
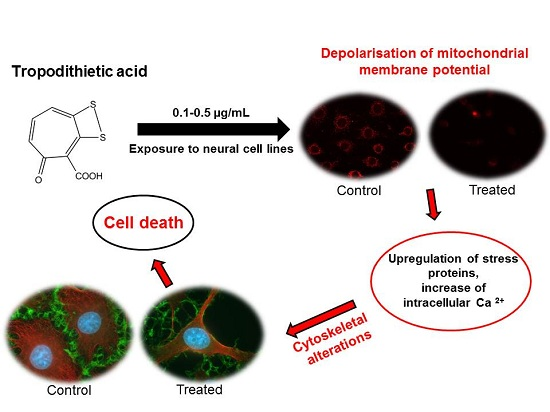
Cytotoxic Effects of Tropodithietic Acid on Mammalian Clonal Cell Lines of Neuronal and Glial Origin


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
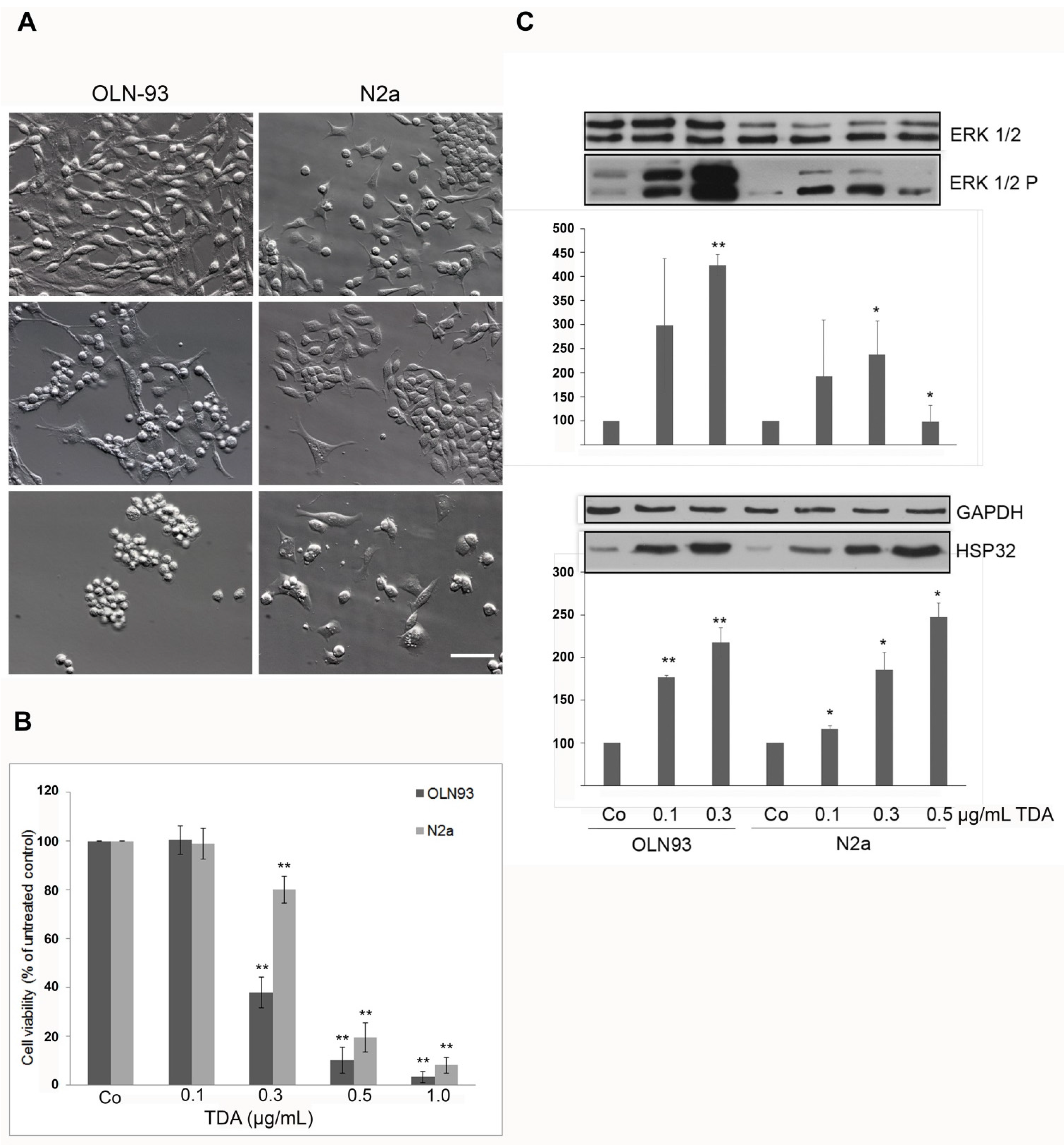
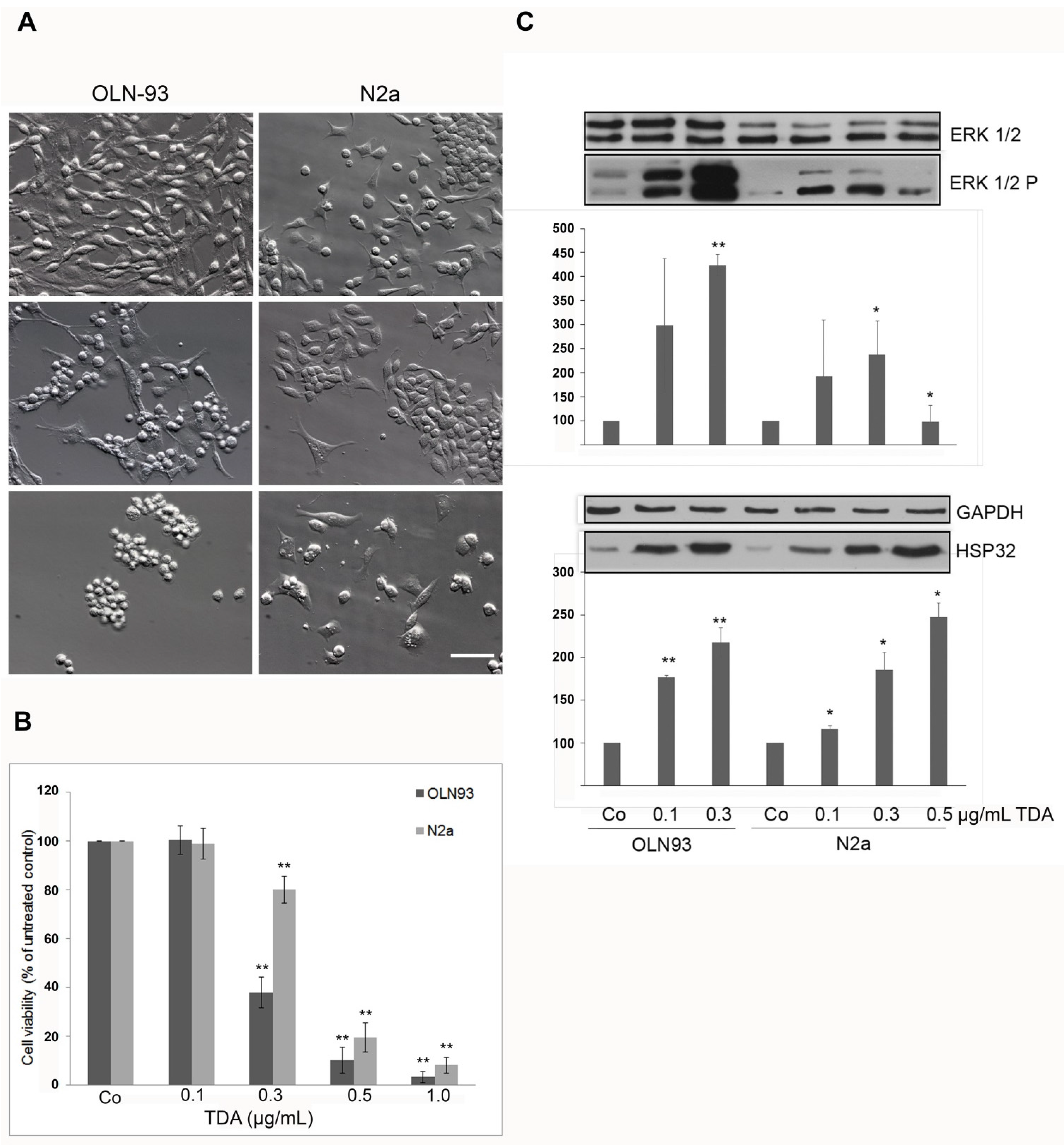
2.1. TDA Induced Cytotoxicity, Upregulation of Hsp32 and ERK1/2 Activation
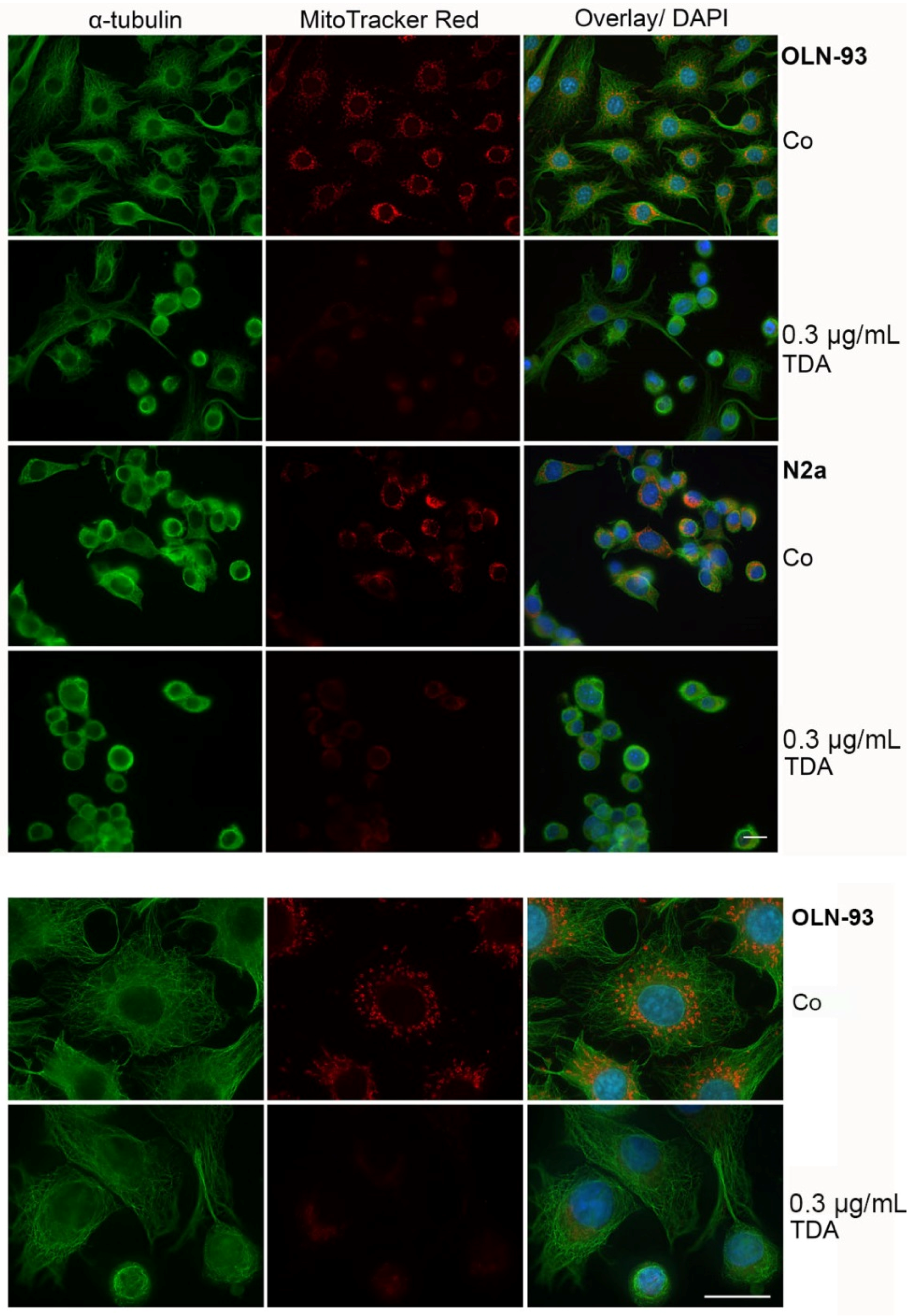
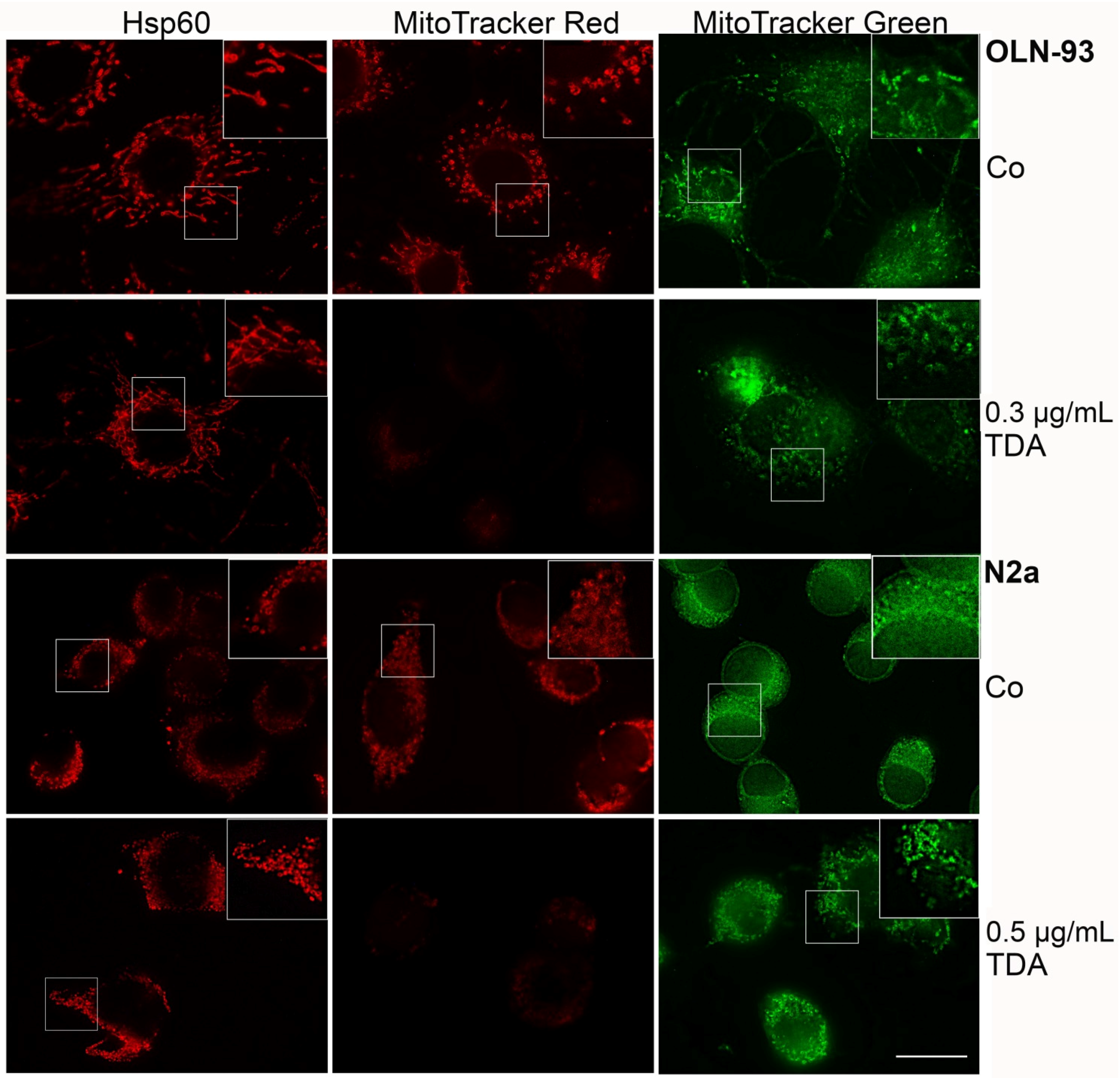
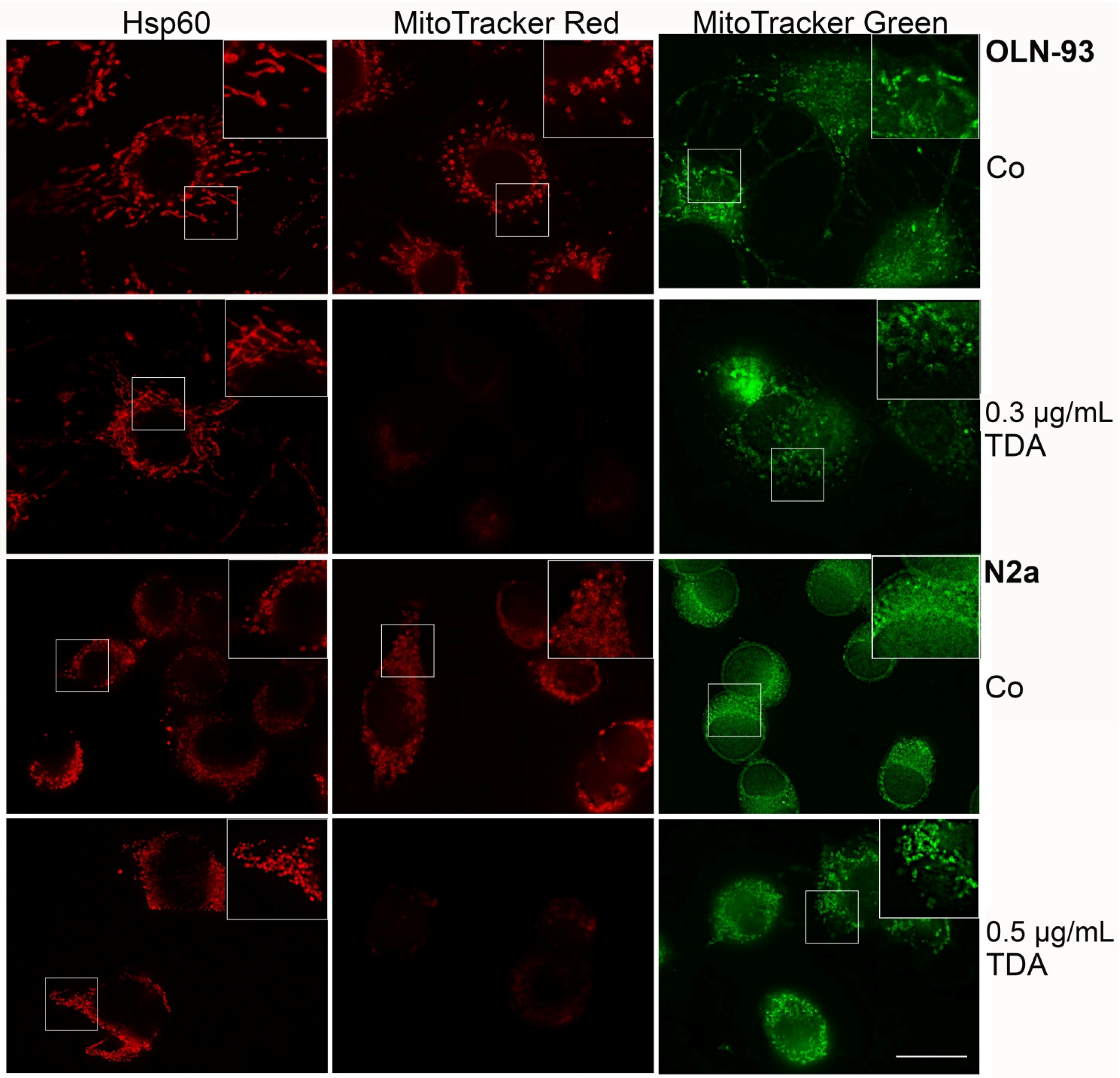
2.2. TDA Affects Microtubule Organization and Causes Mitochondrial Impairment
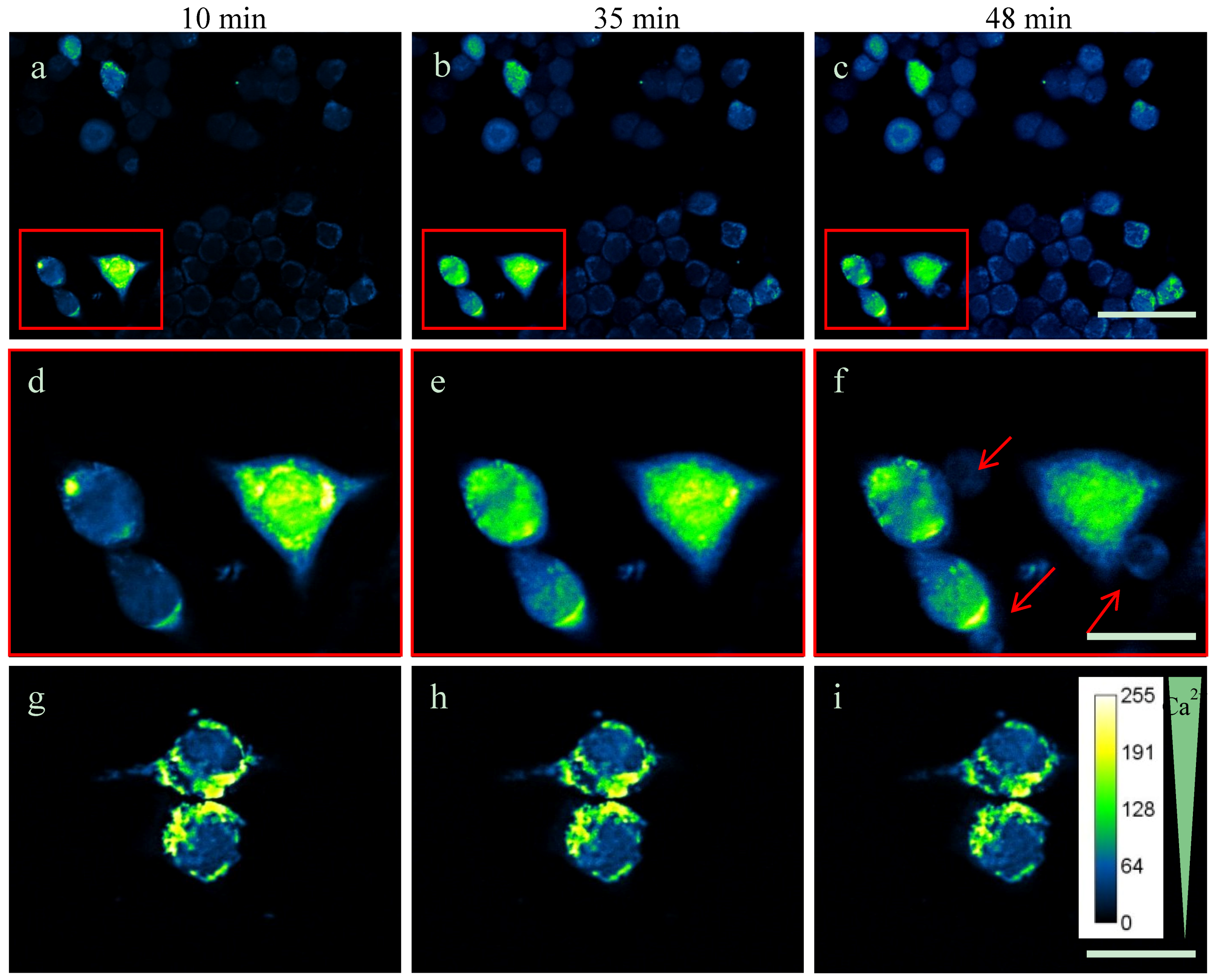
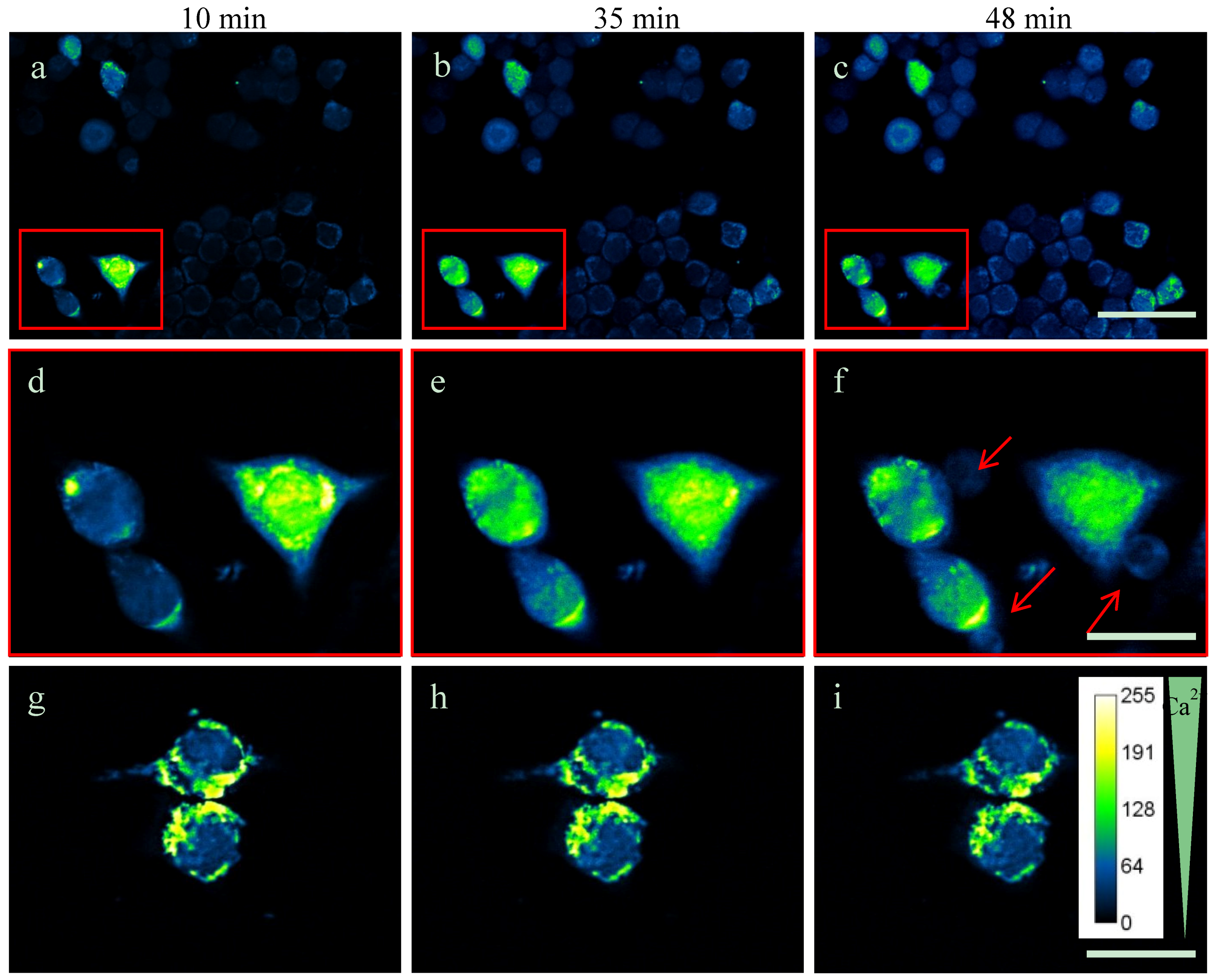
2.3. Intracellular Ca2+-Levels Are Increased after Supplementing TDA
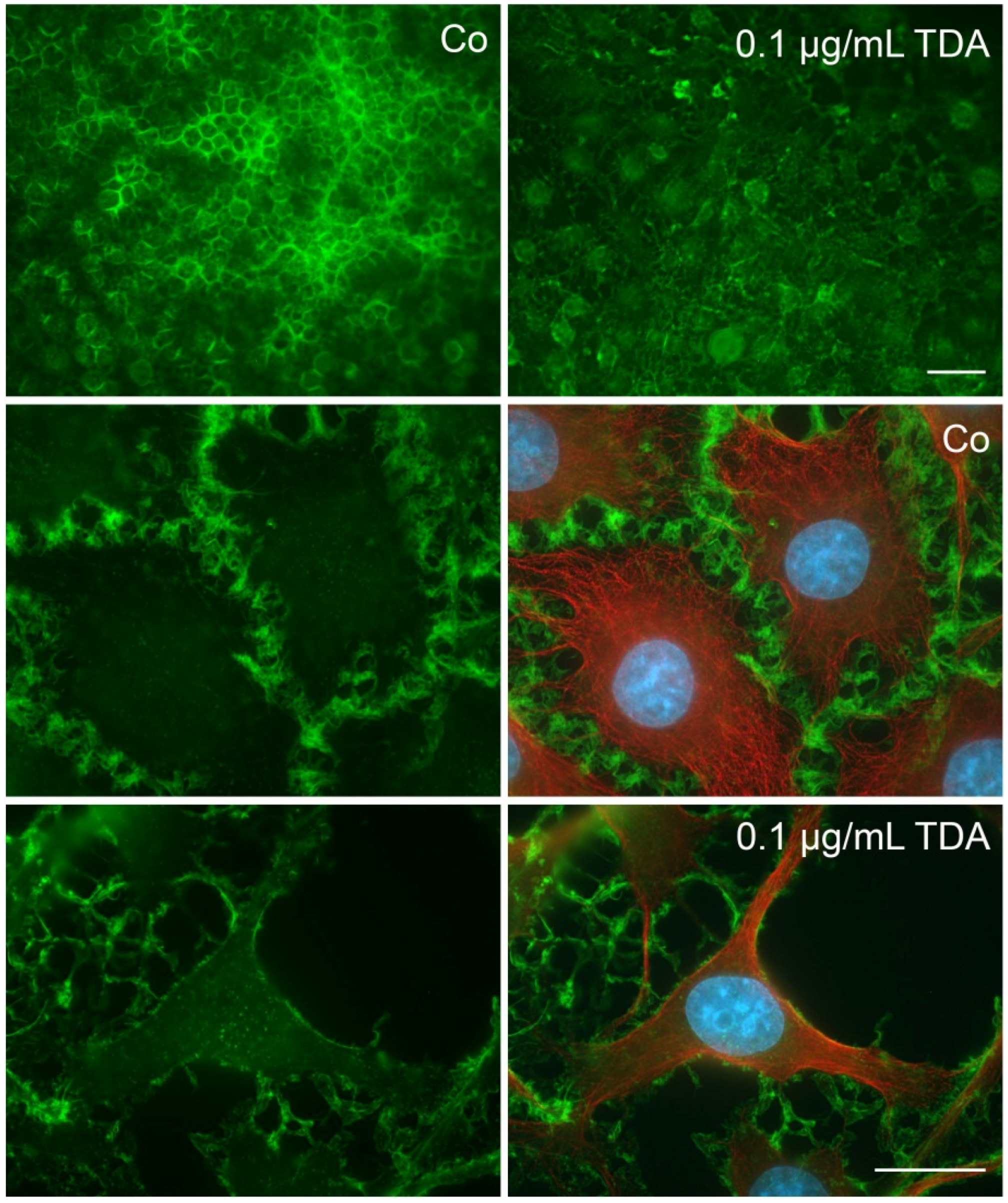
2.4. Influence of TDA on Microfilament Organization

3. Experimental Section
3.1. Materials and Antibodies
3.2. Cell Culture
3.3. Immunoblot Analysis
3.4. Mitochondrial Staining
3.5. Indirect Immunofluorescence
3.6. MTT-Viability Assay
3.7. Fura-2 AM Staining
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sakai, R.; Swanson, G.T. Recent progress in neuroactive marine natural products. Nat. Prod. Rep. 2014, 31, 273–309. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H.; Moore, B.S. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol. 2012, 19, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Penesyan, A.; Gillings, M.; Paulsen, I.T. Antibiotic discovery: Combatting bacterial resistance in cells and in biofilm communities. Molecules 2015, 20, 5286–5298. [Google Scholar] [CrossRef] [PubMed]
- Sagar, S.; Esau, L.; Hikmawan, T.; Antunes, A.; Holtermann, K.; Stingl, U.; Bajic, V.B.; Kaur, M. Cytotoxic and apoptotic evaluations of marine bacteria isolated from brine-seawater interface of the Red Sea. BMC Complement Altern. Med. 2013, 13, 29. [Google Scholar] [CrossRef] [PubMed]
- Cherigo, L.; Lopez, D.; Martinez-Luis, S. Marine natural products as breast cancer resistance protein inhibitors. Mar. Drugs 2015, 13, 2010–2029. [Google Scholar] [CrossRef] [PubMed]
- Brinkhoff, T.; Bach, G.; Heidorn, T.; Liang, L.; Schlingloff, A.; Simon, M. Antibiotic production by a Roseobacter clade-affiliated species from the German Wadden Sea and its antagonistic effects on indigenous isolates. Appl. Environ. Microbiol. 2004, 70, 2560–2565. [Google Scholar] [CrossRef] [PubMed]
- Buchan, A.; González, J.M.; Moran, M.A. Overview of the marine Roseobacter lineage. Appl. Environ. Microbiol. 2005, 71, 5665–5677. [Google Scholar] [CrossRef] [PubMed]
- Selje, N.; Simon, M.; Brinkhoff, T. A newly discovered Roseobacter cluster in temperate and polar oceans. Nature 2004, 427, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Moran, M.A. Evolutionary ecology of the marine Roseobacter clade. Microbiol. Mol. Biol. Rev. 2014, 78, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Martens, T.; Gram, L.; Grossart, H.P.; Kessler, D.; Muller, R.; Simon, M.; Wenzel, S.C.; Brinkhoff, T. Bacteria of the Roseobacter clade show potential for secondary metabolite production. Microb. Ecol. 2007, 54, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Liang, L. Investigation of Secondary Metabolites of North Sea Bacteria: Fermentation, Isolation, Structure Elucidation and Bioactivity. Ph.D. Thesis, University of Göttingen, Göttingen, Germany, 2003. [Google Scholar]
- Berger, M.; Neumann, A.; Schulz, S.; Simon, M.; Brinkhoff, T. Tropodithietic acid production in Phaeobacter gallaeciensis is regulated by N-acyl homoserine lactone-mediated quorum sensing. J. Bacteriol. 2011, 193, 6576–6585. [Google Scholar] [CrossRef] [PubMed]
- Harrington, C.; Reen, F.J.; Mooij, M.J.; Stewart, F.A.; Chabot, J.B.; Guerra, A.F.; Glockner, F.O.; Nielsen, K.F.; Gram, L.; Dobson, A.D.; et al. Characterisation of non-autoinducing tropodithietic acid (TDA) production from marine sponge Pseudovibrio species. Mar. Drugs 2014, 12, 5960–5978. [Google Scholar] [CrossRef] [PubMed]
- Geng, H.; Belas, R. Expression of tropodithietic acid biosynthesis is controlled by a novel autoinducer. J. Bacteriol. 2010, 192, 4377–4387. [Google Scholar] [CrossRef] [PubMed]
- Neu, A.K.; Mansson, M.; Gram, L.; Prol-Garcia, M.J. Toxicity of bioactive and probiotic marine bacteria and their secondary metabolites in Artemia sp. and Caenorhabditis elegans as eukaryotic model organisms. Appl. Environ. Microbiol. 2014, 80, 146–153. [Google Scholar] [PubMed]
- Porsby, C.H.; Webber, M.A.; Nielsen, K.F.; Piddock, L.J.; Gram, L. Resistance and tolerance to tropodithietic acid, an antimicrobial in aquaculture, is hard to select. Antimicrob. Agents Chemother. 2011, 55, 1332–1337. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, R.G.; Sikorska, M.; Sandhu, J.K.; Lanthier, P.; Ribecco-Lutkiewicz, M.; Bani-Yaghoub, M. Differentiation of mouse Neuro 2A cells into dopamine neurons. J. Neurosci. Methods 2010, 186, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Richter-Landsberg, C.; Heinrich, M. OLN-93: A new permanent oligodendroglia cell line derived from primary rat brain glial cultures. J. Neurosci. Res. 1996, 45, 161–173. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, J.B.; Luo, X.Q.; Gong, L.R.; Wang, M.; Cao, X.S.; Dong, S.A.; Yan, Y.M.; Kwon, Y.; He, J. Effect of ERK1/2 signaling pathway in electro-acupuncture mediated up-regulation of heme oxygenase-1 in lungs of rabbits with endotoxic shock. Med. Sci. Monit. 2014, 20, 1452–1460. [Google Scholar] [PubMed]
- Piantadosi, C.A.; Carraway, M.S.; Suliman, H.B. Carbon monoxide, oxidative stress, and mitochondrial permeability pore transition. Free Radic. Biol. Med. 2006, 40, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Stahnke, T.; Stadelmann, C.; Netzler, A.; Bruck, W.; Richter-Landsberg, C. Differential upregulation of heme oxygenase-1 (HSP32) in glial cells after oxidative stress and in demyelinating disorders. J. Mol. Neurosci. 2007, 32, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Mronga, T.; Stahnke, T.; Goldbaum, O.; Richter-Landsberg, C. Mitochondrial pathway is involved in hydrogen-peroxide-induced apoptotic cell death of oligodendrocytes. Glia 2004, 46, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Abdul, H.M.; Butterfield, D.A. Involvement of PI3K/PKG/ERK1/2 signaling pathways in cortical neurons to trigger protection by co-treatment of acetyl-l-carnitine and α-lipoic acid against HNE-mediated oxidative stress and neurotoxicity: Implications for Alzheimer’s disease. Free Radic. Biol. Med. 2007, 42, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Stahnke, T.; Richter-Landsberg, C. Triethyltin-induced stress responses and apoptotic cell death in cultured oligodendrocytes. Glia 2004, 46, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Richter-Landsberg, C. Heat shock proteins: Expression and functional roles in nerve cells and glia. In Heat Shock Proteins in Neural Cells; Richter-Landsberg, C., Ed.; Landes Bioscience: Austin, TX, USA, 2007; pp. 1–12. [Google Scholar]
- Cappello, F.; Conway de Macario, E.; Marino Gammazza, A.; Bonaventura, G.; Carini, F.; Czarnecka, A.M.; Farina, F.; Zummo, G.; Macario, A.J. Hsp60 and human aging: Les liaisons dangereuses. Front. Biosci. (Landmark Ed.) 2013, 18, 626–637. [Google Scholar] [CrossRef] [PubMed]
- Karbowski, M.; Neutzner, A. Neurodegeneration as a consequence of failed mitochondrial maintenance. Acta Neuropathol. 2012, 123, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J.; Theruvath, T.P.; Zhong, Z.; Nieminen, A.L. Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta 2009, 1787, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.S.B.; Boyman, L.; Chikando, A.C.; Khairallah, R.J.; Lederer, W.J. Mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 10479–10486. [Google Scholar] [CrossRef] [PubMed]
- Honda, H.M.; Ping, P. Mitochondrial permeability transition in cardiac cell injury and death. Cardiovasc. Drugs Ther. 2006, 20, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Calcium and mitochondria in the regulation of cell death. Biochem. Biophys. Res. Commun. 2015, 460, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.X.; Thompson, C.B. Necrotic death as a cell fate. Genes Dev. 2006, 20, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Coles, C.H.; Bradke, F. Coordinating neuronal actin-microtubule dynamics. Curr. Biol. 2015, 25, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Hrouzek, P.; Kuzma, M.; Cerny, J.; Novak, P.; Fiser, R.; Simek, P.; Lukesova, A.; Kopecky, J. The cyanobacterial cyclic lipopeptides puwainaphycins F/G are inducing necrosis via cell membrane permeabilization and subsequent unusual actin relocalization. Chem. Res. Toxicol. 2012, 25, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Bacallao, R.; Garfinkel, A.; Monke, S.; Zampighi, G.; Mandel, L.J. ATP depletion: A novel method to study junctional properties in epithelial tissues. I. Rearrangement of the actin cytoskeleton. J. Cell Sci. 1994, 107, 3301–3313. [Google Scholar] [PubMed]
- Neuhoff, V.; Philipp, K.; Zimmer, H.G.; Mesecke, S. A simple, versatile, sensitive and volume-independent method for quantitative protein determination which is independent of other external influences. Hoppe Seylers Z. Physiol. Chem. 1979, 360, 1657–1670. [Google Scholar] [CrossRef] [PubMed]
- Richter-Landsberg, C.; Vollgraf, U. Mode of cell injury and death after hydrogen peroxide exposure in cultured oligodendroglia cells. Exp. Cell Res. 1998, 244, 218–229. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wichmann, H.; Vocke, F.; Brinkhoff, T.; Simon, M.; Richter-Landsberg, C. Cytotoxic Effects of Tropodithietic Acid on Mammalian Clonal Cell Lines of Neuronal and Glial Origin. Mar. Drugs 2015, 13, 7113-7123. https://doi.org/10.3390/md13127058
Wichmann H, Vocke F, Brinkhoff T, Simon M, Richter-Landsberg C. Cytotoxic Effects of Tropodithietic Acid on Mammalian Clonal Cell Lines of Neuronal and Glial Origin. Marine Drugs. 2015; 13(12):7113-7123. https://doi.org/10.3390/md13127058
Chicago/Turabian StyleWichmann, Heidi, Farina Vocke, Thorsten Brinkhoff, Meinhard Simon, and Christiane Richter-Landsberg. 2015. "Cytotoxic Effects of Tropodithietic Acid on Mammalian Clonal Cell Lines of Neuronal and Glial Origin" Marine Drugs 13, no. 12: 7113-7123. https://doi.org/10.3390/md13127058
APA StyleWichmann, H., Vocke, F., Brinkhoff, T., Simon, M., & Richter-Landsberg, C. (2015). Cytotoxic Effects of Tropodithietic Acid on Mammalian Clonal Cell Lines of Neuronal and Glial Origin. Marine Drugs, 13(12), 7113-7123. https://doi.org/10.3390/md13127058