
Molecular Mechanisms by Which a Fucus vesiculosus Extract Mediates Cell Cycle Inhibition and Cell Death in Pancreatic Cancer Cells


 and
and
Abstract
:1. Introduction
2. Results
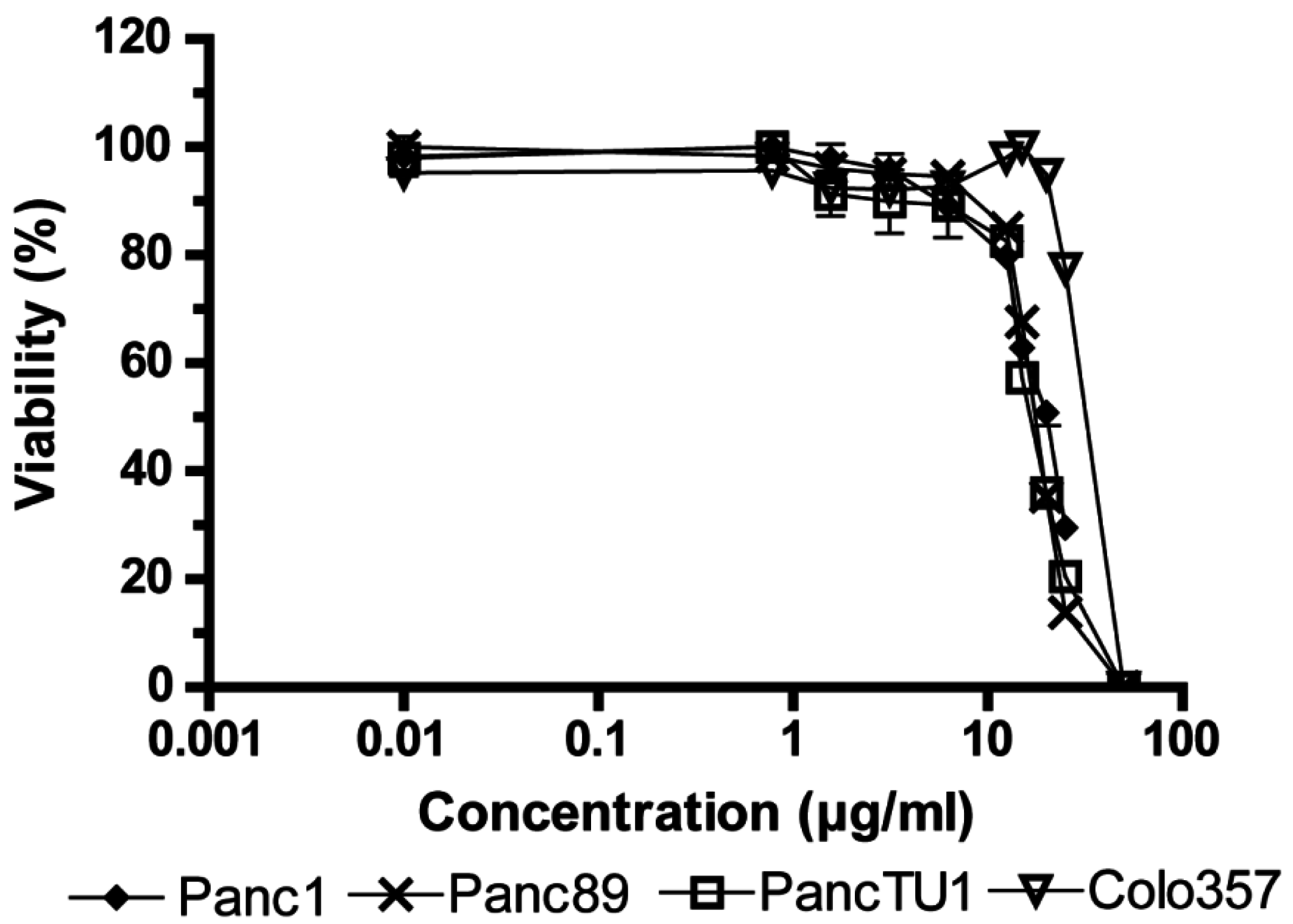
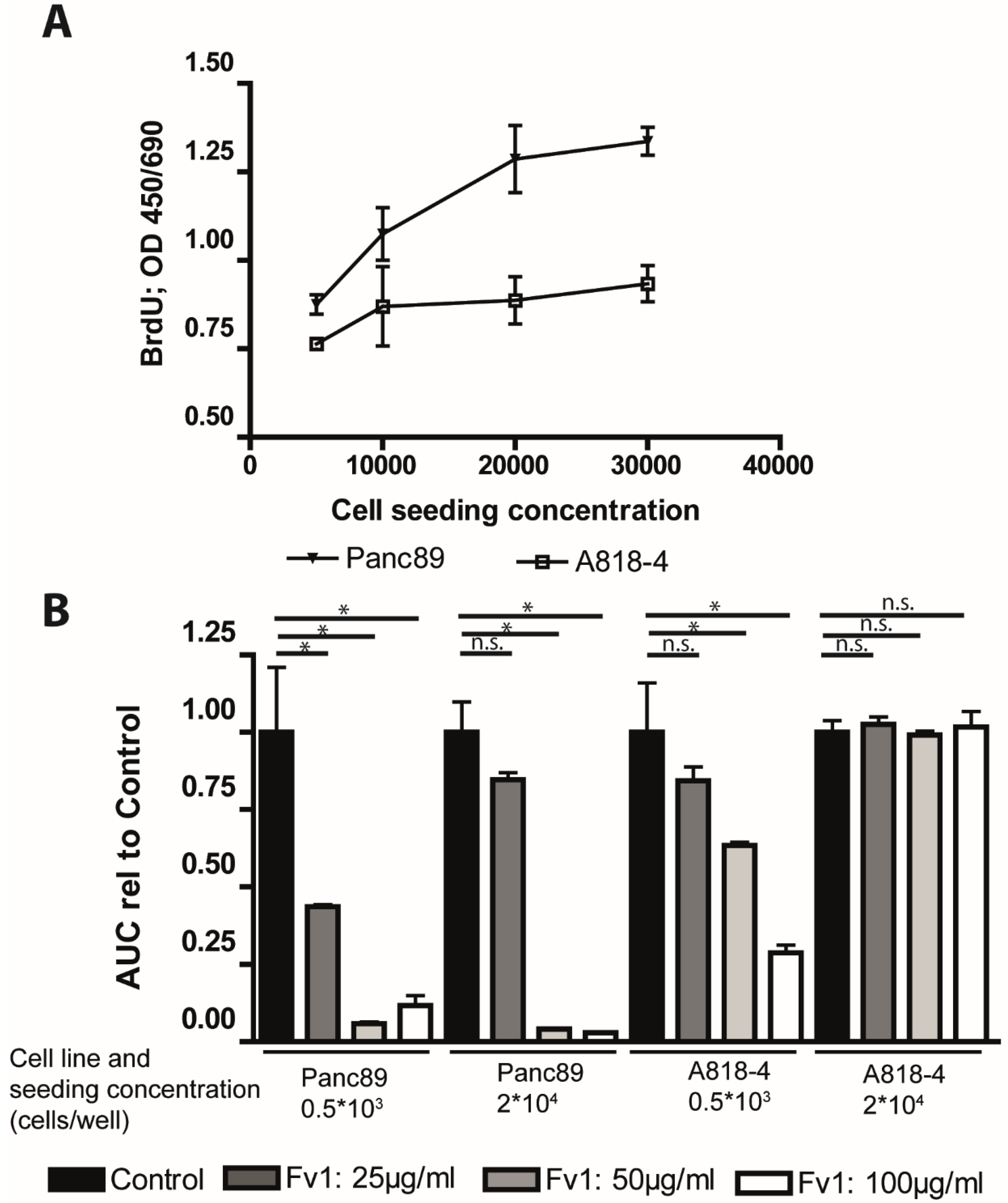
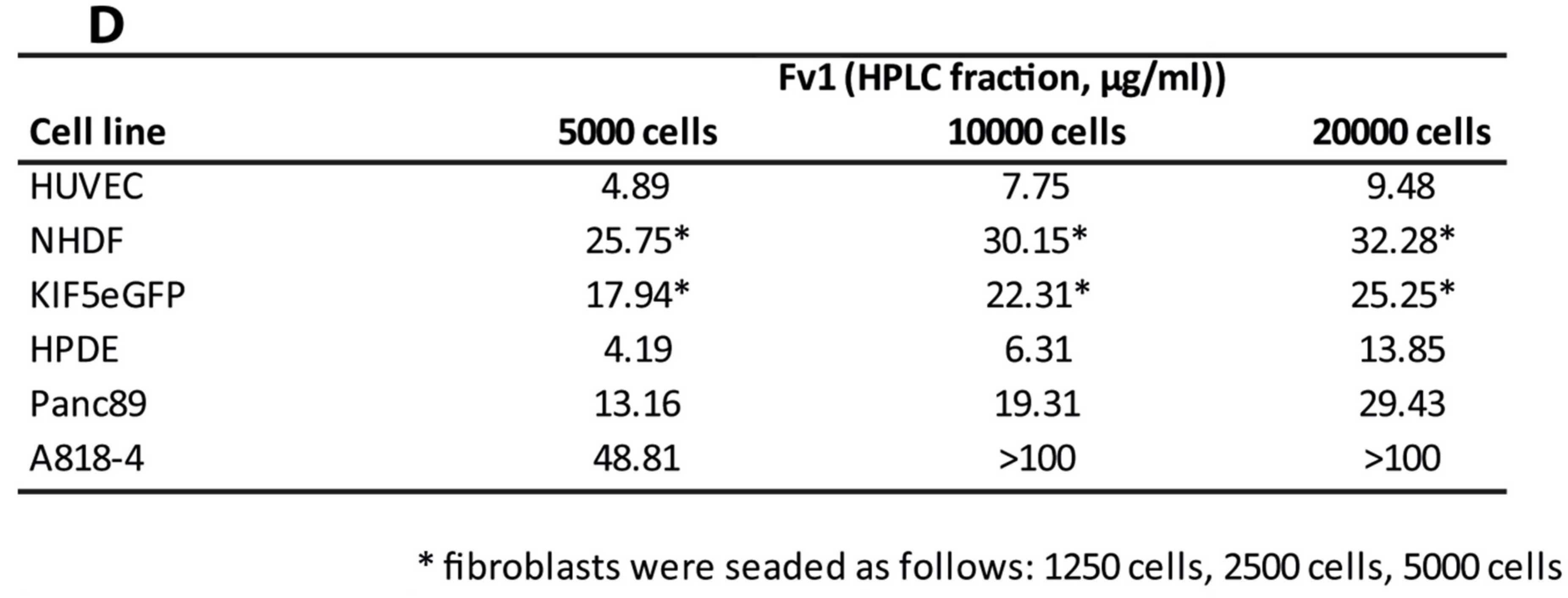
2.1. Influence of Fv1 on Viability of Cancer Cells
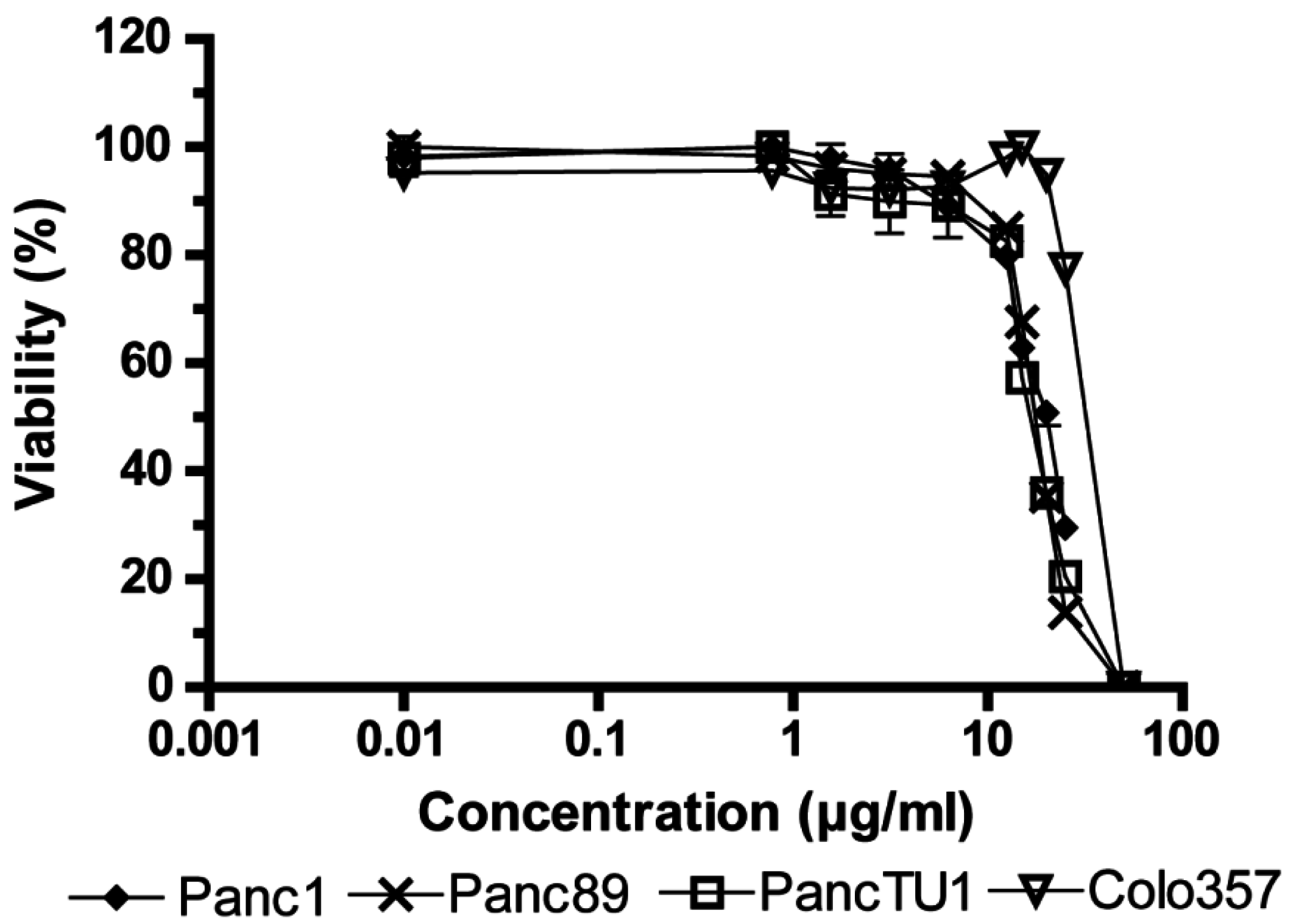
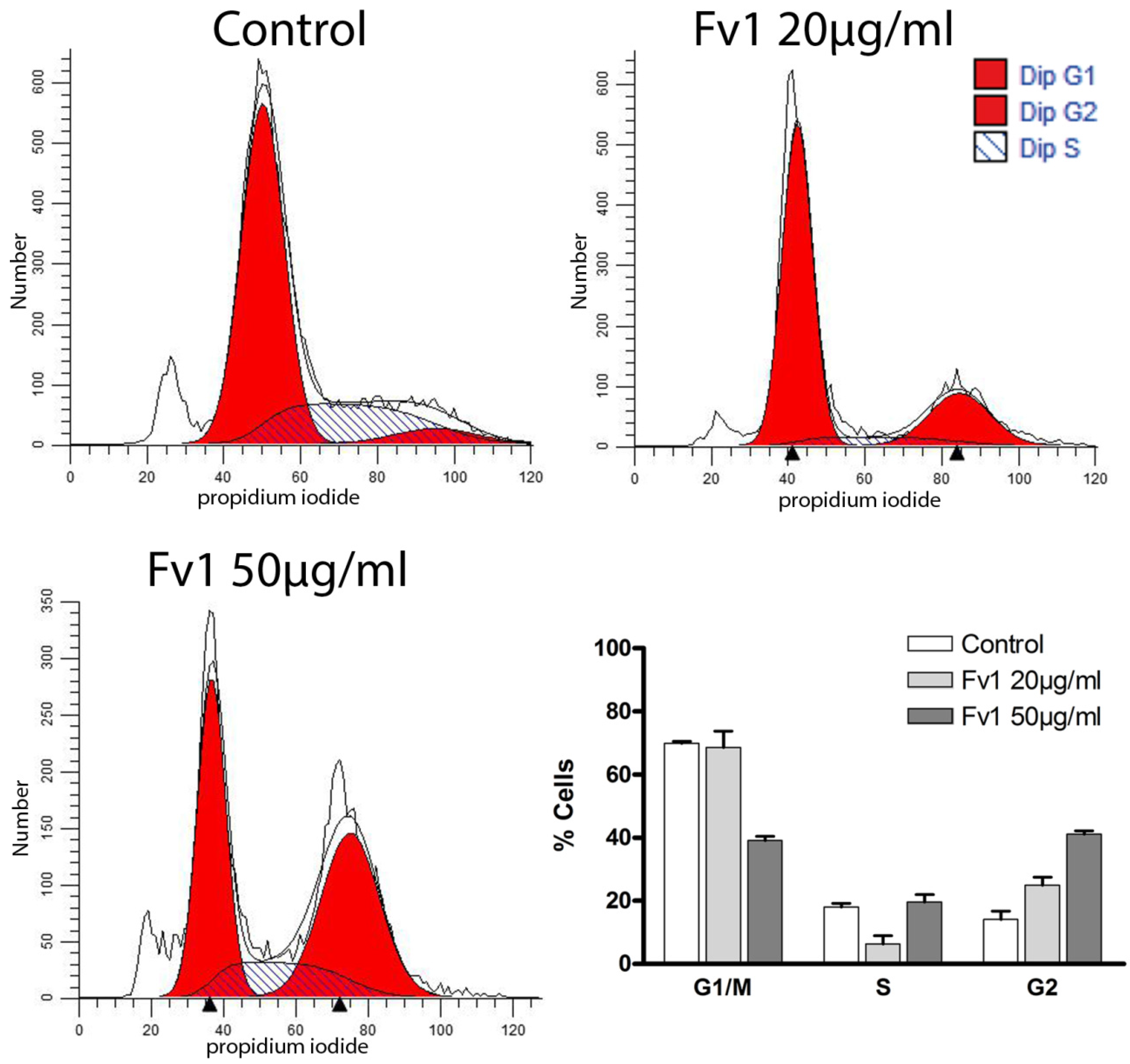
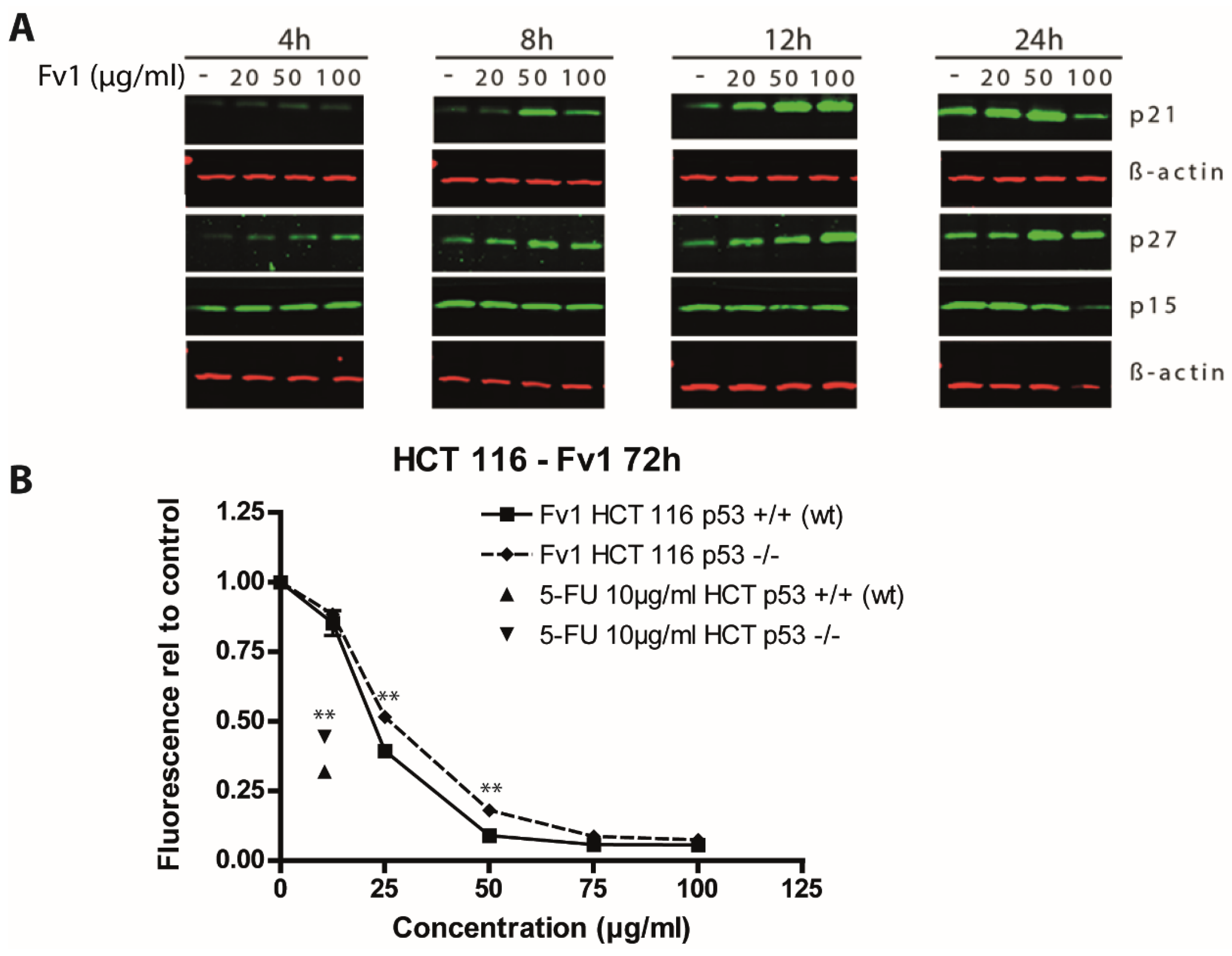
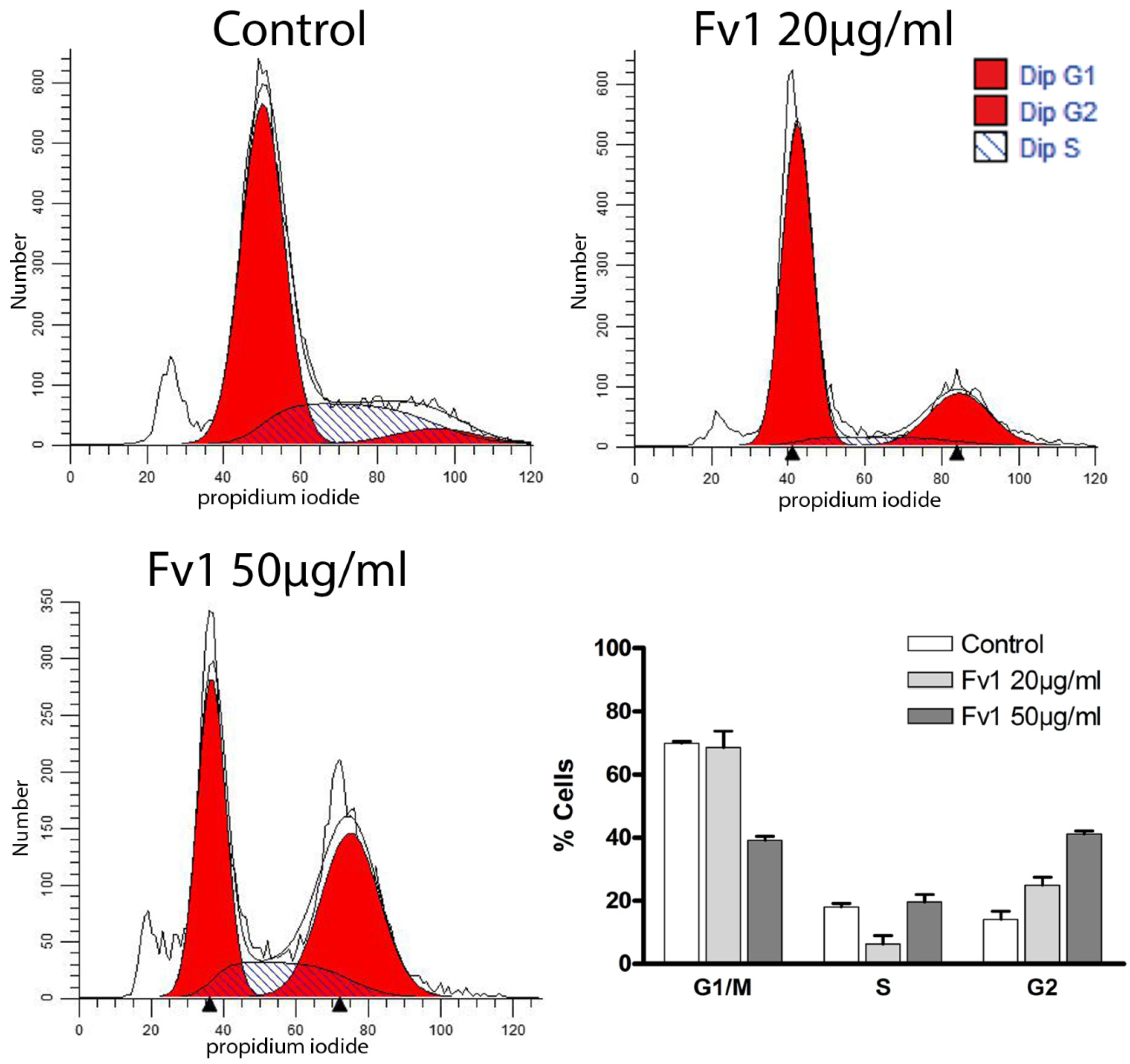
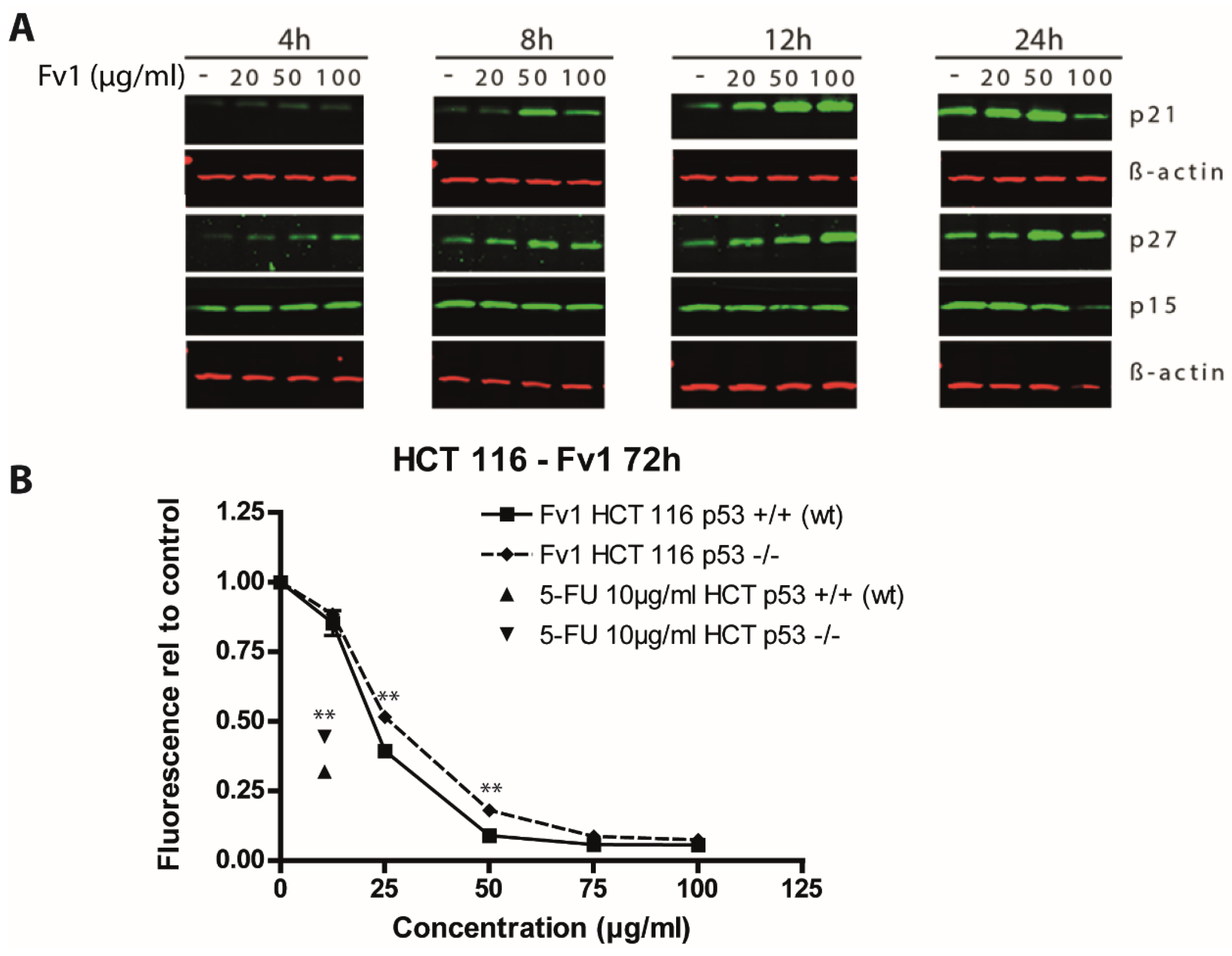
2.2. Effect of Fv1 on Cell Cycle and Cell Cycle Inhibitors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | ||||||||||
| Cell Line | Number of Regulated Genes | |||||||||
| Panc1 | 3951 | |||||||||
| Panc89 | 3909 | |||||||||
| HPDE | 2614 | |||||||||
| Colo357 | 200 | |||||||||
| PancTU1 6 h | 340 | |||||||||
| PancTU1 24 h | 2930 | |||||||||
| B | ||||||||||
| Term | Count | % of Pathway Genes | p-Value (Benjamini-Hochberg-Corrected) | |||||||
| DNA replication | 8 | 6.25 | 0.0000012 | |||||||
| Cell cycle | 11 | 8.59 | 0.0000034 | |||||||
| Base excision repair | 5 | 3.91 | 0.0045238 | |||||||
| Terpenoid backbone biosynthesis | 4 | 3.13 | 0.0038877 | |||||||
| Oocyte meiosis | 5 | 3.91 | 0.1670225 | |||||||
| Lysosome | 5 | 3.91 | 0.1705743 | |||||||
| Pyrimidine metabolism | 4 | 3.13 | 0.3412000 | |||||||
| Nucleotide excision repair | 3 | 2.34 | 0.3292213 | |||||||
| C | ||||||||||
| Target ID | Panc1 | PancTU1 | Panc89 | Gene Symbol | Gene Name | |||||
| Lfc | p-Value | Lfc | p-Value | Lfc | p-Value | |||||
| NM_000076 | 2.0364 | 0.0326 | 1.0465 | 0.0261 | 2.7013 | 0.0347 | CDKN1C | cyclin-dependent kinase inhibitor 1C (p57, Kip2) | ||
| NM_057749 | −1.9238 | 0.0470 | −1.9484 | 0.0003 | −4.3172 | 0.0050 | CCNE2 | cyclin E2 | ||
| NM_003504 | −1.5647 | 0.0243 | −1.3113 | 0.0054 | −3.5616 | 0.0005 | CDC45 | cell division cycle 45 homolog (S. cerevisiae) | ||
| NM_003503 | −1.3615 | 0.0132 | −1.5369 | 0.0012 | −3.7463 | 0.0081 | CDC7 | cell division cycle 7 homolog (S. cerevisiae) | ||
| NM_001789 | −1.9897 | 0.0263 | −1.8791 | 0.0001 | −4.5741 | 0.0359 | CDC25A | cell division cycle 25 homolog A (S. pombe) | ||
| NM_005225 | −1.3352 | 0.0197 | −1.3330 | 0.0004 | −3.2981 | 0.0415 | E2F1 | E2F transcription factor 1 | ||
| NM_002592 | −1.3851 | 0.0047 | −1.1083 | 0.0018 | −2.9858 | 0.0056 | PCNA | proliferating cell nuclear antigen | ||
| NM_033285 | 2.2445 | 0.0023 | 3.9746 | 0.0052 | 5.6604 | 0.0427 | TP53INP1 | tumor protein p53 inducible nuclear protein 1 | ||
| NM_001255 | −1.1762 | 0.0036 | −1.0547 | 0.0160 | −3.2856 | 0.0221 | CDC20 | cell division cycle 20 homolog (S. cerevisiae) | ||
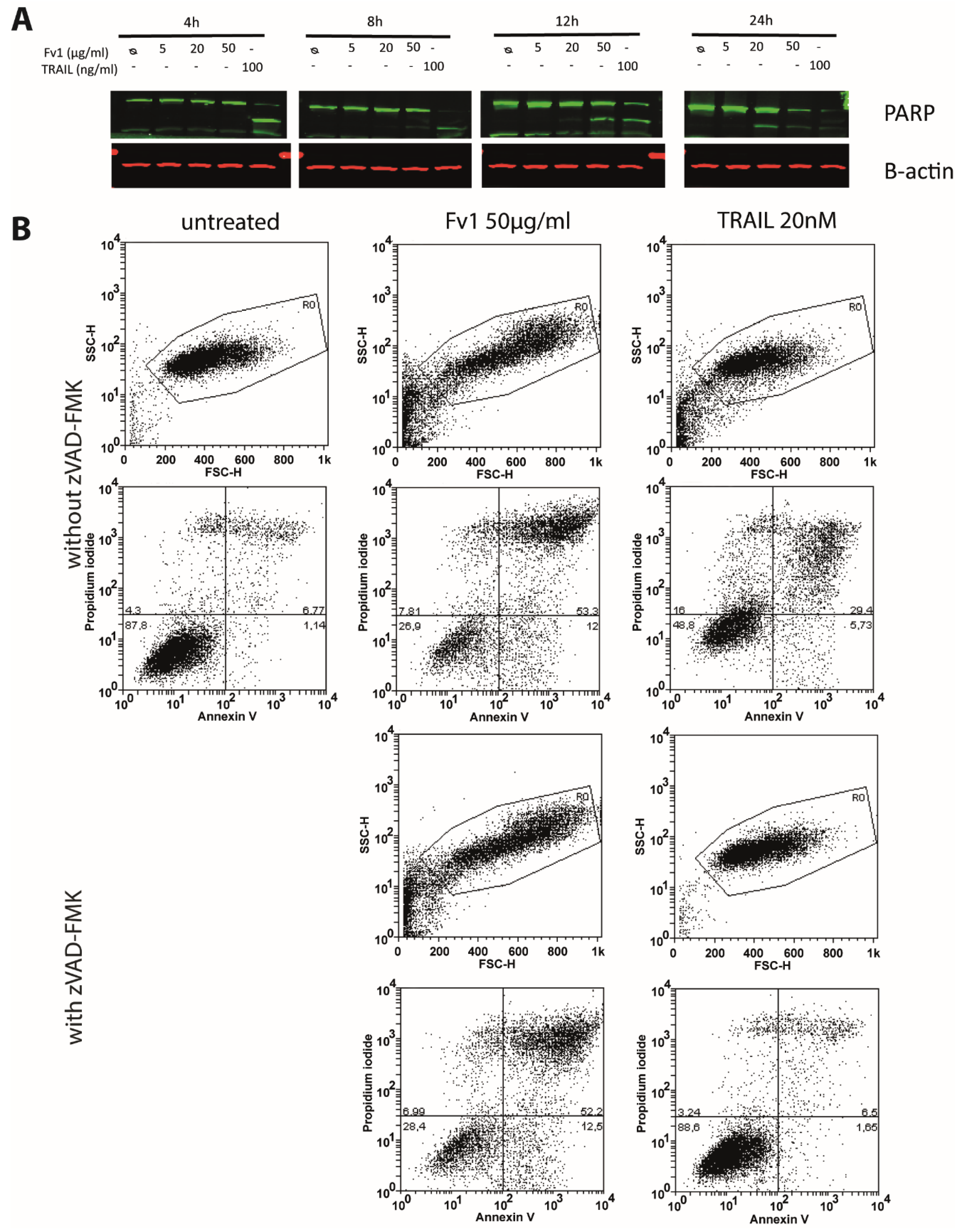
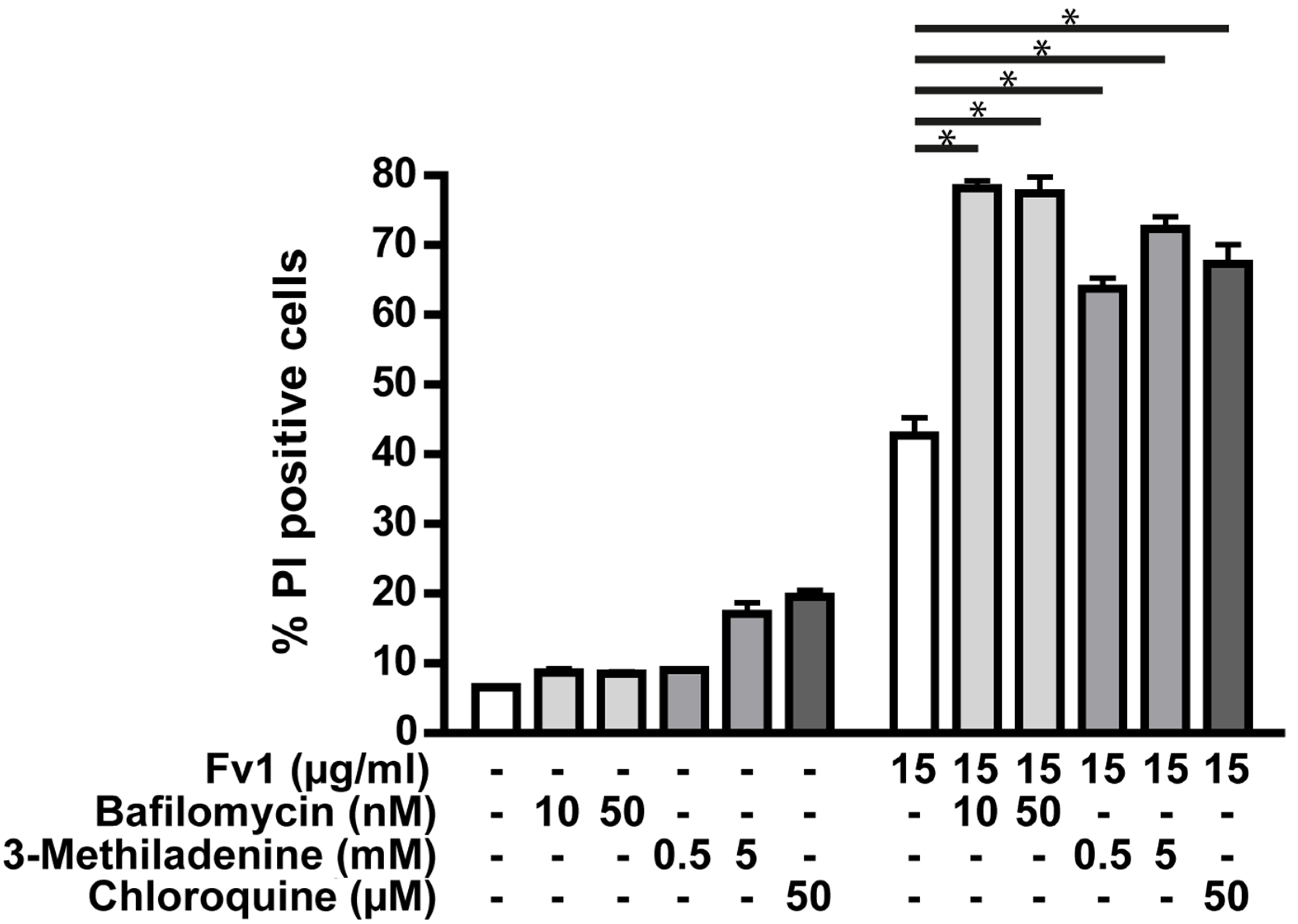
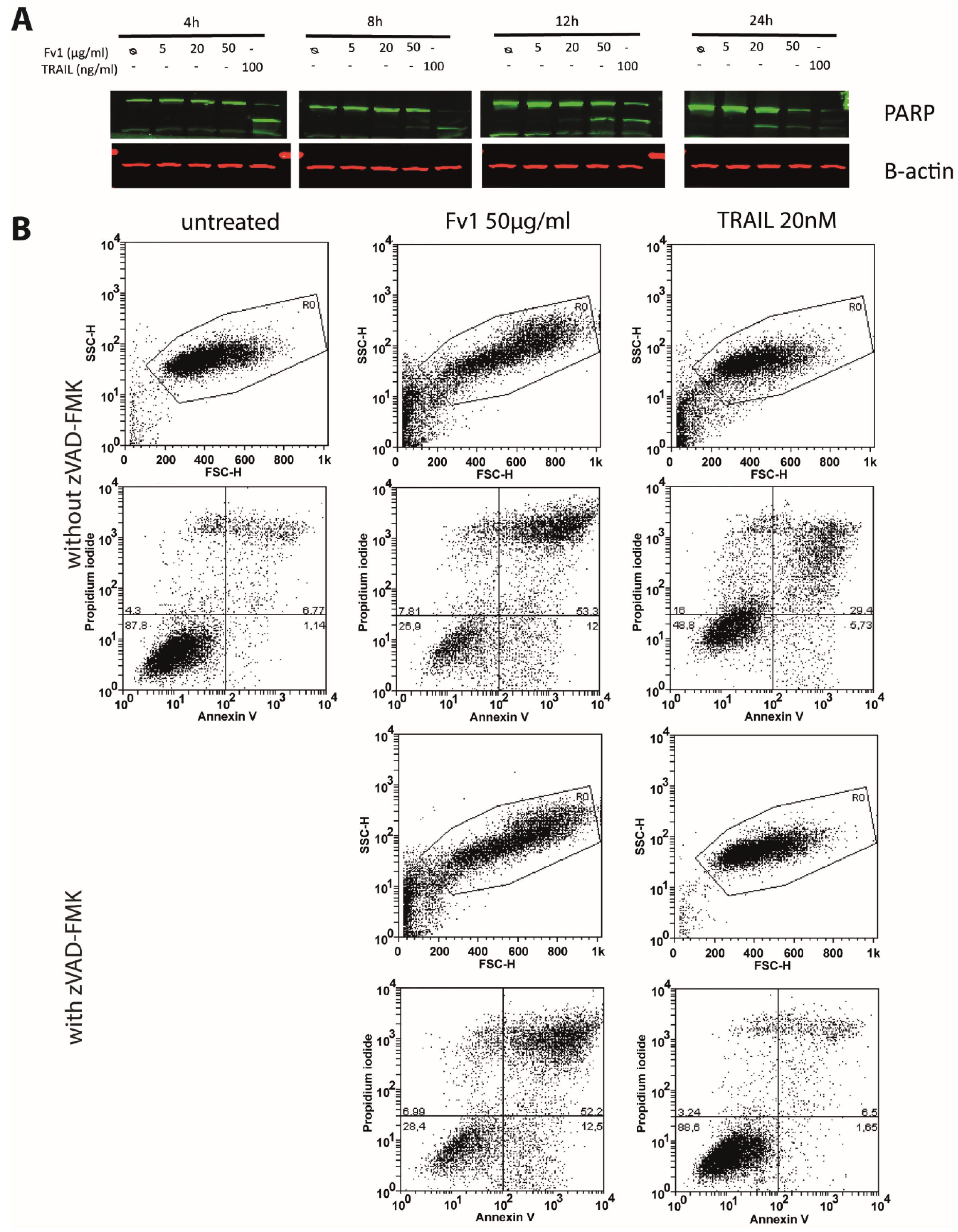
2.3. Impact of Caspase Activity and Autophagy
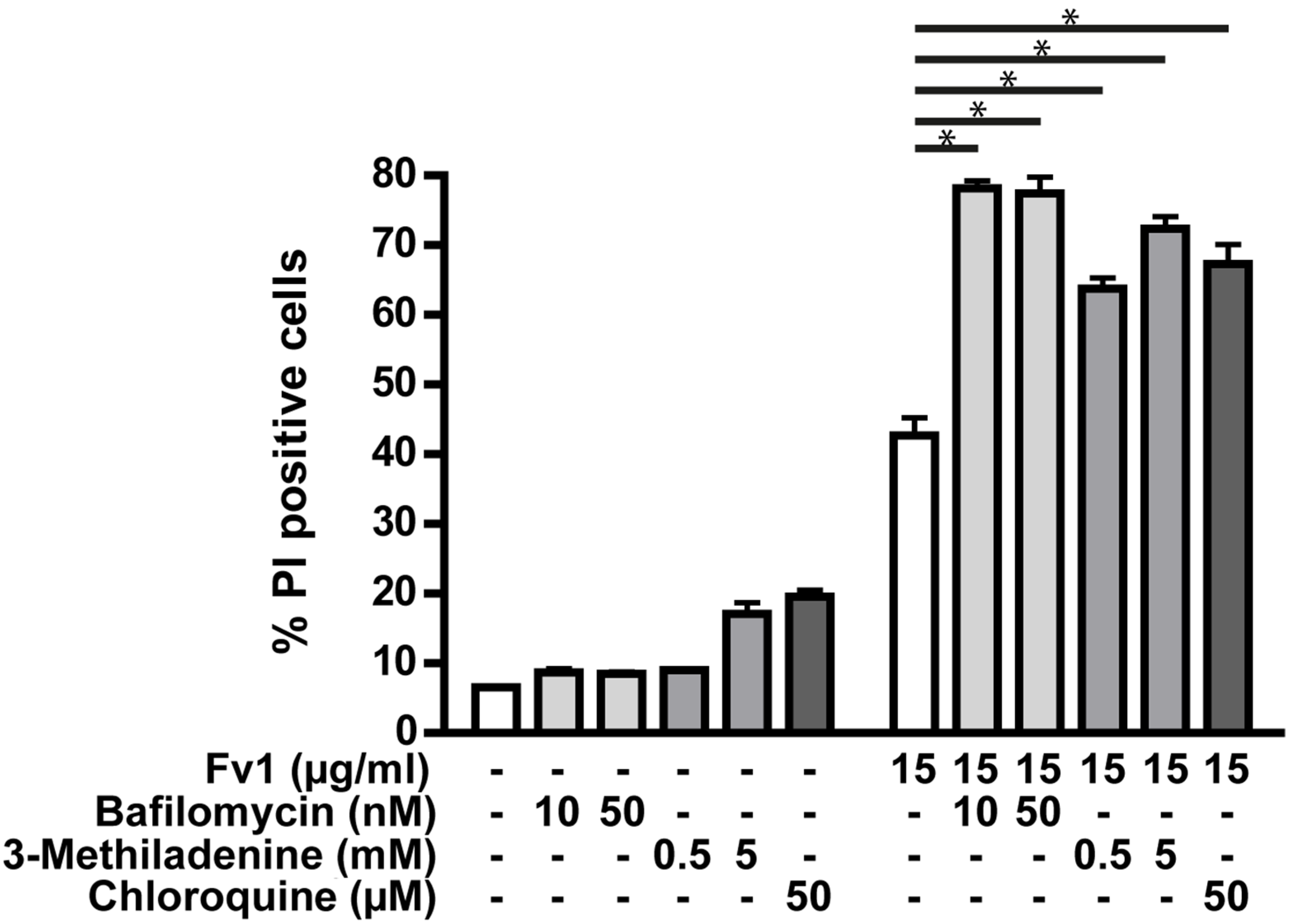
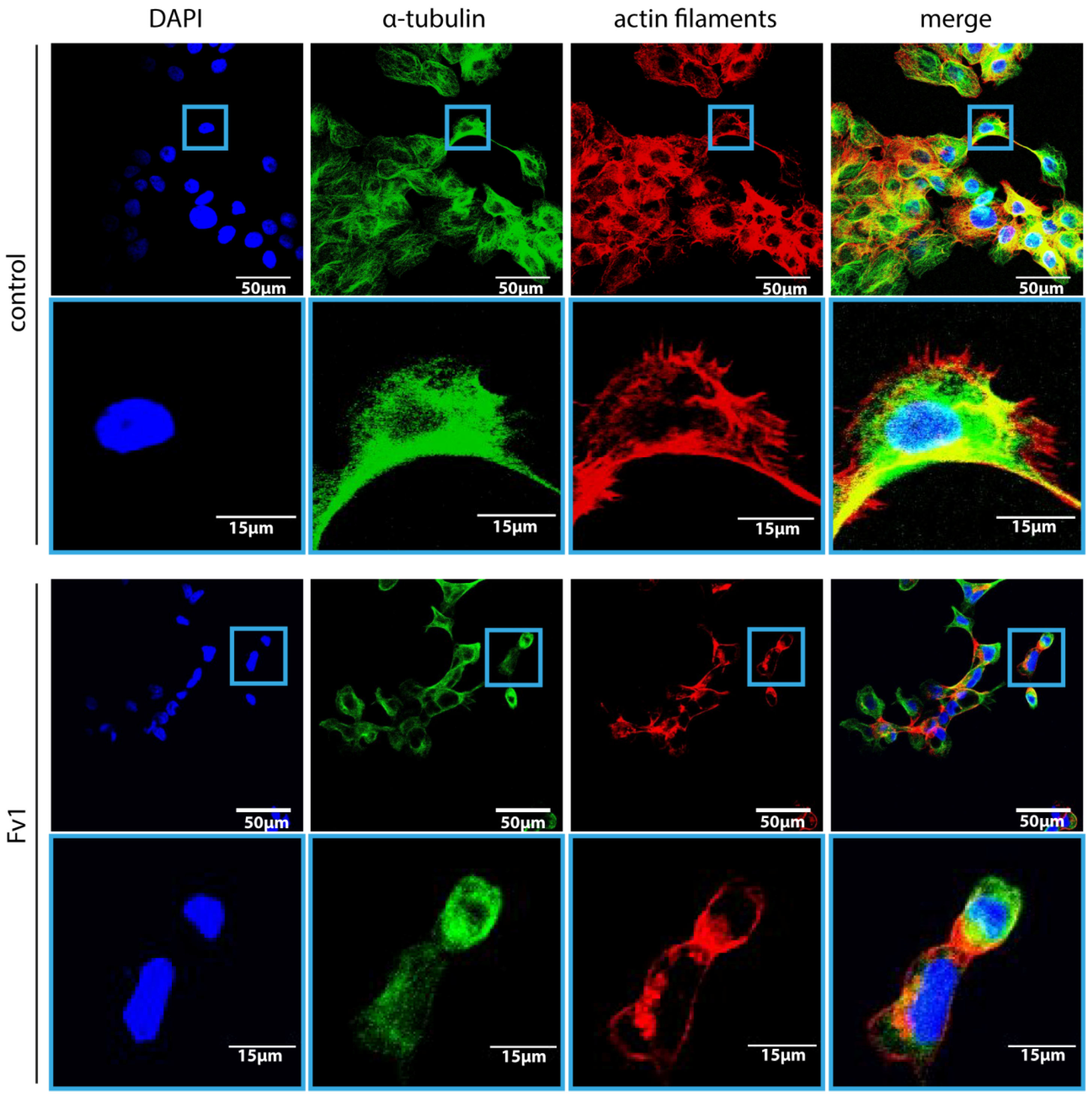
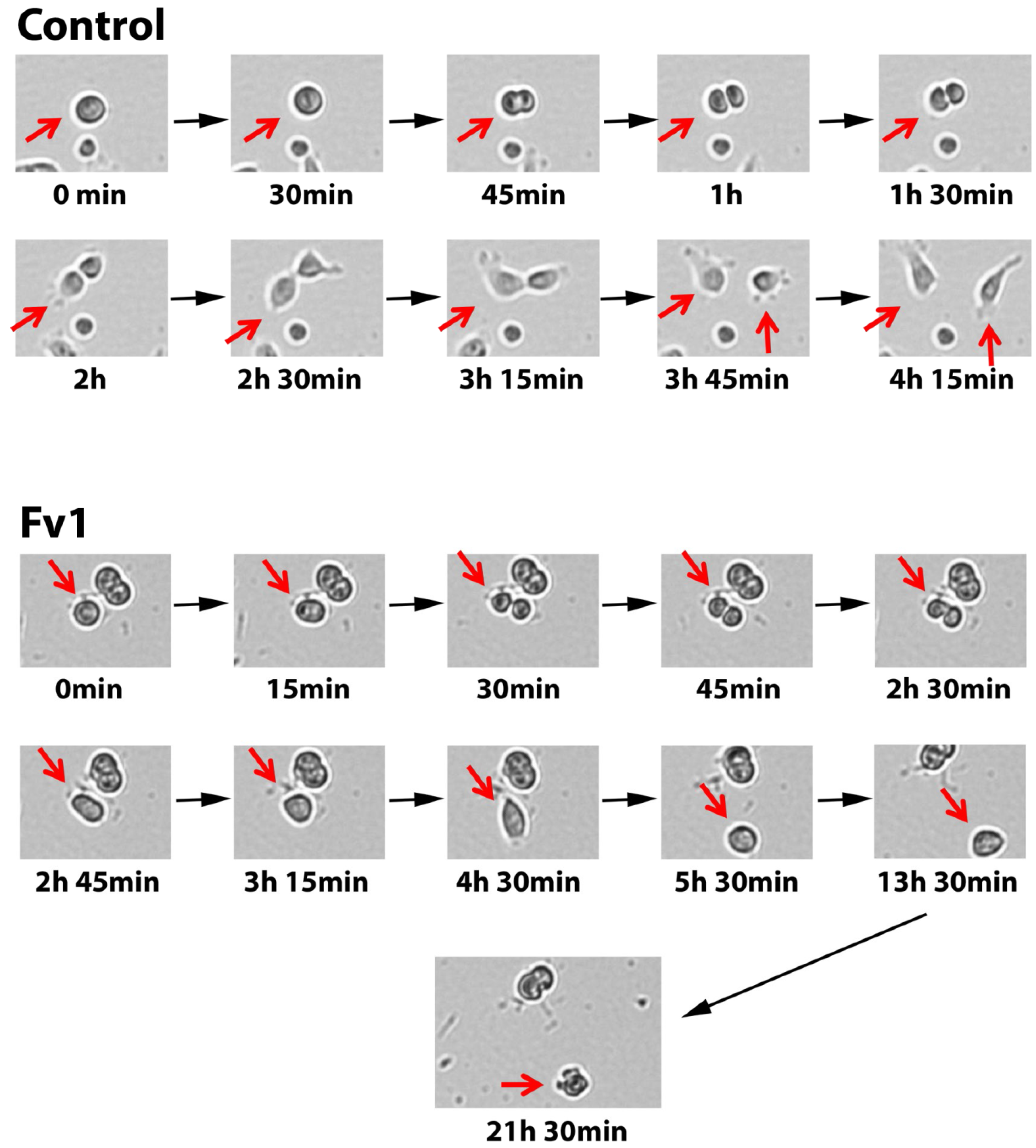
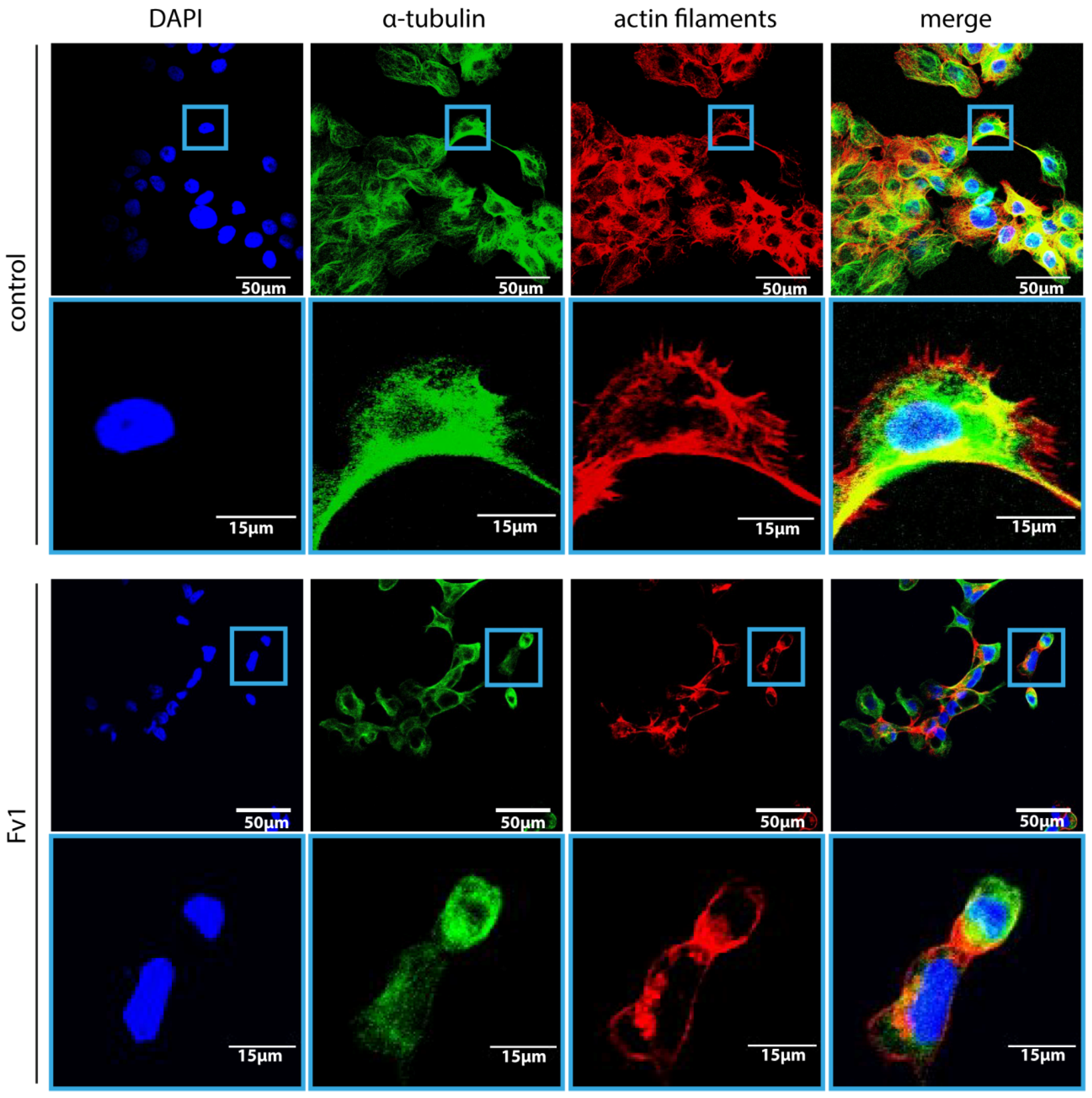
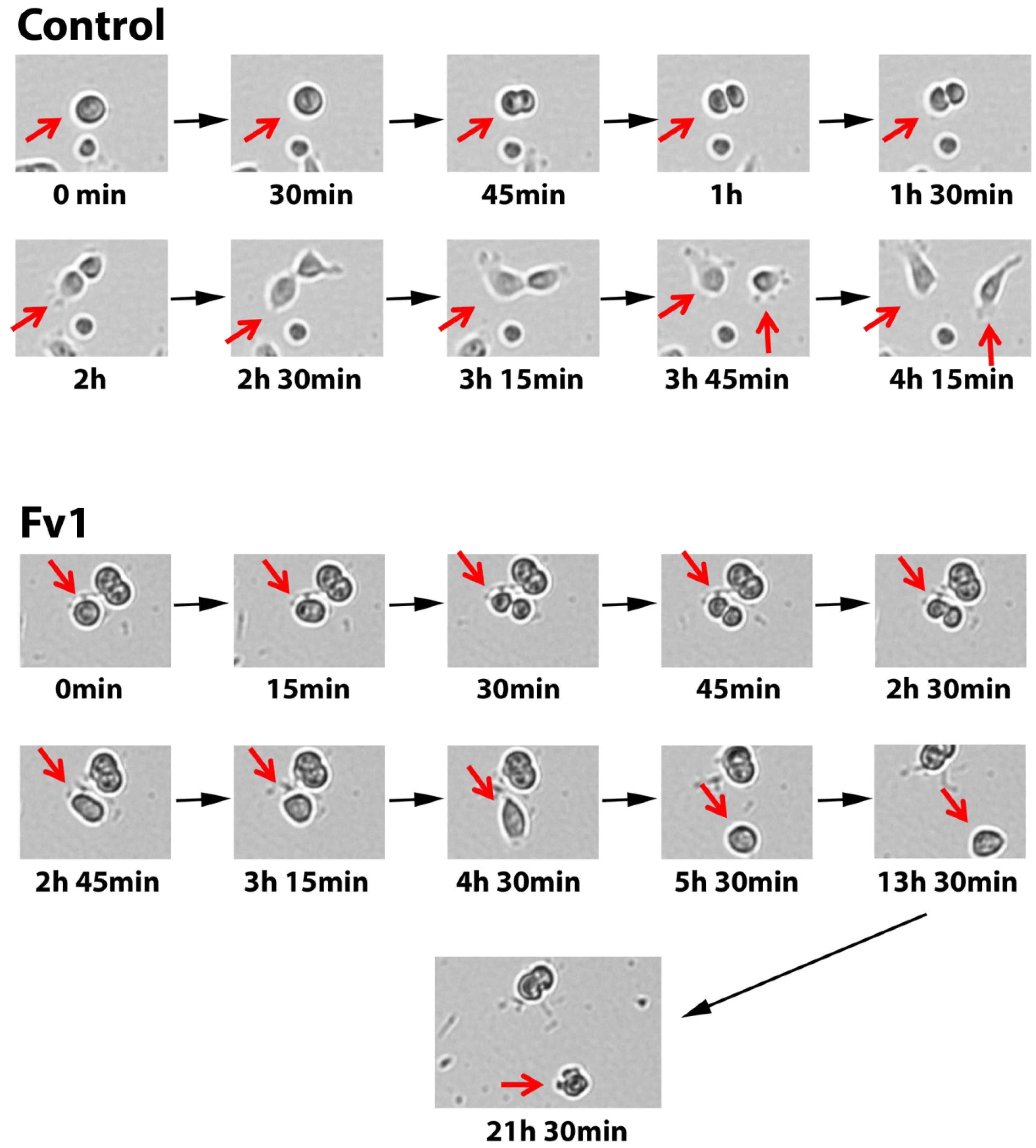
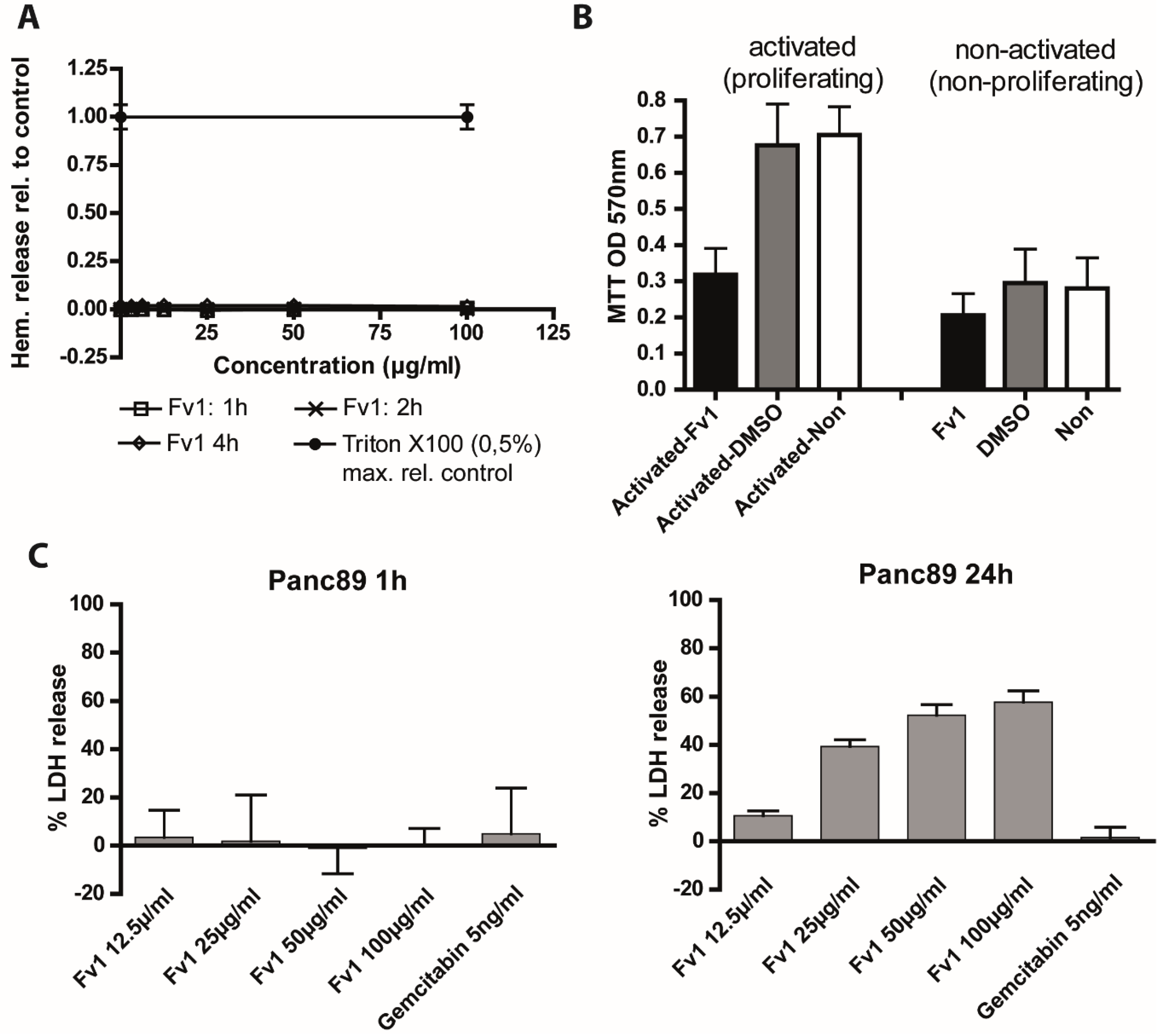
2.4. Analysis of the Fv1 Effect on Non-Dividing Cells
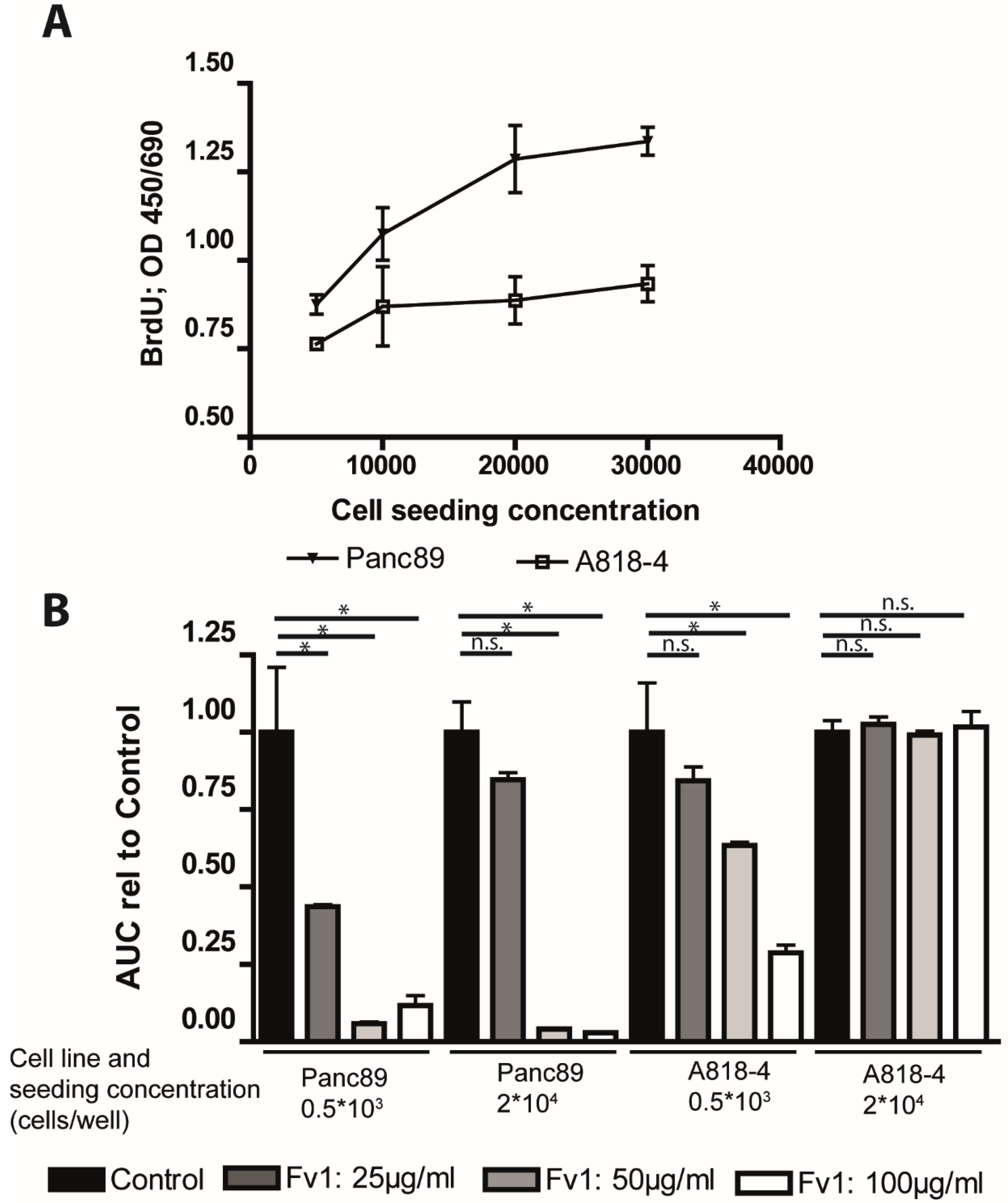
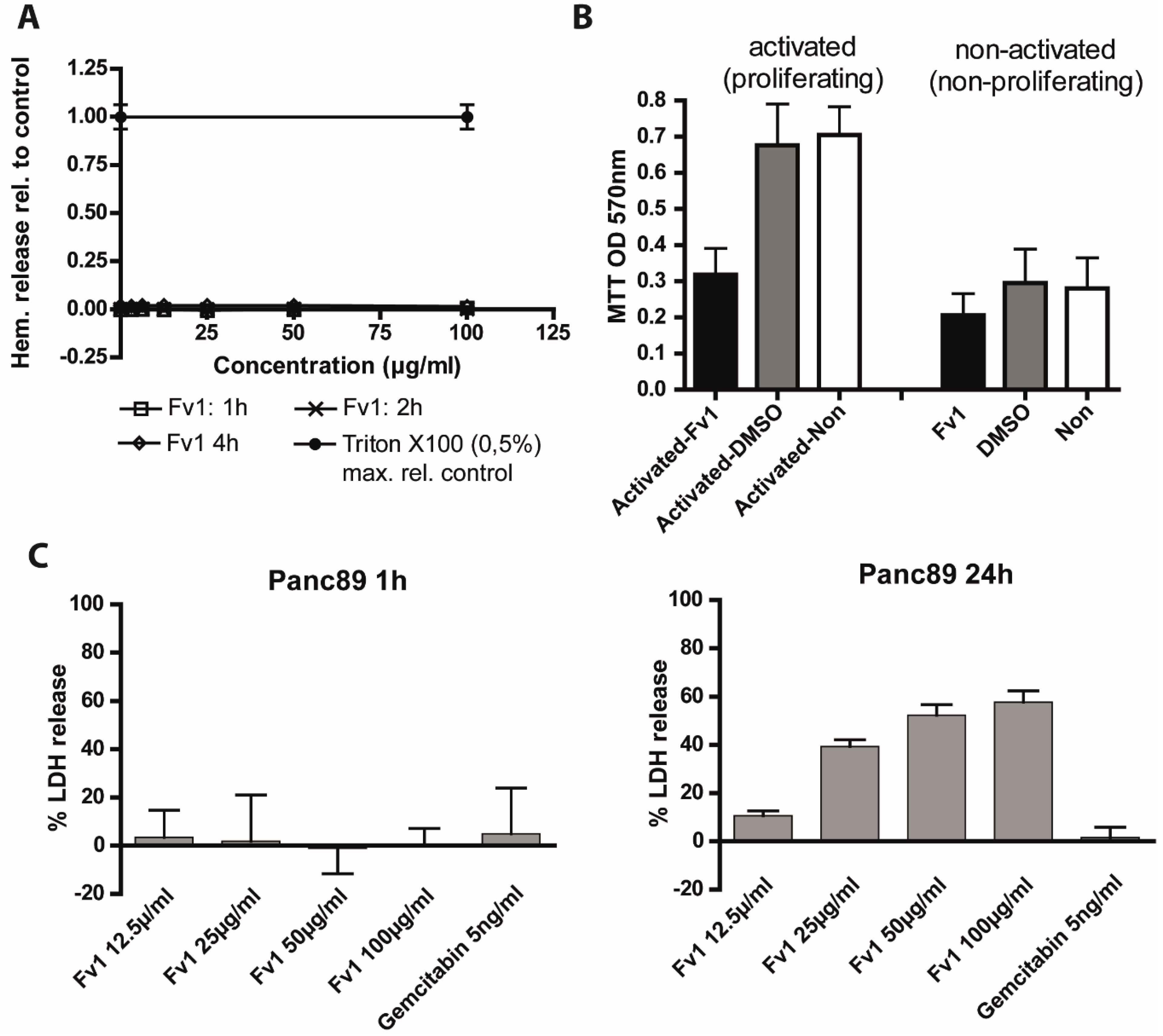
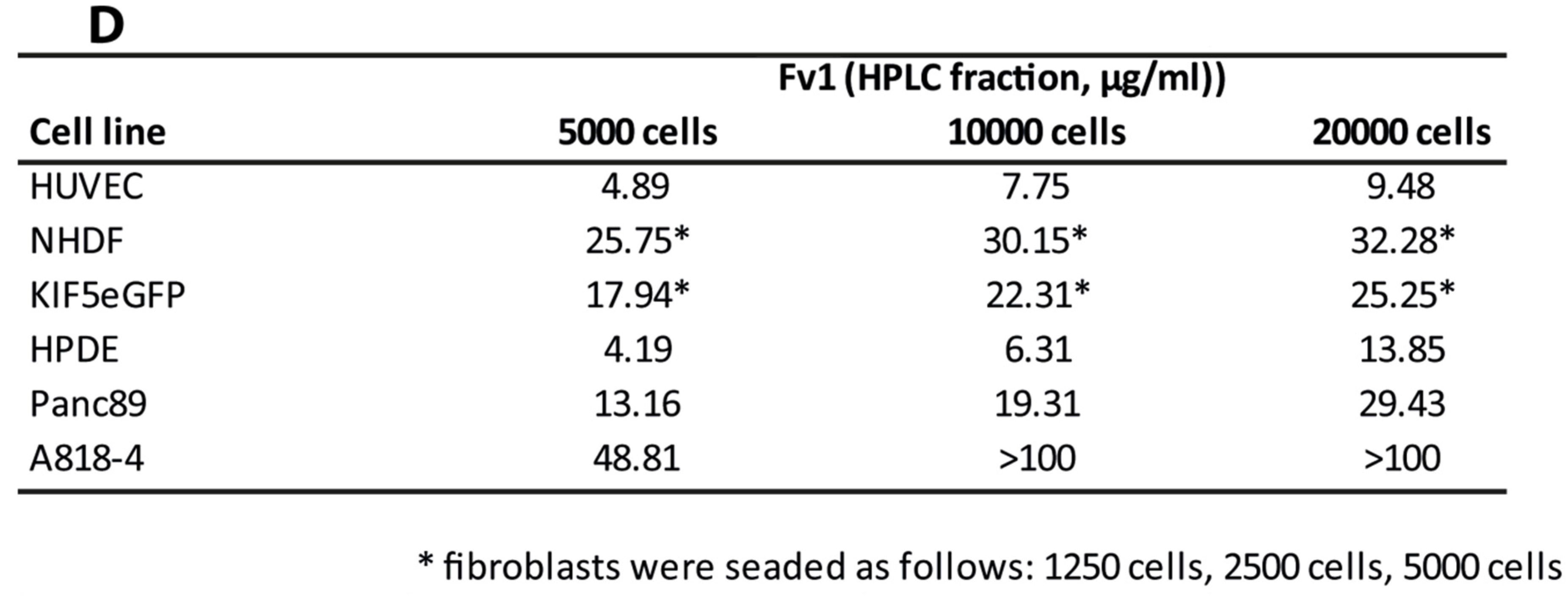
3. Discussion
4. Material and Methods
4.1. Algae Extraction and Fractioning
4.2. Cell Lines, Cell Culture Maintenance and General Experiment Procedure
4.3. Proliferation and Viability Experiments
4.4. Alamar Blue
4.5. BrdU ELISA
4.6. XCelligence Proliferation Measurement
4.7. Cytotoxicity LDH-Release
4.8. Gene Expression Analysis
4.9. Preparation of Cell Lysates and Western Blotting
4.10. FACS Cell Cycle Profiling—Annexin V/PI Staining
4.11. FACS PI Staining and Inhibitor Screening
4.12. Statistical Analysis
Supplementary Materials
- Table S1. Inhibition of cell viability by Fv1 in different cancer cell lines.
- Figure S1. Genes involved in the cell cycle regulation pathway are regulated by Fv1.
- Figure S2. Fv1 does not influence several proteins involved in cell cycle regulation.
- Figure S3. Fv1 does not influence several proteins involved in cell cycle regulation and cell death.
- Figure S4. Supplement to Figure 6. The effect of Fv1 on viability is not caspase dependent.
- Figure S5. Mitosox ROS production of Colo357 cells treated with Fv1.
- Figure S6. Mitosox ROS production of Colo357 cells treated with Antimycin as positive control.
- Figure S7. TMRM membrane potential of Colo357 cells treated with Fv1.
- Figure S8. TMRM membrane potential of Colo357 cells treated with FCCP as positive control.
- Figure S9. Supplement to Figure 8. Fv1 preferentially inhibits proliferating cells.
- Table S2. Fv1 does not induce hemolysis in fresh red blood cells.
- Figure S10. Acetonic algae extracts show additive effects in combinations with different chemotherapeutics.
- Figure S11. Cell cycle profile of Colo357 cells 4 h after treatment with Fv1.
- Figure S12. Cell cycle profile of Panc89 cells 4 h after treatment with Fv1.
- Figure S13. Cell cycle profile of Panc89 cells 24 h after treatment with Fv1.
- Multimedia file S1. Life cell imaging Control.
- Multimedia file S2. Life cell imaging Fv1.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- SEER Cancer Statistics Review, 1975–2011. National Cancer Institute Web site. Available online: http://seer.cancer.gov/csr/1975_2011/ (accessed on 7 November 2014).
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forma, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012: Globocan 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Ale, M.T.; Maruyama, H.; Tamauchi, H.; Mikkelsen, J.D.; Meyer, A.S. Fucose-Containing Sulfated Polysaccharides from Brown Seaweeds iNhibit Proliferation of Melanoma Cells and Induce Apoptosis by Activation of Caspase-3 in vitro. Mar. Drugs 2011, 9, 2605–2621. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.R.; Hosokawa, M.; Miyashita, K. Fucoxanthin: A Marine Carotenoid Exerting Anti-Cancer Effects by Affecting Multiple Mechanisms. Mar. Drugs 2013, 11, 5130–5147. [Google Scholar] [CrossRef] [PubMed]
- Glombitza, K.W.; Hauperich, S.; Keusgen, M. Phlorotannins from the brown algae Cystophora torulosa and Sargassum spinuligerum. Nat. Toxins 1997, 5, 58–63. [Google Scholar] [CrossRef]
- Khotimchenko, Y.S. Antitumor properties of nonstarch polysaccharides: Fucoidans and chitosans. Russ. J. Mar. Biol. 2010, 36, 321–330. [Google Scholar] [CrossRef]
- Park, H.S.; Kim, G.-Y.; Nam, T.-J.; Deuk Kim, N.; Hyun Choi, Y. Antiproliferative Activity of Fucoidan Was Associated with the Induction of Apoptosis and Autophagy in AGS Human Gastric Cancer Cells. J. Food Sci. 2011, 76, T77–T83. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.; Lu, Q.; Zhao, W.; Du, D.; Jin, L.; Liu, Y. Fucoxanthin induces apoptosis in human cervical cancer cell line HeLa via PI3K/Akt pathway. Tumor Biol. 2014, 35, 11261–11267. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.K.; Li, Y.; White, W.L.; Lu, J. Extracts from New Zealand Undaria pinnatifida Containing Fucoxanthin as Potential Functional Biomaterials against Cancer in vitro. J. Funct. Biomater. 2014, 5, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Ale, M.T.; Maruyama, H.; Tamauchi, H.; Mikkelsen, J.D.; Meyer, A.S. Fucoidan from Sargassum sp. and Fucus vesiculosus reduces cell viability of lung carcinoma and melanoma cells in vitro and activates natural killer cells in mice in vivo. Int. J. Biol. Macromol. 2011, 49, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Athukorala, Y.; Ahn, G.N.; Jee, Y.-H.; Kim, G.-Y.; Kim, S.-H.; Ha, J.-H.; Ha, J.-H.; Kang, J.S.; Lee, K.-W.; Jeon, Y.-J. Antiproliferative activity of sulfated polysaccharide isolated from an enzymatic digest of Ecklonia cava on the U-937 cell line. J. Appl. Phycol. 2008, 21, 307–314. [Google Scholar] [CrossRef]
- Peng, J.; Yuan, J.-P.; Wu, C.-F.; Wang, J.-H. Fucoxanthin, a Marine Carotenoid Present in Brown Seaweeds and Diatoms: Metabolism and Bioactivities Relevant to Human Health. Mar. Drugs 2011, 9, 1806–1828. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.S.; Chung, H.Y.; Kim, J.Y.; Son, B.W.; Jung, H.A.; Choi, J.S. Inhibitory phlorotannins from the edible brown alga Ecklonia stolonifera on total reactive oxygen species (ROS) generation. Arch. Pharm. Res. 2004, 27, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.-H.; Yang, Y.-I.; Lee, K.-T.; Choi, J.-H. Dieckol, isolated from the edible brown algae Ecklonia cava, induces apoptosis of ovarian cancer cells and inhibits tumor xenograft growth. J. Cancer Res. Clin. Oncol. 2015, 141, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.-S.; Kasin Yadunandam, A.; Kim, S.-J.; Woo, H.-C.; Kim, H.-R.; Kim, G.-D. Dieckol, isolated from Ecklonia stolonifera, induces apoptosis in human hepatocellular carcinoma Hep3B cells. J. Nat. Med. 2013, 67, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Shirayama, M.; Toth, A.; Galova, M.; Nasmyth, K. APCCdc20 promotes exit from mitosis by destroying the anaphase inhibitor Pds1 and cyclin Clb5. Nature 1999, 402, 203–207. [Google Scholar] [PubMed]
- Sipos, B.; Möser, S.; Kalthoff, H.; Török, V.; Löhr, M.; Klöppel, G. A comprehensive characterization of pancreatic ductal carcinoma cell lines: Towards the establishment of an in vitro research platform. Virchows Arch. 2003, 442, 444–452. [Google Scholar] [PubMed]
- Weinberg, R. The Biology of Cancer, 2nd ed.; Garland Science: New York, NY, USA, 2013. [Google Scholar]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.-T.; Zhou, T.-T.; Liu, B.; Bao, J.K. Programmed cell death pathways in cancer: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Madeo, F.; Zimmermann, A.; Maiuri, M.C.; Kroemer, G. Essential role for autophagy in life span extension. J. Clin. Invest. 2015, 125, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Czarny, P.; Pawlowska, E.; Bialkowska-Warzecha, J.; Kaarniranta, K.; Blasiak, J. Autophagy in DNA Damage Response. Int. J. Mol. Sci. 2015, 16, 2641–2662. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Tang, B.; Xie, X.; Xiao, Y.-F.; Yang, S.-M.; Zhang, J.-W. The Interplay Between DNA Repair and Autophagy in Cancer Therapy. Cancer Biol. Ther. 2015, 16, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Li, H.; Yan, X.-G.; Zhou, Z.-W.; Yi, Z.-G.; He, Z.-X.; Pan, X.T.; Yang, Y.X.; Wang, Z.Z.; Zhang, X.; et al. Alisertib induces cell cycle arrest and autophagy and suppresses epithelial-to-mesenchymal transition involving PI3K/Akt/mTOR and sirtuin 1-mediated signaling pathways in human pancreatic cancer cells. Drug Des. Devel. Ther. 2015, 9, 575–601. [Google Scholar] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel Plus Gem Citabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.-T.; Chen, H.; Wang, Z.-H.; Ni, Z.-L.; Liu, H.-B.; Tong, H.-F.; Guo, H.C.; Liu, D.L.; Lin, S.Z. Enhanced Antitumor Efficacy of Gemcitabine by Evodiamine on Pancreatic Cancer via Regulating PI3K/Akt Pathway. Int. J. Biol. Sci. 2012, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Stadel, D.; Cristofanon, S.; Abhari, B.A.; Deshayes, K.; Zobel, K.; Vucic, D.; Debatin, K.M.; Fulda, S. Requirement of Nuclear Factor κB for Smac Mimetic-Mediated Sensitization of Pancreatic Carcinoma Cells for Gemcitabine-Induced Apoptosis. Neoplasia 2011, 13, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Kong, N.; Wang, X.; Fang, Y.; Hu, X.; Xu, Y.; Chen, W.; Wang, K.; Li, D.; Jin, W.; et al. JNK confers 5-fluorouracil resistance in p53-deficient and mutant p53-expressing colon cancer cells by inducing survival autophagy. Sci. Rep. 2014, 4, 4694. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.; Mitrea, D.M.; Ou, L.; Kriwacki, R.W. Cell cycle regulation by the intrinsically disordered proteins p21 and p27. Biochem. Soc. Trans. 2012, 40, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Seillier, M.; Peuget, S.; Dusetti, N.J.; Carrier, A. Antioxidant Role of p53 and of Its Target TP53INP1. In Antioxidant. Enzyme; El-Missiry, M.A., Ed.; InTech Europe: Rijeka, Croatia, 2012. [Google Scholar]
- Eichhorn, J.M.; Sakurikar, N.; Alford, S.E.; Chu, R.; Chambers, T.C. Critical role of anti-apoptotic Bcl-2 protein phosphorylation in mitotic death. Cell Death Dis. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.T.; Woods, L.K.; Moore, G.E.; Quinn, L.A.; McGavran, L.; Gordon, S.G. Human cell line (COLO 357) of metastatic pancreatic adenocarcinoma. Int. J. Cancer 1980, 25, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Duguid, W.P.; Rosenberg, L.; Viallet, J.; Galloway, D.A.; Tsao, M.S. Long-term culture and immortalization of epithelial cells from normal adult human pancreatic ducts transfected by the E6E7 gene of human papilloma virus 16. Am. J. Pathol. 1996, 148, 1763–1770. [Google Scholar] [PubMed]
- Ouyang, H.; Mou, L.; Luk, C.; Liu, N.; Karaskova, J.; Squire, J.; Tsao, M.-S. Immortal Human Pancreatic Duct Epithelial Cell Lines with Near Normal Genotype and Phenotype. Am. J. Pathol. 2000, 157, 1623–1631. [Google Scholar] [CrossRef]
- Krishan, A. Rapid flow cytofluorometric analysis of mammalian cell cycle by propidium iodide staining. J. Cell Biol. 1975, 66, 188–193. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geisen, U.; Zenthoefer, M.; Peipp, M.; Kerber, J.; Plenge, J.; Managò, A.; Fuhrmann, M.; Geyer, R.; Hennig, S.; Adam, D.; et al. Molecular Mechanisms by Which a Fucus vesiculosus Extract Mediates Cell Cycle Inhibition and Cell Death in Pancreatic Cancer Cells. Mar. Drugs 2015, 13, 4470-4491. https://doi.org/10.3390/md13074470
Geisen U, Zenthoefer M, Peipp M, Kerber J, Plenge J, Managò A, Fuhrmann M, Geyer R, Hennig S, Adam D, et al. Molecular Mechanisms by Which a Fucus vesiculosus Extract Mediates Cell Cycle Inhibition and Cell Death in Pancreatic Cancer Cells. Marine Drugs. 2015; 13(7):4470-4491. https://doi.org/10.3390/md13074470
Chicago/Turabian StyleGeisen, Ulf, Marion Zenthoefer, Matthias Peipp, Jannik Kerber, Johannes Plenge, Antonella Managò, Markus Fuhrmann, Roland Geyer, Steffen Hennig, Dieter Adam, and et al. 2015. "Molecular Mechanisms by Which a Fucus vesiculosus Extract Mediates Cell Cycle Inhibition and Cell Death in Pancreatic Cancer Cells" Marine Drugs 13, no. 7: 4470-4491. https://doi.org/10.3390/md13074470
APA StyleGeisen, U., Zenthoefer, M., Peipp, M., Kerber, J., Plenge, J., Managò, A., Fuhrmann, M., Geyer, R., Hennig, S., Adam, D., Piker, L., Rimbach, G., & Kalthoff, H. (2015). Molecular Mechanisms by Which a Fucus vesiculosus Extract Mediates Cell Cycle Inhibition and Cell Death in Pancreatic Cancer Cells. Marine Drugs, 13(7), 4470-4491. https://doi.org/10.3390/md13074470