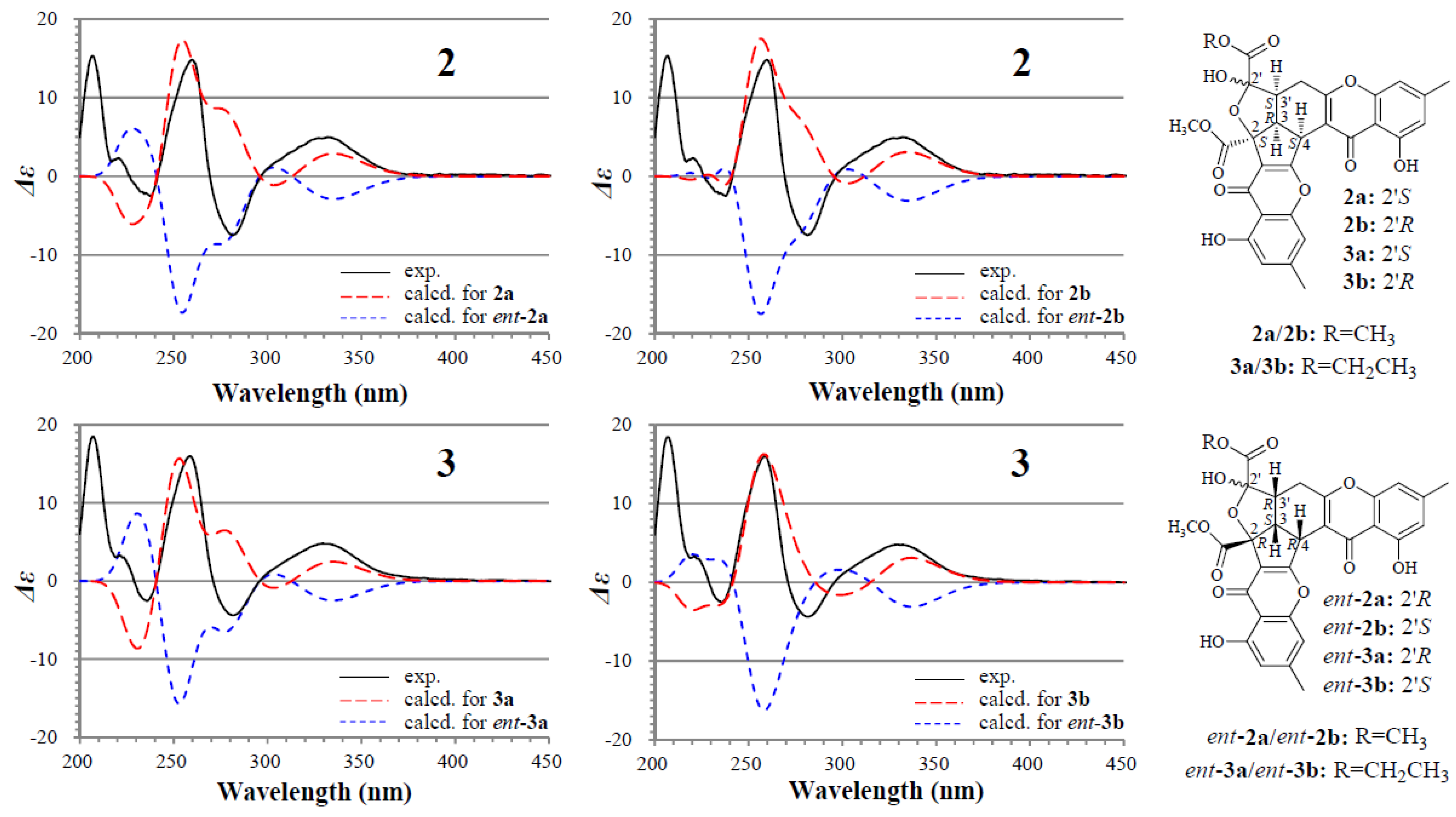
2.2. Structure Determination of New Chromones 2–4
| Proton | 2 | 3 | ||
|---|---|---|---|---|
| 2a | 2b | 3a | 3b | |
| 3 | 3.93 (t, 9.0) | 3.86–3.82 (masked by H-16/16′) | 3.95 (t, 9.0) | 3.90 (t, 9.0) |
| 4 | 5.19 (d, 9.0) | 5.22 (d, 9.0) | 5.21 (d, 9.0) | 5.22 (d, 9.0) |
| 8 | 6.70 (s) | 6.70 (s) | 6.70 (s) | 6.70 (s) |
| 10 | 6.60 (s) | 6.60 (s) | 6.60 (s) | 6.60 (s) |
| 15 | 2.32 (s) | 2.30 (s) | 2.33 (s) | 2.32 (s) |
| 16 | 3.79 (s) | 3.82 (s) | 3.79 (s) | 3.84 (s) |
| 3′ | 3.01 (ddd, 11.7, 9.0, 5.8) | 2.93 (ddd, 12.8, 8.2, 5.2) | 2.98 (ddd, 11.9, 9.0, 5.7) | 2.92 (ddd, 12.7, 9.0, 5.4) |
| 4′α | 2.87 (dd, 16.7, 5.8) | 2.81 (dd, 15.9, 5.2) | 2.88 (dd, 16.6, 5.7) | 2.81 (dd,15.9, 5.4) |
| β | 2.62 (dd, 16.7, 11.7) | 2.46 (dd, 15.9, 12.8) | 2.62 (dd, 16.6, 11.9) | 2.49 (dd,15.9, 12.7) |
| 8′ | 6.71 (s) | 6.70 (s) | 6.72 (s) | 6.71 (s) |
| 10′ | 6.68 (s) | 6.68 (s) | 6.69 (s) | 6.69 (s) |
| 15′ | 2.42 (s) | 2.42 (s) | 2.42 (s) | 2.42 (s) |
| 16′ | 3.87 (s) | 3.85 (s) | 4.37-4.29 (m) | 4.35-4.27 (m) |
| 17′ | — | — | 1.35 (t, 7.1) | 1.37 (t, 7.0) |
| 11–OH | 12.04 (s) | 11.95 (s) | 12.06 (s) | 11.98 (s) |
| 2′–OH | 4.64 (br s) | 4.64 (br s) | 4.64 (br s) | 4.64 (br s) |
| 11′–OH | 12.35 (s) | 12.30 (s) | 12.36 (s) | 12.31 (s) |
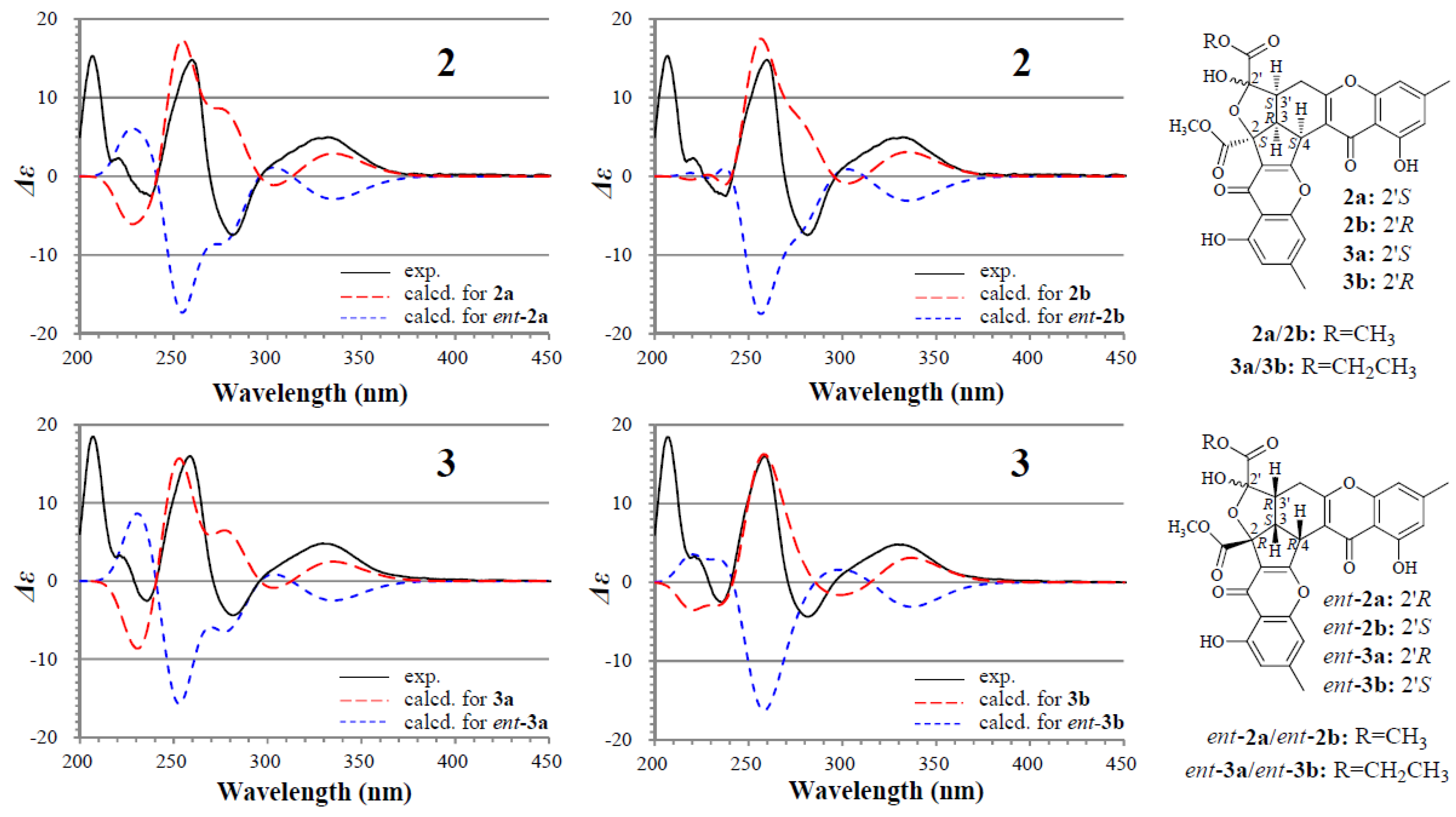
2.3. Detection of 1–6 in the Mutant AD-1-2 Extract by HPLC-PDAD-UV/HPLC-ESI-MS Analyses
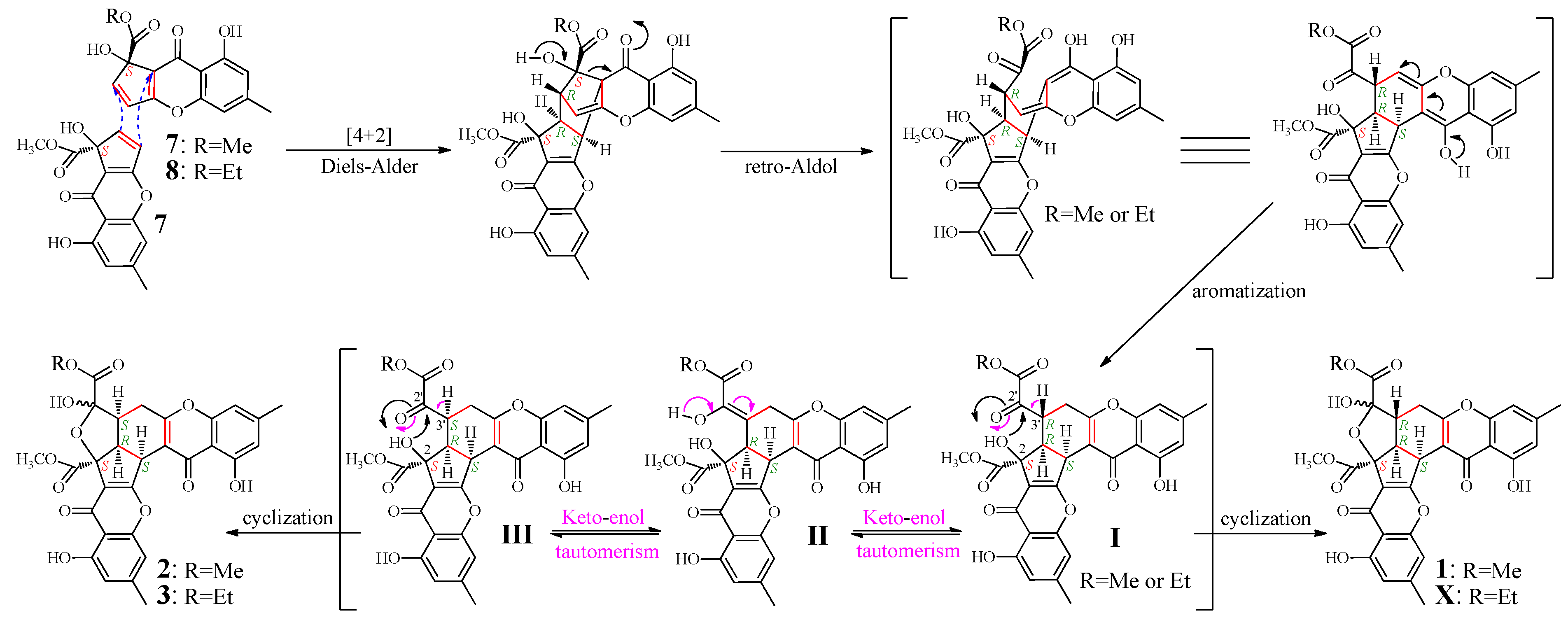
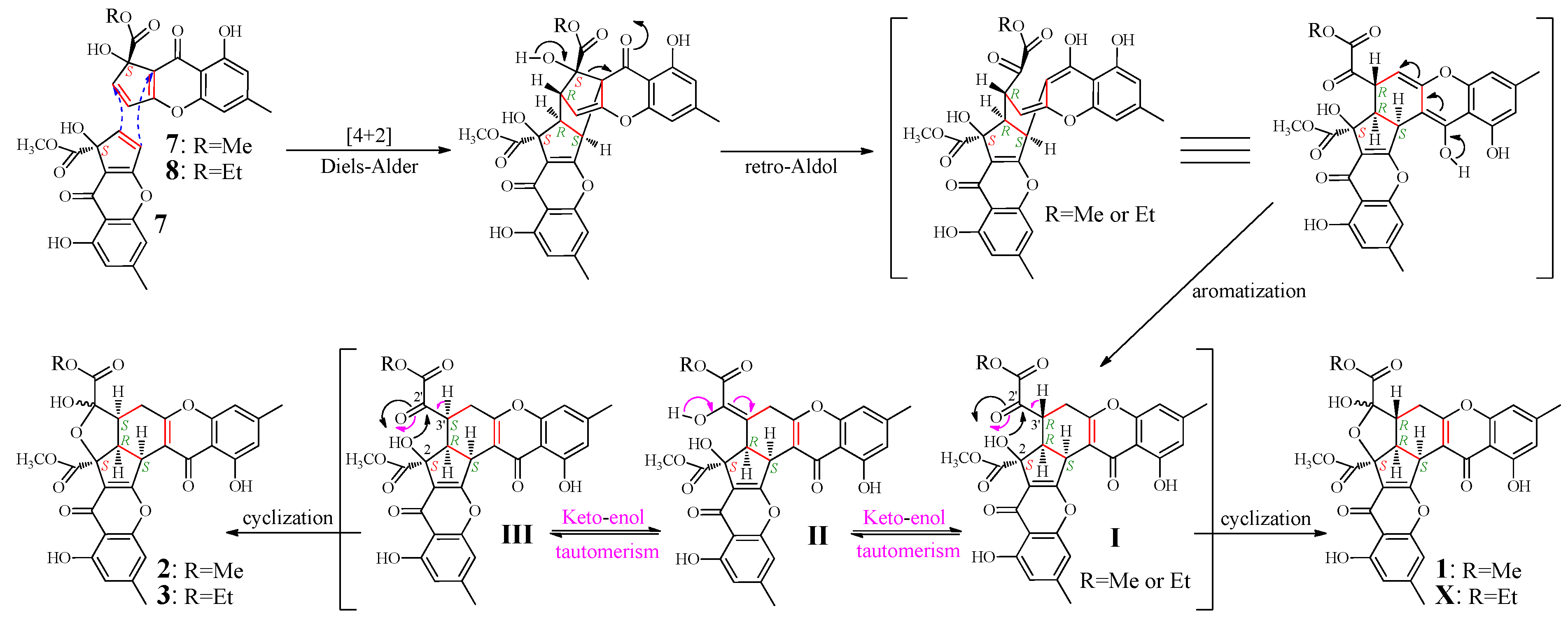
2.4. Mechanism of 1–3 Formation
2.5. Inhibitory Effects of 1–6 on Several Human Cancer Cell Lines
| Compound | K562 | HL-60 | HeLa | BGC-823 |
|---|---|---|---|---|
| 1 | 64.0% | 71.6% | 35.7% | 36.8% |
| 2 | 73.1% | 77.4% | 44.6% | 41.4% |
| 3 | 74.0% | 80.0% | 45.7% | 46.8% |
| 4 | 20.4% | 26.0% | 11.9% | — |
| 5 | 36.1% | 62.4% | 13.9% | 11.4% |
| 6 | 38.8% | 46.3% | 29.1% | 27.8% |
| Decotaxol | 49.2% | 46.9% | 41.7% | 44.1% |
2.6. Discussion
3. Experimental Section
3.1. General Experimental
3.2. MTT Assay
3.3. Fermentation and Isolation of Compounds 1–6
3.3.1. Parent Fungal Strain and Its Mutant AD-1-2 the 1–8 Producing Strain
3.3.2. Fermentation, Extraction, and Preparation of Targeted Bioactive Fraction
3.3.3. Isolation of 1–6
3.4. Physicochemical and Spectroscopic Data of 1–6
3.5. HPLC-PDAD-UV Analysis of the AD-1-2 and G59 Extracts for Detecting 1–8
3.6. HPLC-ESI-MS Analysis of the AD-1-2 and G59 Extracts for Detecting 1–8
3.7. Computation Section for ECD Calculation
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, H.J.; Gloer, J.B.; Scott, J.A.; Malloch, D. Coniochaetones A and B: New antifungal benzopyranones from the coprophilous fungus Coniochaeta saccardoi. Tetrahedron Lett. 1995, 36, 5847–5850. [Google Scholar] [CrossRef]
- Fujimoto, H.; Inagaki, M.; Satoh, Y.; Yoshida, E.; Yamazaki, M. Monoamine oxidase-inhibitory components from an ascomycete, Coniochaeta tetraspora. Chem. Pharm. Bull. 1996, 44, 1090–1092. [Google Scholar] [CrossRef]
- Khamthong, N.; Rukachaisirikul, V.; Phongpaichit, S.; Preedanon, S.; Sakayaroj, J. Bioactive polyketides from the sea fan-derived fungus Penicillium citrinum PSU-F51. Tetrahedron 2012, 68, 8245–8250. [Google Scholar] [CrossRef]
- Deng, L.; Niu, S.; Liu, X.; Che, Y.; Li, E. Coniochaetones E–I, new 4H-chromen-4-one derivatives from the Cordyceps-colonizing fungus Fimetariella sp. Fitoterapia 2013, 89, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Ondeyka, J.G.; Zink, D.; Basilio, A.; Vicente, F.; Bills, G.; Diez, M.T.; Motyl, M.; Dezeny, G.; Byrne, K.; Singh, S.B. Coniothyrione, a chlorocyclopentandienylbenzopyrone as a bacterial protein synthesis inhibitor discovered by antisense technology. J. Nat. Prod. 2007, 70, 668–670. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Zhu, T.; Pan, W.; Tsao, R.; Pagano, T.G.; Nguyen, B.; Marquez, B. WYE-120318, a ring contraction product of methylnaltrexone, and structure revision of coniothyrione. Magn. Reson. Chem. 2012, 50, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Bungihan, M.E.; Tan, M.A.; Kitajima, M.; Kogure, N.; Franzblau, S.G.; deLa Cruz, T.E.E.; Takayama, H.; Nonato, M.G. Bioactive metabolites of Diaporthe sp. P133, an endophytic fungus isolated from Pandanus amaryllifolius. J. Nat. Med. 2011, 65, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Li, L.; Niu, S.; Si, Y.; Guo, L.; Jiang, X.; Che, Y. A thiopyranchromenone and other chromone derivatives from an endolichenic fungus, Preussia africana. J. Nat. Prod. 2012, 75, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Saleem, M.; Tousif, M.I.; Riaz, N.; Ahmed, I.; Schulz, B.; Ashraf, M.; Nasar, R.; Pescitelli, G.; Hussain, H.; Jabbar, A.; Shafiq, N.; Krohn, K. Cryptosporioptide: A bioactive polyketide produced by an endophytic fungus Cryptosporiopsis sp. Phytochemistry 2013, 93, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Carter, G.T. Remisporine B, a novel dimeric chromenone derived from spontaneous Diels–Alder reaction of remisporine A. Tetrahedron Lett. 2003, 44, 3119–3122. [Google Scholar] [CrossRef]
- Hertweck, C. Hidden biosynthetic treasures brought to light. Nat. Chem. Biol. 2009, 5, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Cichewicz, R.H.; Henrikson, J.C.; Wang, X.; Branscum, K.M. Strategies for accessing microbial secondary metabolites from silent biosynthetic pathways. In Manual of Industrial Microbiology and Biotechnology, 3rd ed.; Baltz, R.H., Davies, J.E., Demain, A.L., Bull, A.T., Junker, B., Katz, L., Lynd, L.R., Masurekar, P.C., Reeves, D., Zhao, H., Eds.; ASM Press: Washington, DC, USA, 2010; pp. 78–95. [Google Scholar]
- Brakhage, A.A.; Schroeckh, V. Fungal secondary metabolites—Strategies to activate silent gene clusters. Fungal Genet. Biol. 2011, 48, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, J.A.; Teles, A.P.C.; Bracarense, A.A.P.; Gomes, D.C. Classical and epigenetic approaches to metabolite diversification in filamentous fungi. Phytochem. Rev. 2013, 12, 773–789. [Google Scholar] [CrossRef]
- Bode, H.B.; Bethe, B.; Höfs, R.; Zeeck, A. Big effects from small changes: Possible ways to explore nature’s chemical diversity. ChemBioChem 2002, 3, 619–627. [Google Scholar] [CrossRef]
- Marmann, A.; Aly, A.H.; Lin, W.; Wang, B.; Proksch, P. Co-cultivation—A powerful emerging tool for enhancing the chemical diversity of microorganisms. Mar. Drugs 2014, 12, 1043–1065. [Google Scholar] [CrossRef] [PubMed]
- Henrikson, J.C.; Hoover, A.R.; Joyner, P.M.; Cichewicz, R.H. A chemical epigenetics approach for engineering the in situ biosynthesis of a cryptic natural product from Aspergillus niger. Org. Biomol. Chem. 2009, 7, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Ochi, K.; Okamoto, S.; Tozawa, Y.; Inaoka, T.; Hosaka, T.; Xu, J.; Kurosawa, K. Ribosome engineering and secondary metabolite production. Adv. Appl. Microbiol. 2004, 56, 155–184. [Google Scholar] [PubMed]
- Ochi, K. From microbial differentiation to ribosome engineering. Biosci. Biothenol. Biochem. 2007, 71, 1373–1386. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, T.; Ohnishi-Kameyama, M.; Muramatsu, H.; Murakami, K.; Tsurumi, Y.; Kodani, S.; Yoshida, M.; Fujie, A.; Ochi, K. Antibacterial discovery in actinomycetes strains with mutations in RNA polymerase or ribosomal protein S12. Nat. Biotechnol. 2009, 27, 462–464. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.; Jamison, M.; La, S.; MacMillan, J.B. Inducamides A–C, chlorinated alkaloids from an RNA polymerase mutant strain of Streptomyces sp. Org. Lett. 2014, 16, 5656–5659. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.-B. A new approach for exploiting microbial new strain resources for drug screening. J. Int. Pharm. Res. 2010, 37, 1–7. [Google Scholar]
- Wu, C.-J.; Cui, C.-B.; Tian, C.-K.; Li, C.-W. Antitumor metabolites produced by two Penicillium purpurogenum G59 mutants. J. Int. Pharm. Res. 2010, 37, 122–126. [Google Scholar]
- Chai, Y.-J.; Cui, C.-B.; Li, C.-W.; Hua, W. Antitumor metabolites newly produced by a gentamicin-resistant mutant of Penicillium purpurogenum G59. J. Int. Pharm. Res. 2011, 38, 216–222. [Google Scholar]
- Chai, Y.-J.; Cui, C.-B.; Li, C.-W.; Wu, C.-J.; Tian, C.-K.; Hua, W. Activation of the dormant secondary metabolite production by introducing gentamicin-resistance in a marine-derived Penicillium purpurogenum G59. Mar. Drugs 2012, 10, 559–582. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-J.; Yi, L.; Cui, C.-B.; Li, C.-W.; Wang, N.; Han, X. Activation of the silent secondary metabolite production by introducing neomycin-resistance in a marine-derived Penicillium purpurogenum G59. Mar. Drugs 2015, 13, 2465–2487. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Cui, C.-B.; Li, C.-W.; Hua, W.; Wu, C.-J.; Zhu, T.-J.; Gu, Q.-Q. Activation of dormant secondary metabolite production by introducing neomycin resistance into the deep-sea fungus, Aspergillus versicolor ZBY-3. Mar. Drugs 2014, 12, 4326–4352. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.-M.; Wu, C.-J.; Li, C.-W.; Cui, C.-B. A practical strategy to discover new antitumor compounds by activating silent metabolite production in fungi by diethyl sulphate mutagenesis. Mar. Drugs 2014, 12, 1788–1814. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.-M.; Cui, C.-B.; Li, C.-W.; Wu, C.-J.; Zhang, Z.-J.; Li, L.; Huang, X.-J.; Ye, W.-C. Purpurogemutantin and purpurogemutantidin, new drimenyl cyclohexenone derivatives produced by a mutant obtained by diethyl sulfate mutagenesis of a marine-derived Penicillium purpurogenum G59. Mar. Drugs 2012, 10, 1266–1287. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.-W.; Cui, C.-B.; Li, C.-W.; Wu, C.-J. Three new and eleven known unusual C25 steroids: Activated production of silent metabolites in a marine-derived fungus by chemical mutagenesis strategy using diethyl sulphate. Mar. Drugs 2014, 12, 1545–1568. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-J.; Li, C.-W.; Cui, C.-B. Seven new and two known lipopeptides as well as five known polyketides: The activated production of silent metabolites in a marine-derived fungus by chemical mutagenesis strategy using diethyl sulphate. Mar. Drugs 2014, 12, 1815–1838. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.-K.; Cui, C.-B.; Han, X.-X. Isolation of fungal strains in unusual environment and screening for their antitumor activity. J. Int. Pharm. Res. 2008, 35, 401–405. [Google Scholar]
- Shao, C.; Wang, C.; Wei, M.; Gu, Y.; Xia, X.; She, Z.; Lin, Y. Structure elucidation of two new xanthone derivatives from the marine fungus Penicillium sp. (ZZF 32#) from the South China Sea. Magn. Reson. Chem. 2008, 46, 1066–1069. [Google Scholar] [PubMed]
- Spartan′14; Wavefunction Inc.: Irvine, CA, USA, 2013.
- Gaussian 09; Revision A.01; Gaussian Inc.: Wallingford, CT, USA, 2009.
- Bruhn, T.; Hemberger, Y.; Schaumlöffel, A.; Bringmann, G. SpecDis; Version 1.53; University of Wuerzburg: Wuerzburg, Germany, 2011. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, M.-W.; Cui, C.-B.; Li, C.-W.; Wu, C.-J.; Peng, J.-X.; Li, D.-H. Rare Chromones from a Fungal Mutant of the Marine-Derived Penicillium purpurogenum G59. Mar. Drugs 2015, 13, 5219-5236. https://doi.org/10.3390/md13085219
Xia M-W, Cui C-B, Li C-W, Wu C-J, Peng J-X, Li D-H. Rare Chromones from a Fungal Mutant of the Marine-Derived Penicillium purpurogenum G59. Marine Drugs. 2015; 13(8):5219-5236. https://doi.org/10.3390/md13085219
Chicago/Turabian StyleXia, Ming-Wen, Cheng-Bin Cui, Chang-Wei Li, Chang-Jing Wu, Ji-Xing Peng, and De-Hai Li. 2015. "Rare Chromones from a Fungal Mutant of the Marine-Derived Penicillium purpurogenum G59" Marine Drugs 13, no. 8: 5219-5236. https://doi.org/10.3390/md13085219
APA StyleXia, M.-W., Cui, C.-B., Li, C.-W., Wu, C.-J., Peng, J.-X., & Li, D.-H. (2015). Rare Chromones from a Fungal Mutant of the Marine-Derived Penicillium purpurogenum G59. Marine Drugs, 13(8), 5219-5236. https://doi.org/10.3390/md13085219