
Inhibition of N-Type Calcium Channels by Fluorophenoxyanilide Derivatives


 ,
,
Abstract
:
1. Introduction
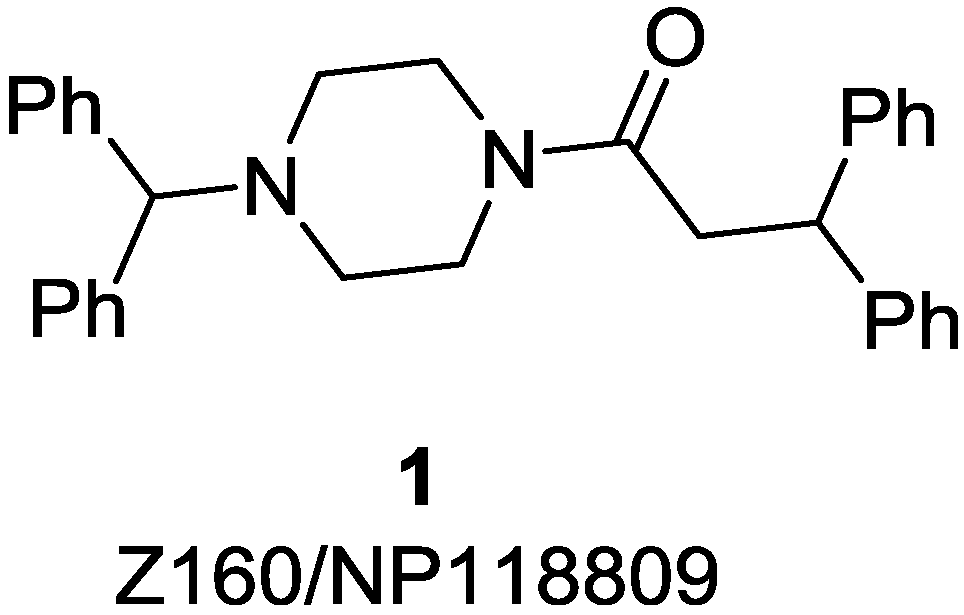
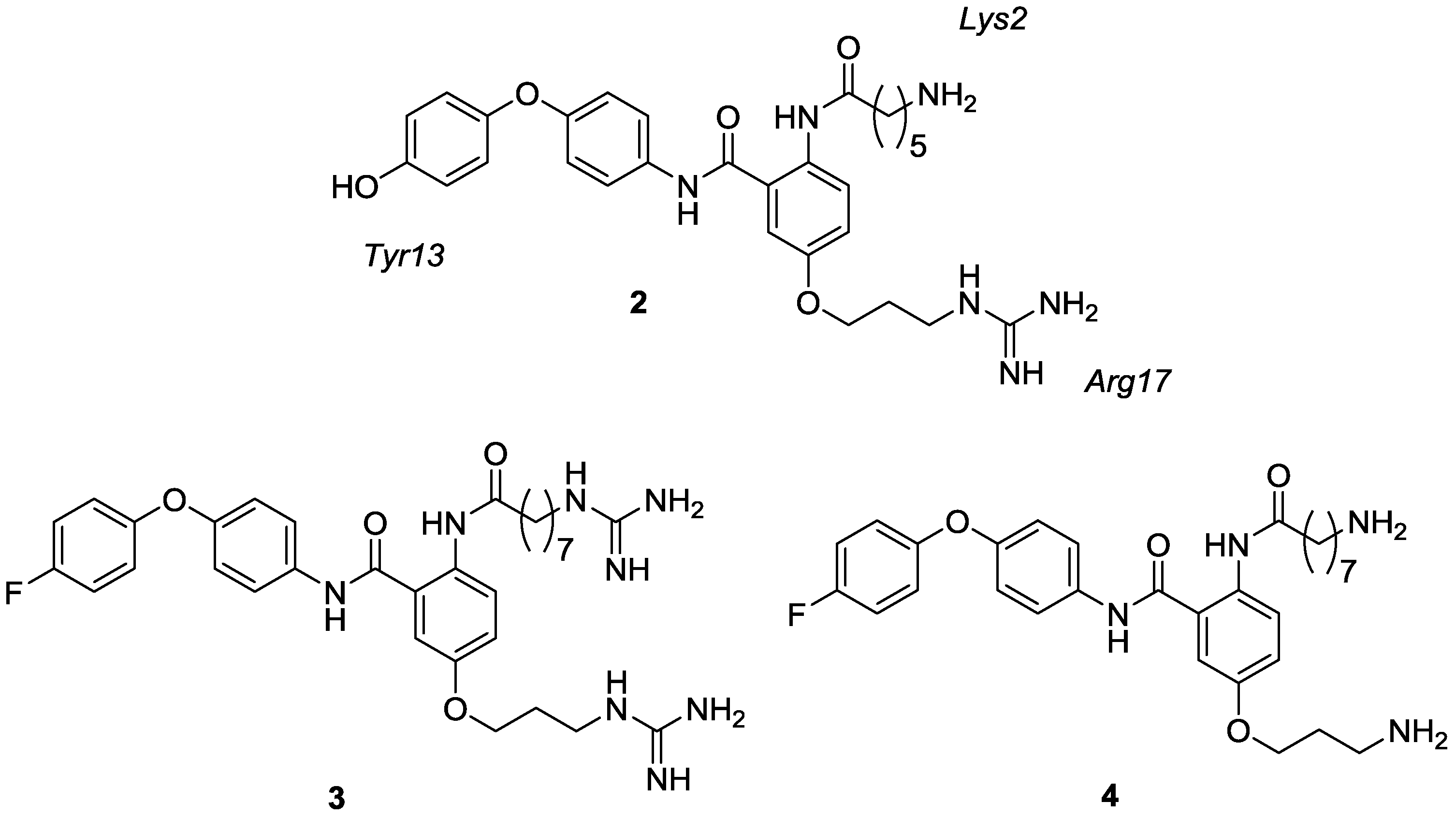
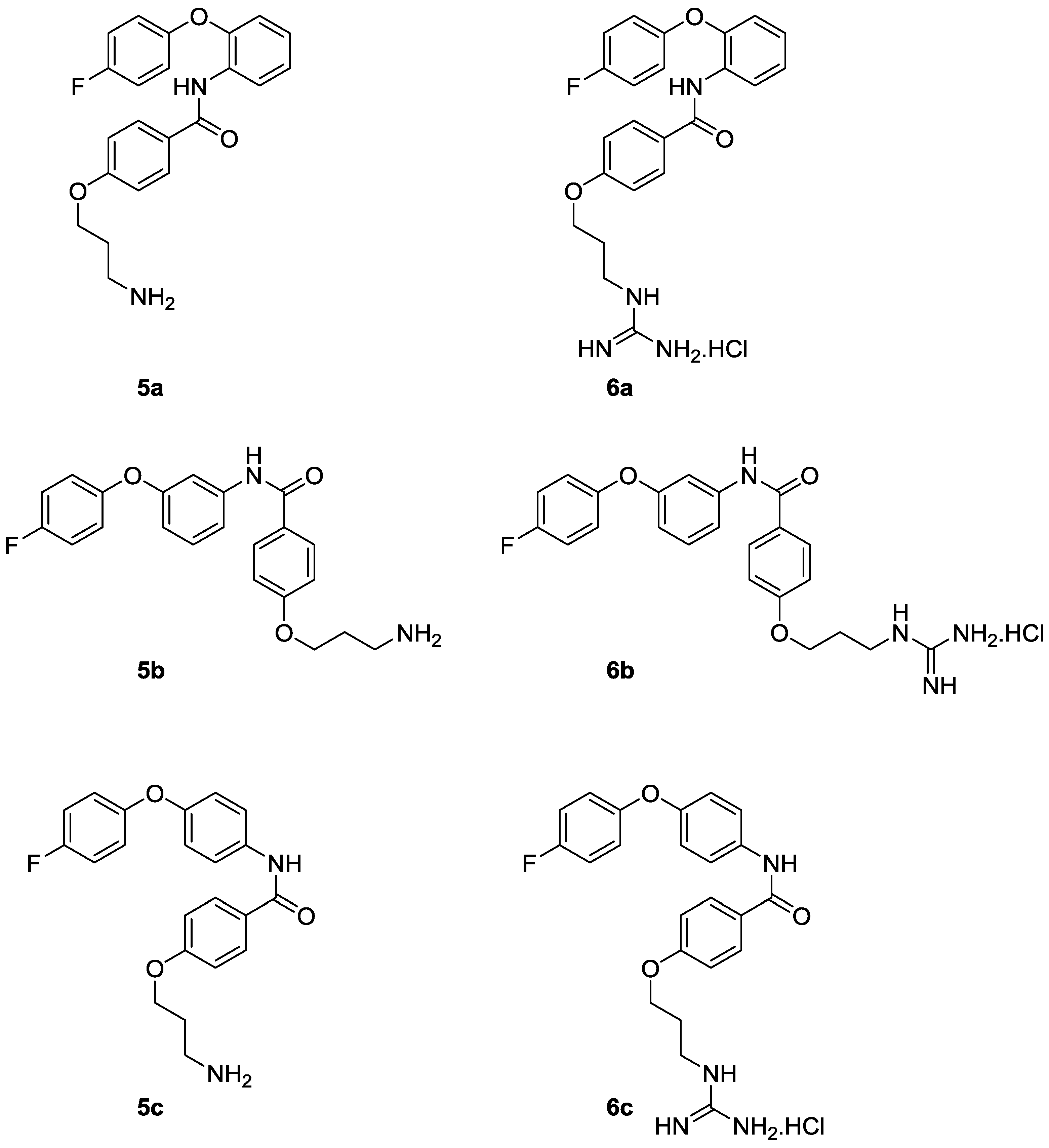
2. Results and Discussion
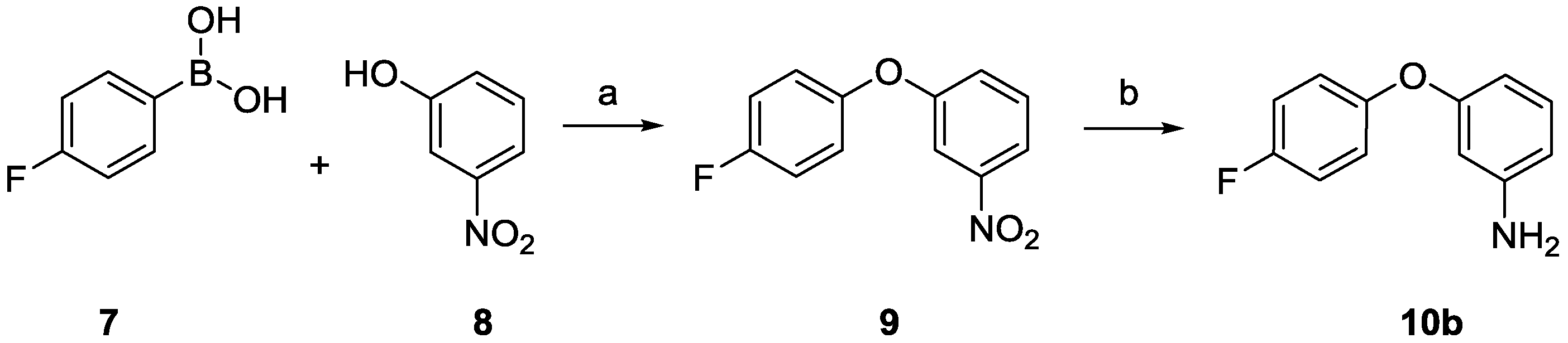
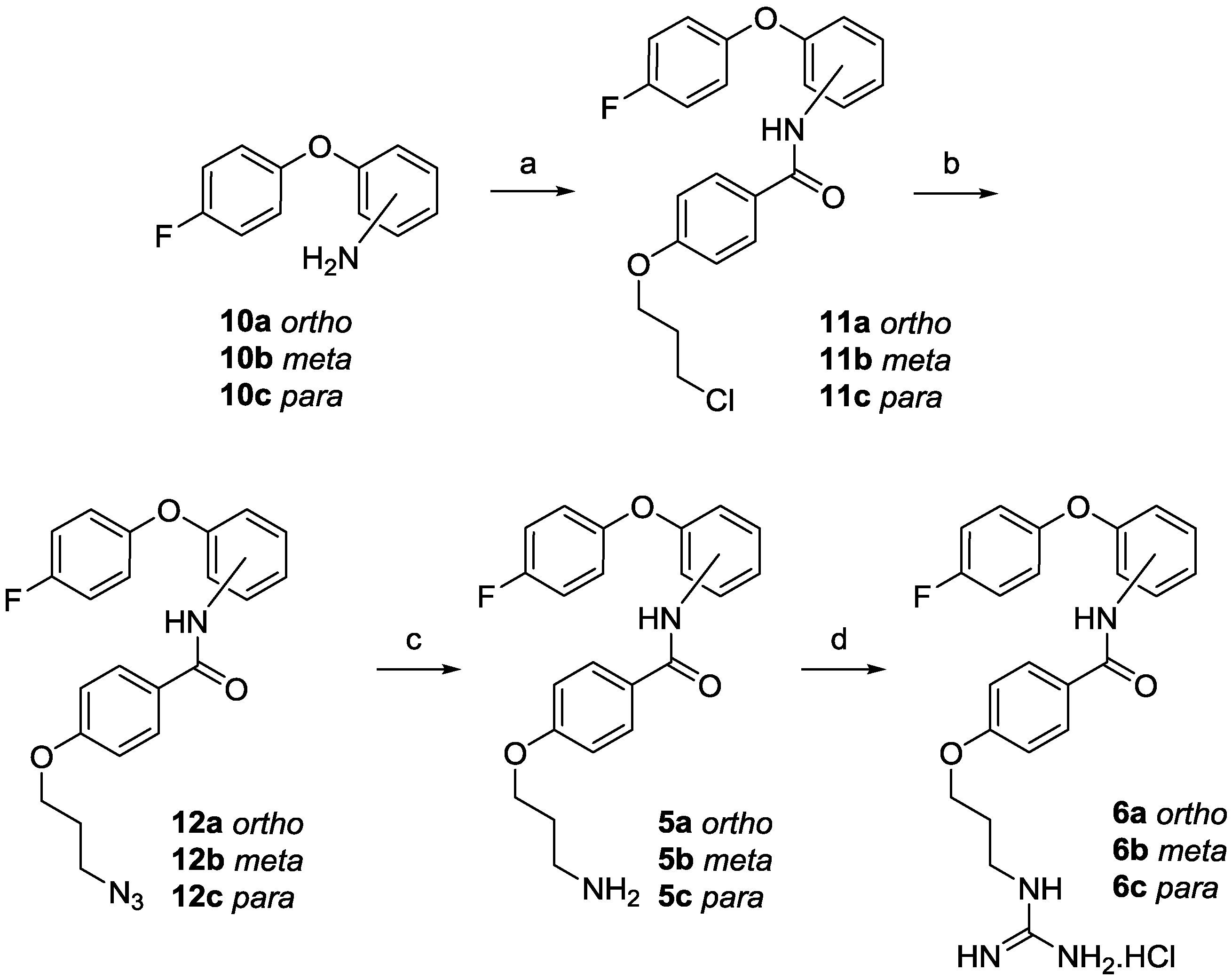
2.1. Chemistry
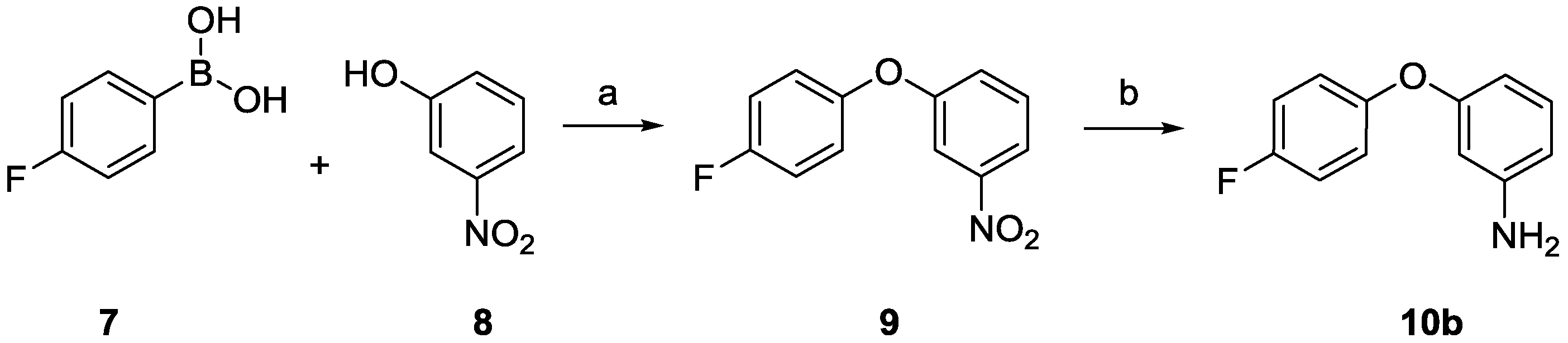

2.2. Biology

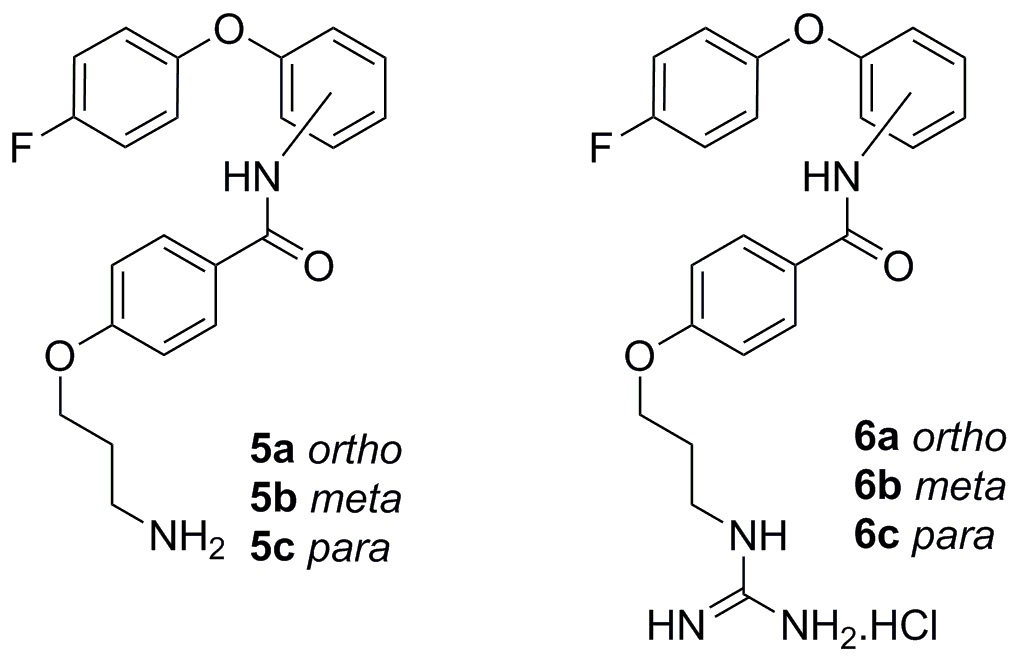{kind=link}
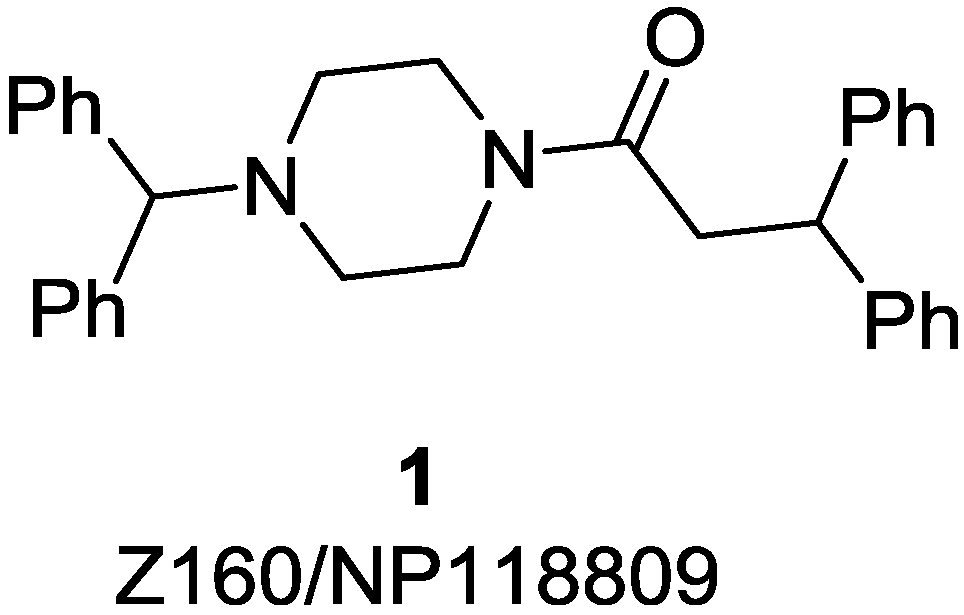{kind=link}
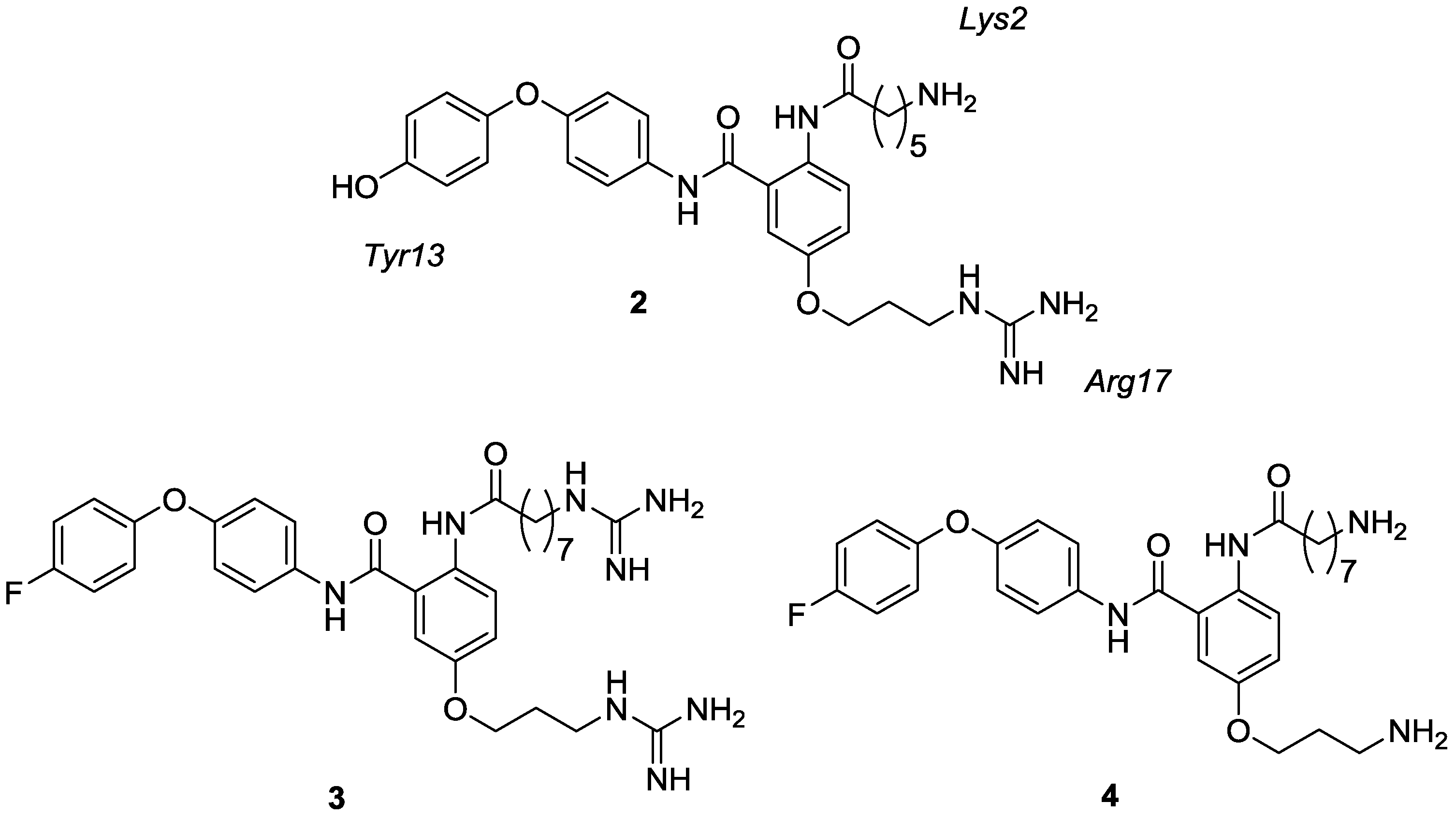{kind=link}
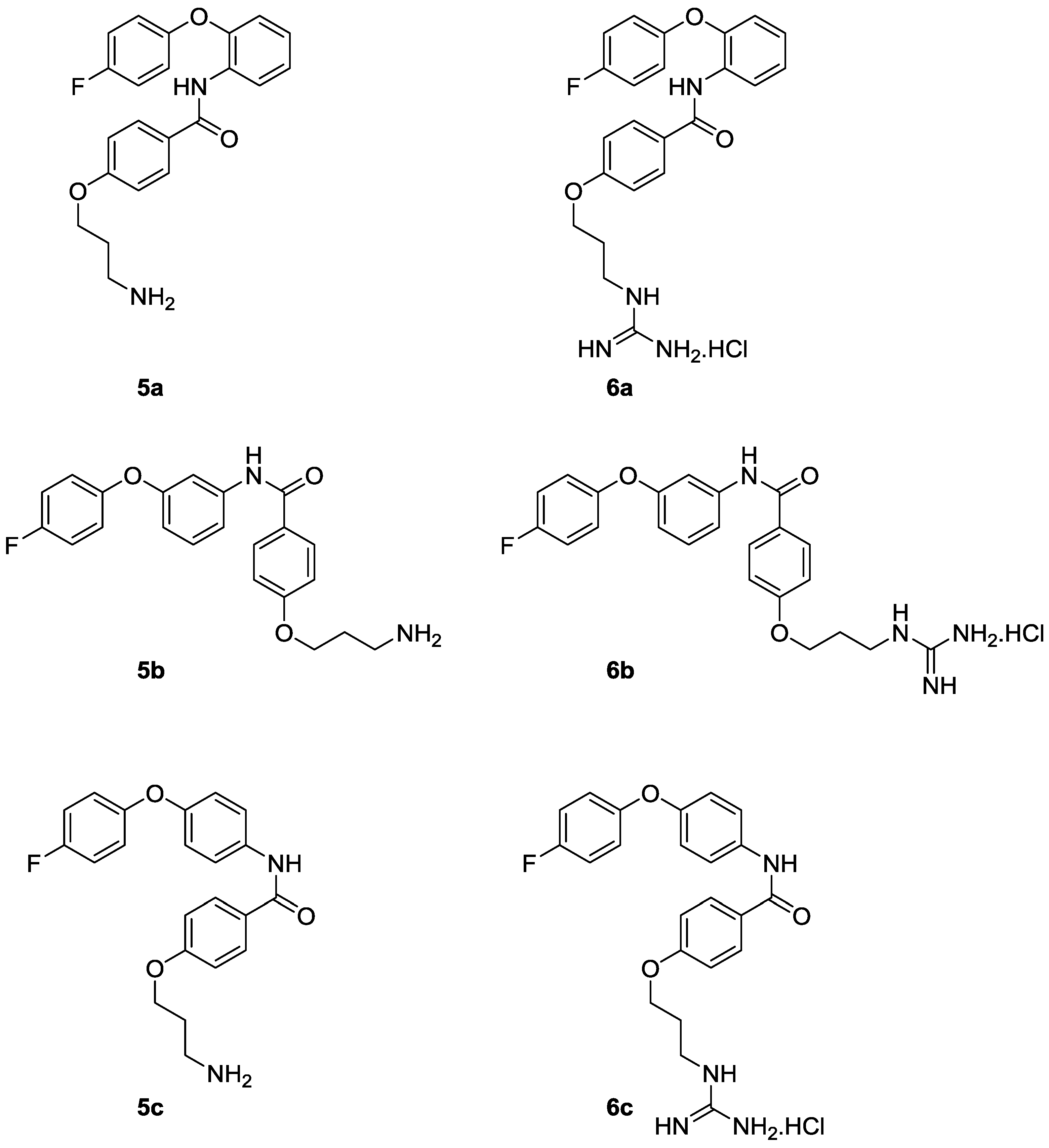{kind=link}
{kind=link}
{kind=link}
| Compound | logIC50 | SEM | IC50 (µM) | 95% CI (µM) |
|---|---|---|---|---|
| 3 a | ~–2.84 | 0.29 | 1452 b | 380–5550 |
| 5a | –4.34 | 0.01 | 46 | 44–48 |
| 6a | –3.90 | 0.03 | 124 c | 107–150 |
| 5b | –4.45 | 0.01 | 35 | 33–37 |
| 6b | –3.73 | 0.02 | 185 | 169–203 |
| 5c | ~–3.12 | 0.06 | 764 d | 575–1020 |
| 6c | –3.14 | 0.10 | 723 e | 447–1170 |
3. Experimental Section
3.1. Chemistry
3.1.1. General Experimental Procedures
3.1.2. Synthesis
4-(3-Chloropropoxy)-N-(2-(4-fluorophenoxy)phenyl)benzamide (11a)
4-(3-Azidopropoxy)-N-(2-(4-fluorophenoxy)phenyl)benzamide (12a)
4-(3-Aminopropoxy)-N-(2-(4-fluorophenoxy)phenyl)benzamide (5a)
N-(2-(4-Fluorophenoxy)phenyl)-4-(3-guanidinopropoxy)benzamide hydrochloride (6a)
1-(4-Fluorophenoxy)-3-nitrobenzene (9)
3-(4-Fluorophenoxy)aniline (10b)
4-(3-Chloropropoxy)-N-(3-(4-fluorophenoxy)phenyl)benzamide (11b)
4-(3-Azidopropoxy)-N-(3-(4-fluorophenoxy)phenyl)benzamide (12b)
4-(3-Aminopropoxy)-N-(3-(4-fluorophenoxy)phenyl)benzamide (5b)
N-(3-(4-Fluorophenoxy)phenyl)-4-(3-guanidinopropoxy)benzamide hydrochloride (6b)
4-(3-Chloropropoxy)-N-(4-(4-fluorophenoxy)phenyl)benzamide (11c)
4-(3-Azidopropoxy)-N-(4-(4-fluorophenoxy)phenyl)benzamide (12c)
4-(3-Aminopropoxy)-N-(4-(4-fluorophenoxy)phenyl)benzamide (5c)
N-(4-(4-Fluorophenoxy)phenyl)-4-(3-guanidinopropoxy)benzamide hydrochloride (6c)
3.2. Biology
Fluorescence Measurement of Calcium Responses
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vink, S.; Alewood, P.F. Targeting voltage-gated calcium channels: Developments in peptide and small-molecule inhibitors for the treatment of neuropathic pain. Br. J. Pharmacol. 2012, 167, 970–989. [Google Scholar] [CrossRef] [PubMed]
- Altier, C.; Zamponi, G.W. Targeting Ca2+ channels to treat pain: T-type versus N-type. Trends Pharmacol. Sci. 2004, 25, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S. Chapter four—Recent progress in the discovery and development of N-type calcium channel modulators for the treatment of pain. In Progress in Medicinal Chemistry; Lawton, G., Witty, D.R., Eds.; Elsevier: Oxford, UK, 2014; Volume 53, pp. 147–186. [Google Scholar]
- Perret, D.; Luo, Z.D. Targeting voltage-gated calcium channels for neuropathic pain management. NeuroRX 2009, 6, 679–692. [Google Scholar]
- Pope, J.E.; Deer, T.R. Ziconotide: A clinical update and pharmacologic review. Exp. Opin. Pharmacother. 2013, 14, 957–966. [Google Scholar] [CrossRef]
- Bear, B.; Asgian, J.; Termin, A.; Zimmermann, N. Small molecules targeting sodium and calcium channels for neuropathic pain. Curr. Opin. Drug Discov. Dev. 2009, 12, 543–561. [Google Scholar]
- Yamamoto, T.; Takahara, A. Recent updates of N-type calcium channel blockers with therapeutic potential for neuropathic pain and stroke. Curr. Top. Med. Chem. 2009, 9, 377–395. [Google Scholar] [CrossRef] [PubMed]
- Zamponi, G.W.; Feng, Z.-P.; Zhang, L.; Pajouhesh, H.; Ding, Y.; Belardetti, F.; Pajouhesh, H.; Dolphin, D.; Mitscher, L.A.; Snutch, T.P. Scaffold-based design and synthesis of potent N-type calcium channel blockers. Bioorg. Med. Chem. Lett. 2009, 19, 6467–6472. [Google Scholar] [CrossRef] [PubMed]
- Abbadie, C.; McManus, O.B.; Sun, S.-Y.; Bugianesi, R.M.; Dai, G.; Haedo, R.J.; Herrington, J.B.; Kaczorowski, G.J.; Smith, M.M.; Swensen, A.M.; et al. Analgesic effects of a substituted N-triazole oxindole (Trox-1), a state-dependent, voltage-gated calcium channel 2 blocker. J. Pharmacol. Exp. Ther. 2010, 334, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Pajouhesh, H.; Feng, Z.-P.; Ding, Y.; Zhang, L.; Pajouhesh, H.; Morrison, J.-L.; Belardetti, F.; Tringham, E.; Simonson, E.; Vanderah, T.W.; et al. Structure-activity relationships of diphenylpiperazine N-type calcium channel inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 1378–1383. [Google Scholar] [CrossRef] [PubMed]
- Tyagarajan, S.; Chakravarty, P.K.; Park, M.; Zhou, B.; Herrington, J.B.; Ratliff, K.; Bugianesi, R.M.; Williams, B.; Haedo, R.J.; Swensen, A.M.; et al. A potent and selective indole N-type calcium channel (Cav2.2) blocker for the treatment of pain. Bioorg. Med. Chem. Lett. 2011, 21, 869–873. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Ohno, S.; Niwa, S.; Tokumasu, M.; Hagihara, M.; Koganei, H.; Fujita, S.-I.; Takeda, T.; Saitou, Y.; Iwayama, S.; et al. Asymmetric synthesis and biological evaluations of (+)- and (−)-6-dimethoxymethyl-1,4-dihydropyridine-3-carboxylic acid derivatives blocking N-type calcium channels. Bioorg. Med. Chem. Lett. 2011, 21, 3317–3319. [Google Scholar] [CrossRef] [PubMed]
- Beebe, X.; Darczak, D.; Henry, R.F.; Vortherms, T.; Janis, R.; Namovic, M.; Donnelly-Roberts, D.; Kage, K.L.; Surowy, C.; Milicic, I.; et al. Synthesis and SAR of 4-aminocyclopentapyrrolidines as N-type Ca2+ channel blockers with analgesic activity. Bioorg. Med. Chem. 2012, 20, 4128–4139. [Google Scholar] [CrossRef] [PubMed]
- Scott, V.E.; Vortherms, T.A.; Niforatos, W.; Swensen, A.M.; Neelands, T.; Milicic, I.; Banfor, P.N.; King, A.; Zhong, C.; Simler, G.; et al. A-1048400 is a novel, orally active, state-dependent neuronal calcium channel blocker that produces dose-dependent antinociception without altering hemodynamic function in rats. Biochem. Pharmacol. 2012, 83, 406–418. [Google Scholar] [CrossRef] [PubMed]
- Subasinghe, N.L.; Wall, M.J.; Winters, M.P.; Qin, N.; Lubin, M.L.; Finley, M.F.A.; Brandt, M.R.; Neeper, M.P.; Schneider, C.R.; Colburn, R.W.; et al. A novel series of pyrazolylpiperidine N-type calcium channel blockers. Bioorg. Med. Chem. Lett. 2012, 22, 4080–4083. [Google Scholar] [CrossRef] [PubMed]
- Shao, P.P.; Ye, F.; Chakravarty, P.K.; Varughese, D.J.; Herrington, J.B.; Dai, G.; Bugianesi, R.M.; Haedo, R.J.; Swensen, A.M.; Warren, V.A.; et al. Aminopiperidine sulfonamide Cav2.2 channel inhibitors for the treatment of chronic pain. J. Med. Chem. 2012, 55, 9847–9855. [Google Scholar] [CrossRef] [PubMed]
- Pajouhesh, H.; Feng, Z.-P.; Zhang, L.; Pajouhesh, H.; Jiang, X.; Hendricson, A.; Dong, H.; Tringham, E.; Ding, Y.; Vanderah, T.W.; et al. Structure-activity relationships of trimethoxybenzyl piperazine N-type calcium channel inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 4153–4158. [Google Scholar] [CrossRef] [PubMed]
- Beebe, X.; Yeung, C.M.; Darczak, D.; Shekhar, S.; Vortherms, T.A.; Miller, L.; Milicic, I.; Swensen, A.M.; Zhu, C.Z.; Banfor, P.; et al. Synthesis and SAR of 4-aminocyclopentapyrrolidines as orally active N-type calcium channel inhibitors for inflammatory and neuropathic pain. Bioorg. Med. Chem. Lett. 2013, 23, 4857–4861. [Google Scholar] [CrossRef] [PubMed]
- Borzenko, A.; Pajouhesh, H.; Morrison, J.-L.; Tringham, E.; Snutch, T.P.; Schafer, L.L. Modular, efficient synthesis of asymmetrically substituted piperazine scaffolds as potent calcium channel blockers. Bioorg. Med. Chem. Lett. 2013, 23, 3257–3261. [Google Scholar] [CrossRef] [PubMed]
- Shao, P.P.; Ye, F.; Chakravarty, P.K.; Herrington, J.B.; Dai, G.; Bugianesi, R.M.; Haedo, R.J.; Swensen, A.M.; Warren, V.A.; Smith, M.M.; et al. Improved Cav2.2 channel inhibitors through a gem-dimethylsulfone bioisostere replacement of a labile sulfonamide. ACS Med. Chem. Lett. 2013, 4, 1064–1068. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chu, K.L.; Zhu, C.Z.; Niforatos, W.; Swensen, A.; Searle, X.; Lee, L.; Jarvis, M.F.; McGaraughty, S. A mixed Ca2+ channel blocker, A-1264087, utilizes peripheral and spinal mechanisms to inhibit spinal nociceptive transmission in a rat model of neuropathic pain. J. Neurophysiol. 2014, 111, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.Z.; Vortherms, T.A.; Zhang, M.; Xu, J.; Swensen, A.M.; Niforatos, W.; Neelands, T.; Milicic, I.; Lewis, L.G.; Zhong, C.; et al. Mechanistic insights into the analgesic efficacy of A-1264087, a novel neuronal Ca2+ channel blocker that reduces nociception in rat preclinical pain models. J. Pain 2014, 15, 387.e1–387.e14. [Google Scholar] [CrossRef] [PubMed]
- Mollica, A.; Costante, R.; Novellino, E.; Stefanucci, A.; Pieretti, S.; Zador, F.; Samavati, R.; Borsodi, A.; Benyhe, S.; Vetter, I.; et al. Design, synthesis and biological evaluation of two opioid agonist and Cav2.2 blocker multitarget ligands. Chem. Biol. Drug Des. 2014. [Google Scholar] [CrossRef]
- Duggan, P.J.; Faber, J.M.; Graham, J.E.; Lewis, R.J.; Lumsden, N.G.; Tuck, K.L. Synthesis and Cav2.2 binding data for non-peptide mimetics of ω-conotoxin GVIA based on a 5-amino-anthranilamide core. Aust. J. Chem. 2008, 61, 11–15. [Google Scholar] [CrossRef]
- Andersson, A.; Baell, J.B.; Duggan, P.J.; Graham, J.E.; Lewis, R.J.; Lumsden, N.G.; Tranberg, C.E.; Tuck, K.L.; Yang, A. ω-Conotoxin GVIA mimetics based on an anthranilamide core: Effect of variation in ammonium side chain lengths and incorporation of fluorine. Bioorg. Med. Chem. 2009, 17, 6659–6670. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Duggan, P.J.; Forsyth, S.A.; Lewis, R.J.; Lok, Y.P.; Schroeder, C.I. Synthesis and biological evaluation of nonpeptide mimetics of ω-conotoxin GVIA. Bioorg. Med. Chem. 2004, 12, 4025–4037. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Duggan, P.J.; Forsyth, S.A.; Lewis, R.J.; Lok, Y.P.; Schroeder, C.I.; Shepherd, N.E. Synthesis and biological evaluation of anthranilamide-based non-peptide mimetics of ω-conotoxin GVIA. Tetrahedron 2006, 62, 7284–7292. [Google Scholar] [CrossRef]
- Duggan, P.J.; Lewis, R.J.; Lok, Y.P.; Lumsden, N.G.; Tuck, K.L.; Yang, A. Low molecular weight non-peptide mimics of ω-conotoxin GVIA. Bioorg. Med. Chem. Lett. 2009, 19, 2763–2765. [Google Scholar] [CrossRef] [PubMed]
- Tranberg, C.E.; Yang, A.; Vetter, I.; McArthur, J.R.; Baell, J.B.; Lewis, R.J.; Tuck, K.L.; Duggan, P.J. ω-Conotoxin GVIA mimetics that bind and inhibit neuronal Cav2.2 ion channels. Mar. Drugs 2012, 10, 2349–2368. [Google Scholar] [CrossRef] [PubMed]
- Press release investors and media—zalicus. Available online: http://phx.corporate-ir.net/phoenix.zhtml?c=148036&p=irol-newsArticle&ID=1874737&highlight= (accessed on 30 January 2015).
- Lauri, G.; Bartlett, P. Caveat: A program to facilitate the design of organic molecules. J. Comput. -Aided. Mol. Des. 1994, 8, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.; Forsyth, S.; Gable, R.; Norton, R.; Mulder, R. Design and synthesis of type-III mimetics of ω-conotoxin GVIA. J. Comput. -Aided. Mol. Des. 2001, 15, 1119–1136. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, M.-I.; Purcell, A.W.; Devi, R.; Lew, R.; Rossjohn, J.; Smith, A.I.; Perlmutter, P. β-amino acid-containing hybrid peptides-new opportunities in peptidomimetics. Org. Biomol. Chem. 2007, 5, 2884–2890. [Google Scholar] [CrossRef] [PubMed]
- Wen, F.; Zhang, H.; Yu, Z.; Jin, H.; Yang, Q.; Hou, T. Design, synthesis and antifungal/insecticidal evaluation of novel nicotinamide derivatives. Pestic. Biochem. Physiol. 2010, 98, 248–253. [Google Scholar] [CrossRef]
- Bertrand, I.; Capet, M.; Lecomte, J.-M.; Levoin, N.; Ligneau, X.; Poupardin-Olivier, O.; Robert, P.; Schwartz, J.-C.; Labeeuw, O. Preparation of pyrrolidinyl- and piperidinylpropoxyarenes as histamine H3 receptor ligands. WO2006117609 A2, 9 November 2006. [Google Scholar]
- Genzer, J.D.; Huttrer, C.P.; Wessem, G.C.V. Phenoxy- and benzyloxyalkyl thiocyanates. J. Am. Chem. Soc. 1951, 73, 3159–3162. [Google Scholar] [CrossRef]
- Altin, J.G.; Banwell, M.G.; Coghlan, P.A.; Easton, C.J.; Nairn, M.R.; Offermann, D.A. Synthesis of NTA3-DTDA—A chelator-lipid that promotes stable binding of His-tagged proteins to membranes. Aust. J. Chem. 2006, 59, 302–306. [Google Scholar] [CrossRef]
- Bernatowicz, M.S.; Wu, Y.; Matsueda, G.R. 1H-Pyrazole-1-carboxamidine hydrochloride an attractive reagent for guanylation of amines and its application to peptide synthesis. J. Org. Chem. 1992, 57, 2497–2502. [Google Scholar] [CrossRef]
- Sousa, S.R.; Vetter, I.; Ragnarsson, L.; Lewis, R.J. Expression and pharmacology of endogenous Cav channels in SH-SY5Y human neuroblastoma cells. PLOS ONE 2013, 8, e59293. [Google Scholar] [CrossRef] [PubMed]
- McArthur, J.R.; Adams, D.J.; RMIT University, Melbourne, Victoria, Australia. Personal Communication, 2015. The SH-SY5Y FLIPR assay has previously been shown to be a good predictor of Cav activity in other functional assays such as whole-cell patch clamp electrophysiology measurements. Correspondingly, compound 6a was tested for its ability to block calcium currents in HEK293 cells stably expressing human Cav2.2 channels, giving an IC50 of 36.3 ± 2.4 µM (n = 4–7).
- Alvarez, S.G.; Alvarez, M.T. A practical procedure for the synthesis of alkyl azides at ambient temperature in dimethyl sulfoxide in high purity and yield. Synthesis 1997, 1997, 413–414. [Google Scholar] [CrossRef]
- Evans, D.A.; Katz, J.L.; West, T.R. Synthesis of diaryl ethers through the copper-promoted arylation of phenols with arylboronic acids. An expedient synthesis of thyroxine. Tetrahedron Lett. 1998, 39, 2937–2940. [Google Scholar] [CrossRef]
- Bavin, P.M.G. The preparation of amines and hydrozo compounds using hydrazine and palladized charcoal. Can. J. Chem. 1958, 36, 238–241. [Google Scholar] [CrossRef]
- Maiti, D.; Buchwald, S.L. Orthogonal Cu- and Pd-based catalyst systems for the O- and N-arylation of aminophenols. J. Am. Chem. Soc. 2009, 131, 17423–17429. [Google Scholar] [CrossRef] [PubMed]
- Pajouhesh, H.; Lenz, G. Medicinal chemical properties of successful central nervous system drugs. Neurotherapeutics 2005, 2, 541–553. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gleeson, E.C.; Graham, J.E.; Spiller, S.; Vetter, I.; Lewis, R.J.; Duggan, P.J.; Tuck, K.L. Inhibition of N-Type Calcium Channels by Fluorophenoxyanilide Derivatives. Mar. Drugs 2015, 13, 2030-2045. https://doi.org/10.3390/md13042030
Gleeson EC, Graham JE, Spiller S, Vetter I, Lewis RJ, Duggan PJ, Tuck KL. Inhibition of N-Type Calcium Channels by Fluorophenoxyanilide Derivatives. Marine Drugs. 2015; 13(4):2030-2045. https://doi.org/10.3390/md13042030
Chicago/Turabian StyleGleeson, Ellen C., Janease E. Graham, Sandro Spiller, Irina Vetter, Richard J. Lewis, Peter J. Duggan, and Kellie L. Tuck. 2015. "Inhibition of N-Type Calcium Channels by Fluorophenoxyanilide Derivatives" Marine Drugs 13, no. 4: 2030-2045. https://doi.org/10.3390/md13042030
APA StyleGleeson, E. C., Graham, J. E., Spiller, S., Vetter, I., Lewis, R. J., Duggan, P. J., & Tuck, K. L. (2015). Inhibition of N-Type Calcium Channels by Fluorophenoxyanilide Derivatives. Marine Drugs, 13(4), 2030-2045. https://doi.org/10.3390/md13042030