
Preparative Separation of Sulfur-Containing Diketopiperazines from Marine Fungus Cladosporium sp. Using High-Speed Counter-Current Chromatography in Stepwise Elution Mode

 ,
, 
Abstract
:1. Introduction
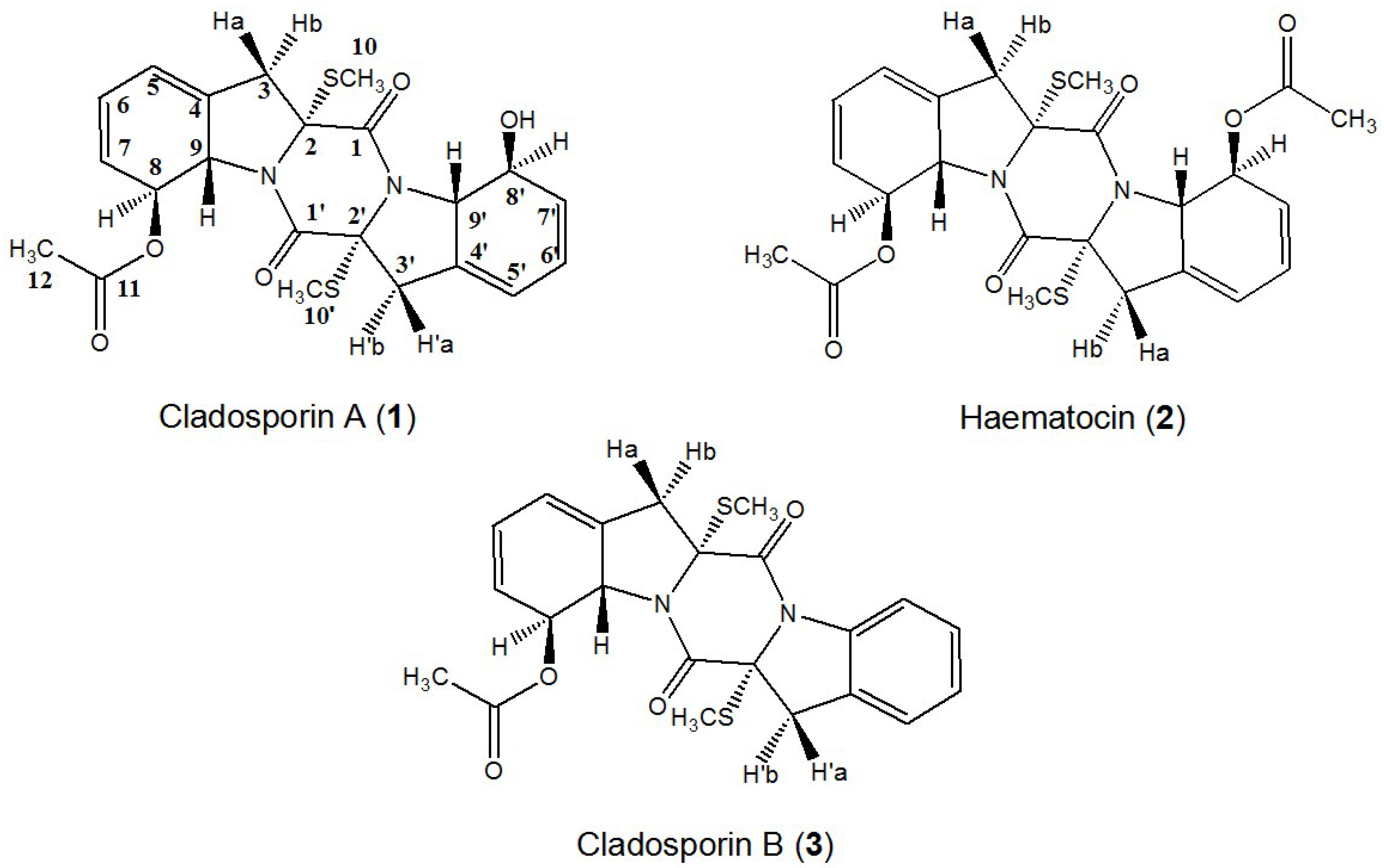
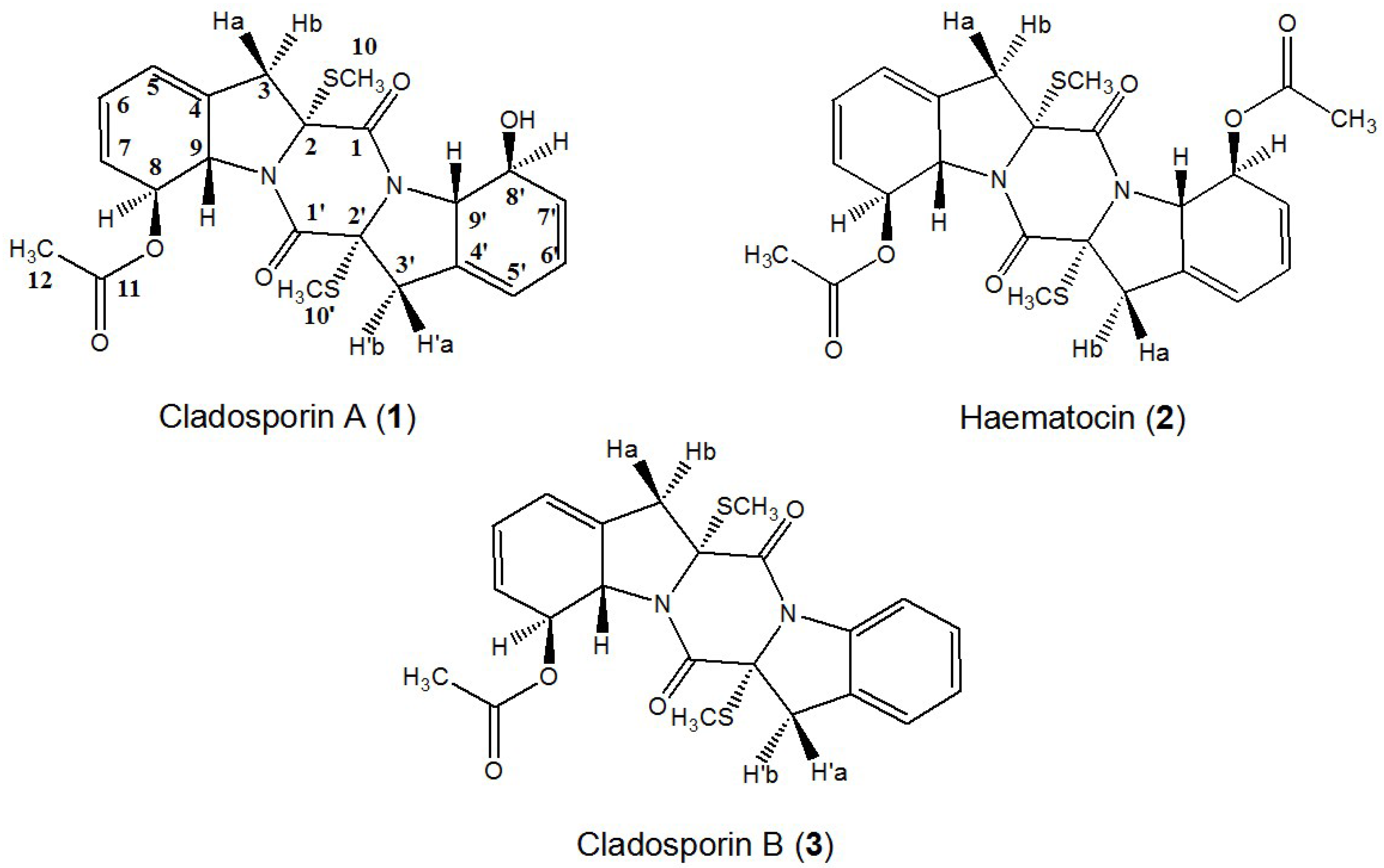
2. Results and Discussion
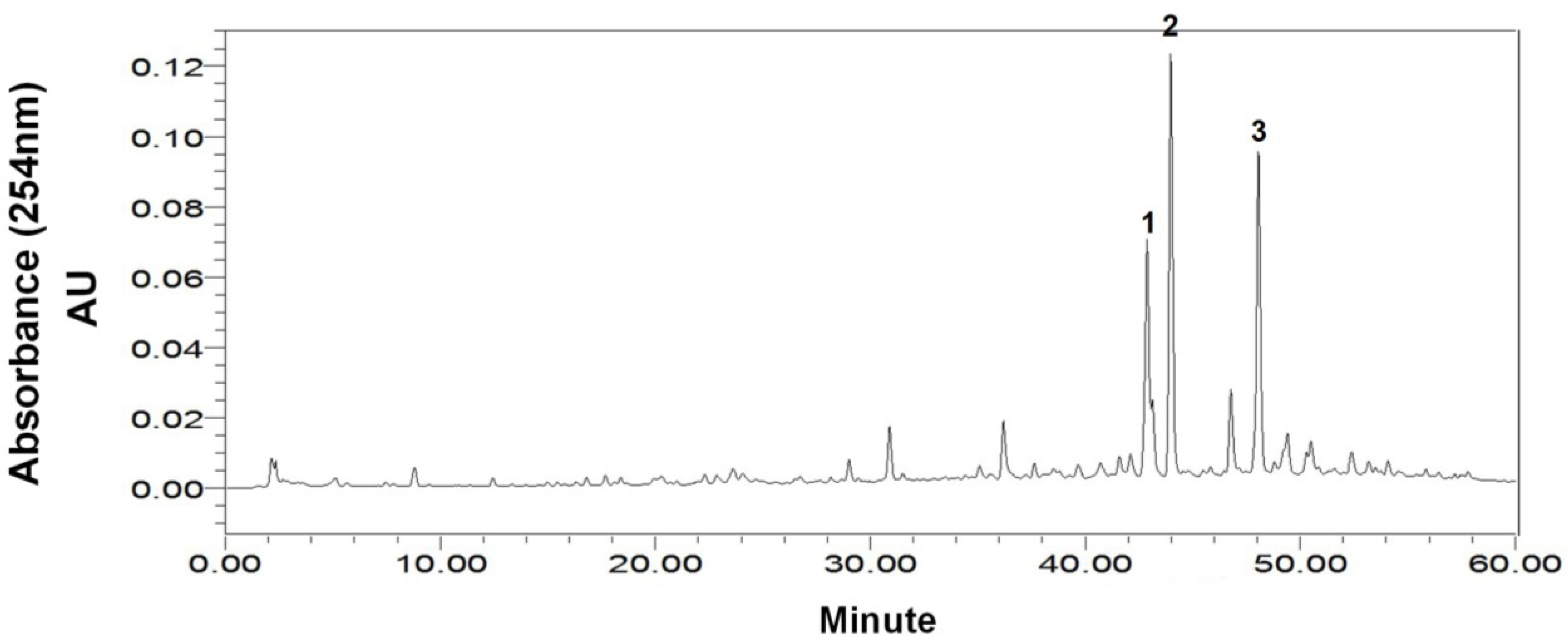
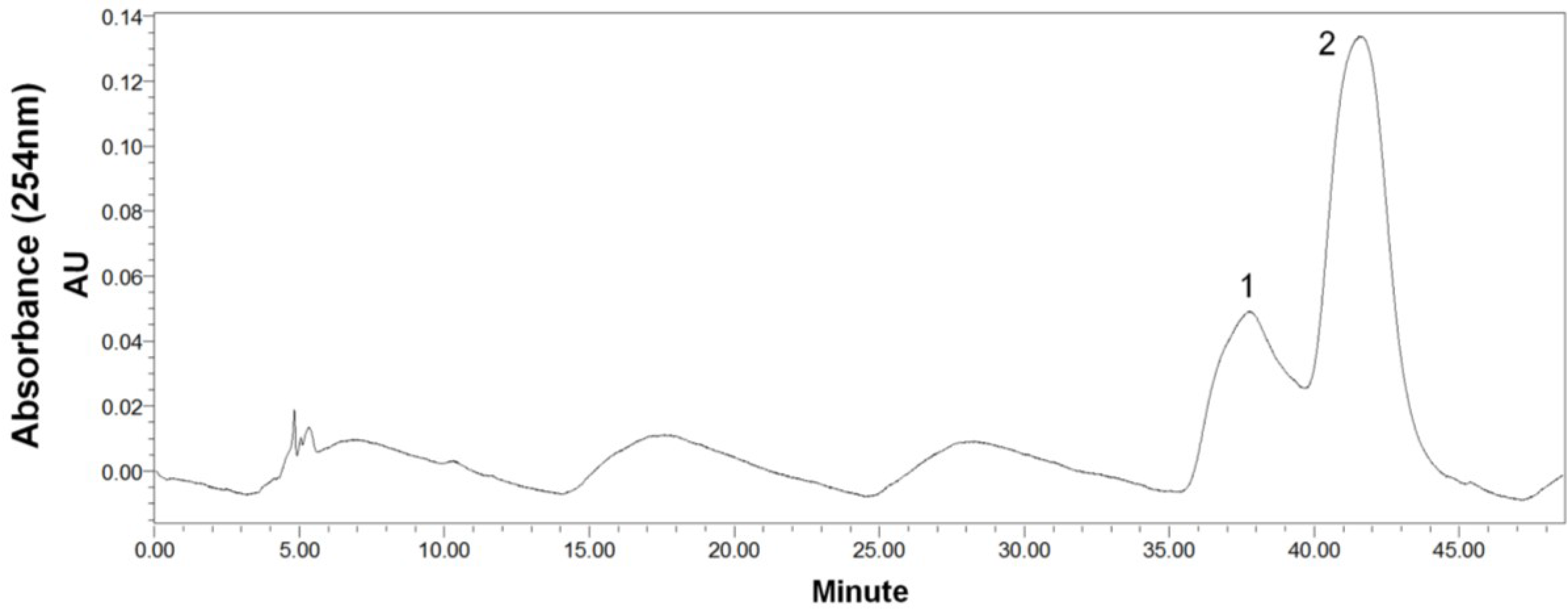
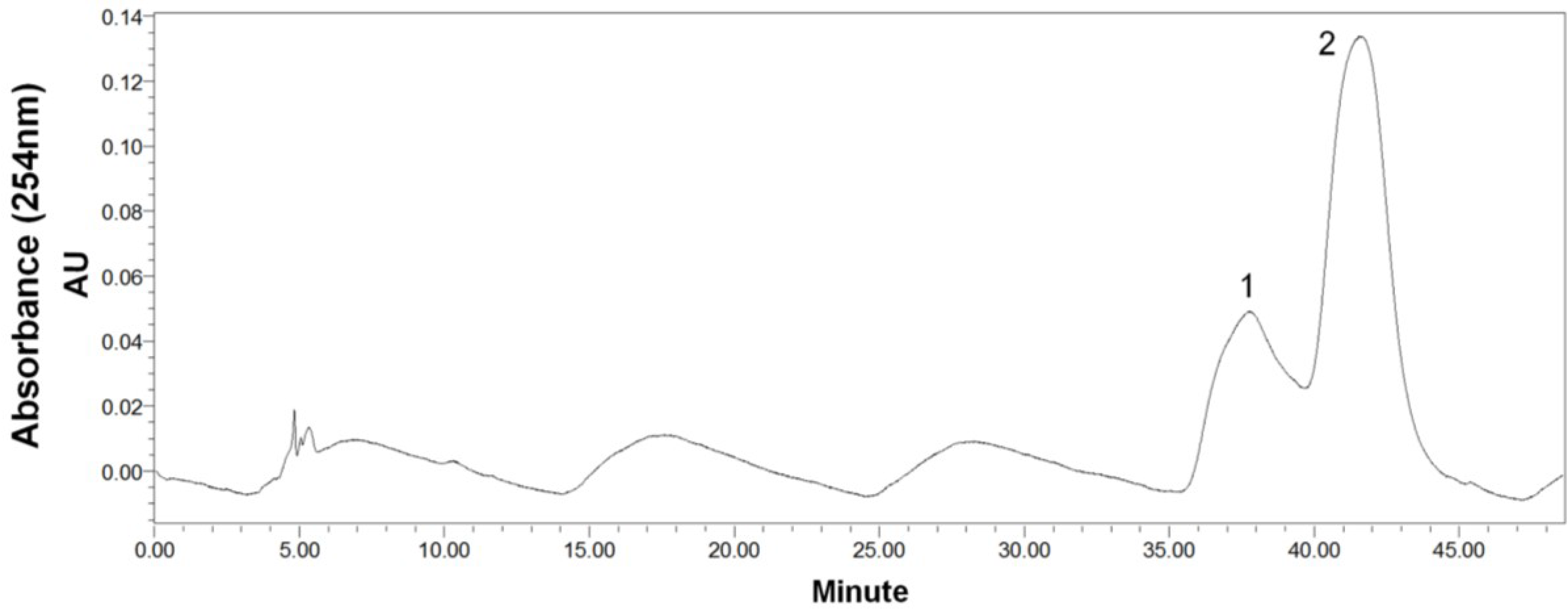
2.1. HPLC Analysis of the Crude Sample
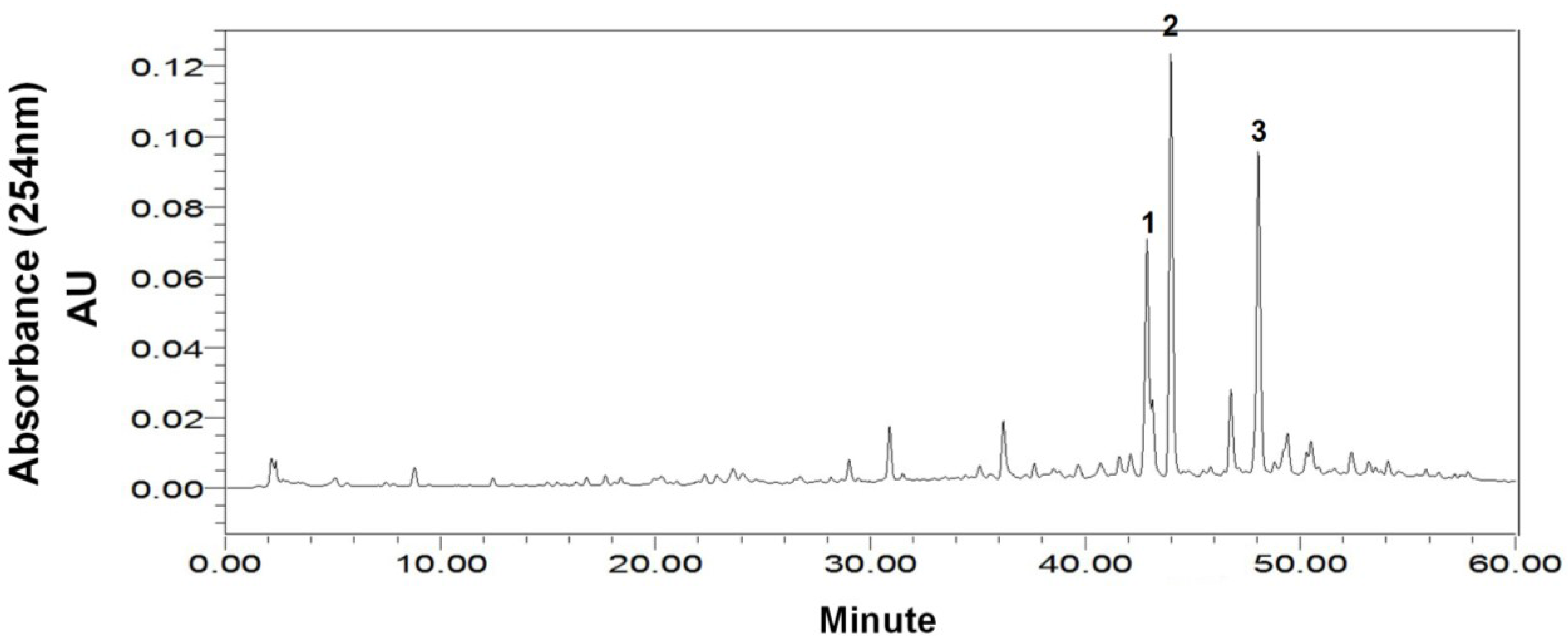
2.2. Selection of the HSCCC Two-Phase Solvent System

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n-Hexane–ethyl Acetate–methanol–water | KD | ||
|---|---|---|---|
| Compound 1 | Compound 2 | Compound 3 | |
| 0.8:1:0.8:1 | 1.80 | 2.01 | 6.37 |
| 1:1:1:1 | 1.37 | 1.57 | 5.22 |
| 1.5:1:1.5:1 | 0.67 | 0.76 | 2.39 |
| 2:1:2:1 | 0.22 | 0.24 | 0.84 |
| 2.5:1:2.5:1 | 0.11 | 0.13 | 0.52 |
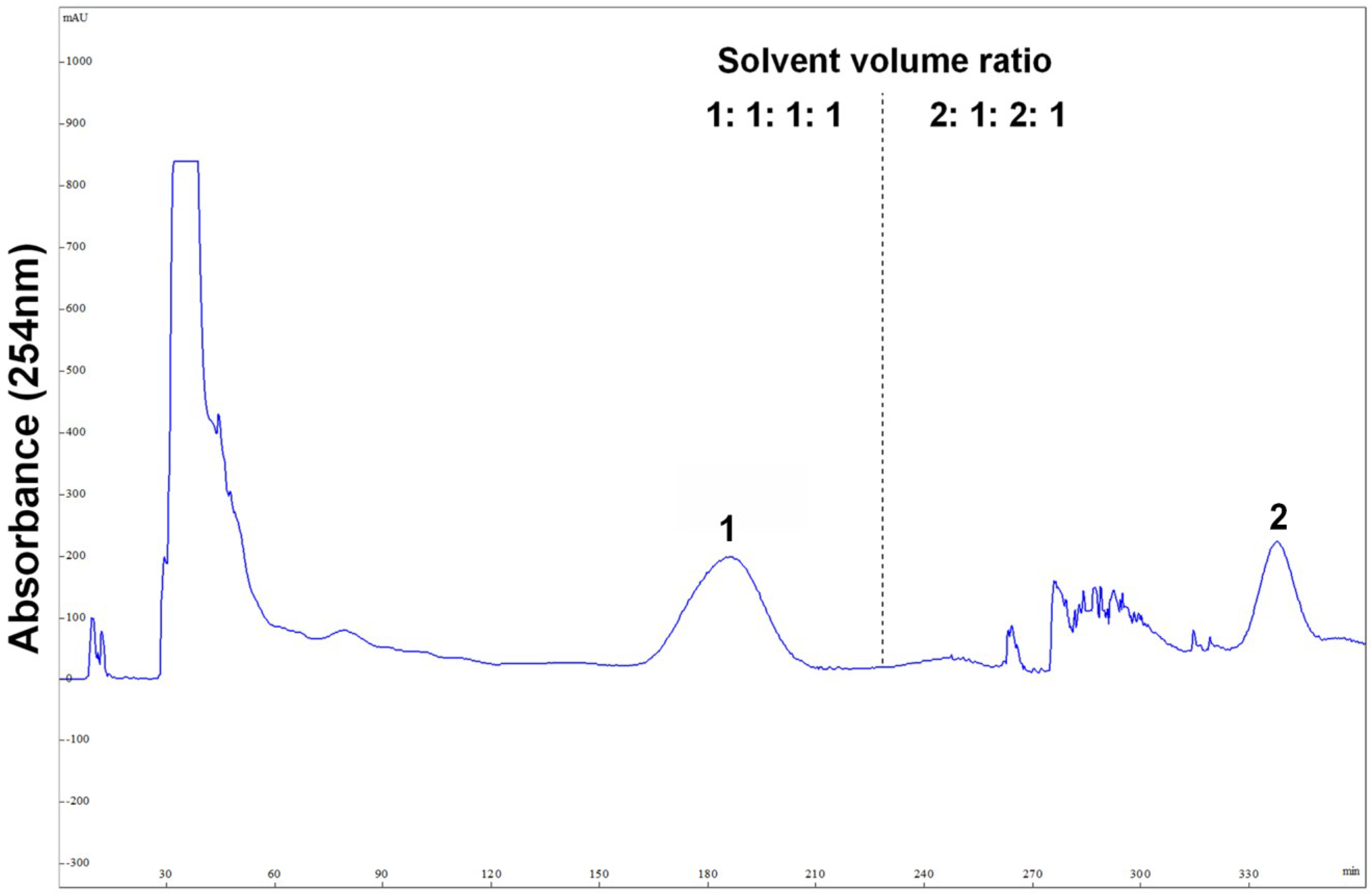
2.3. Stepwise HSCCC Separation
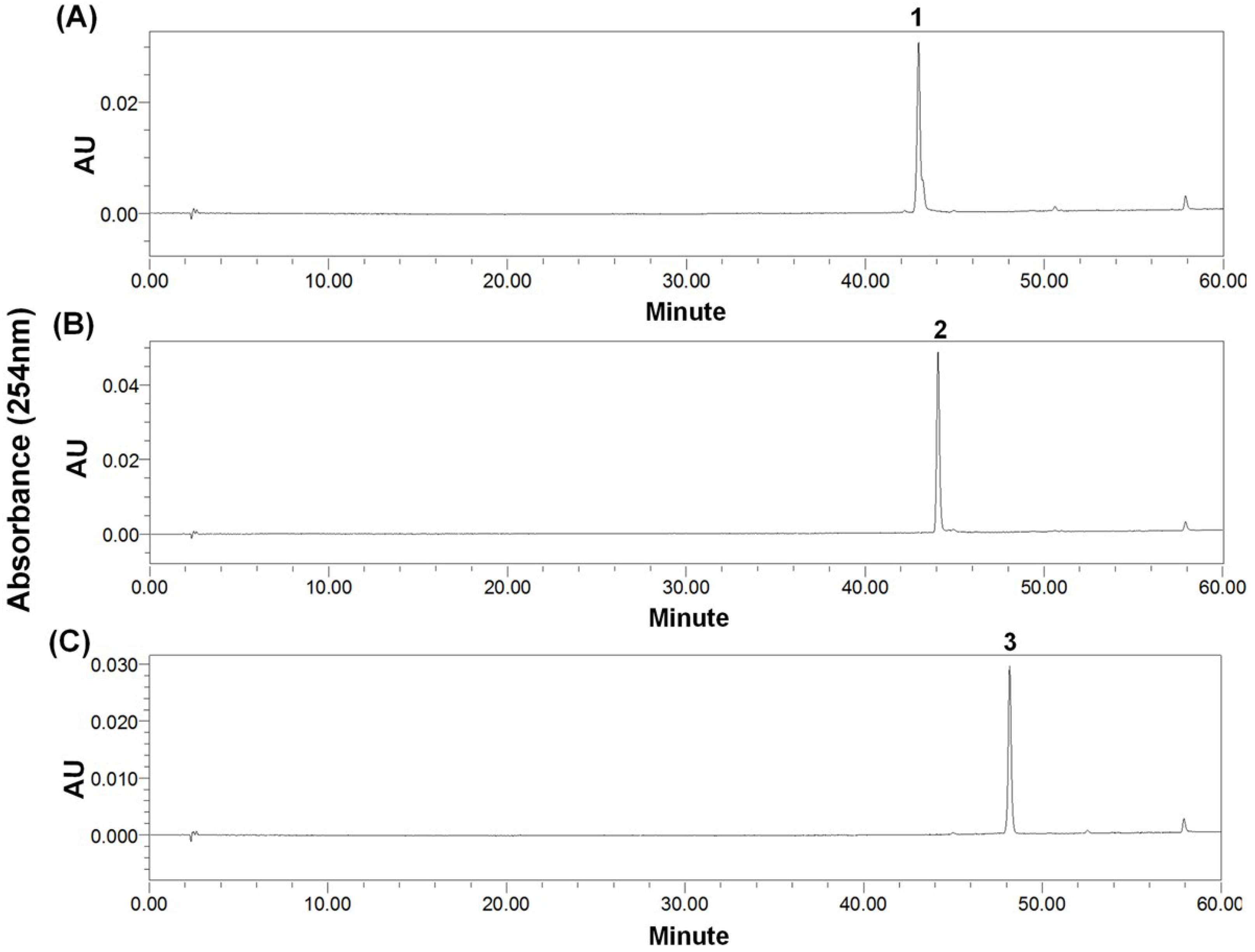
2.4. Preparative-HPLC Separation
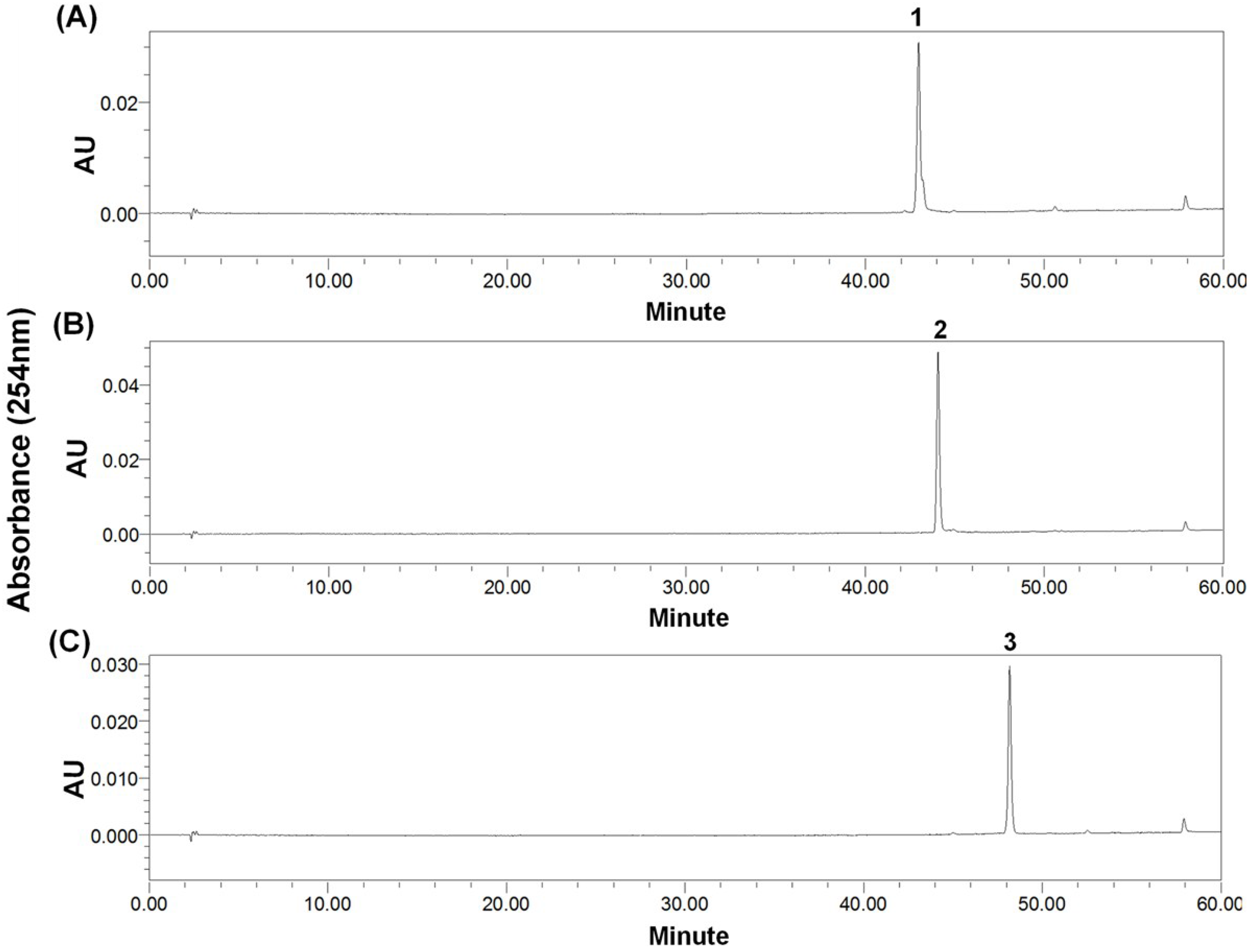
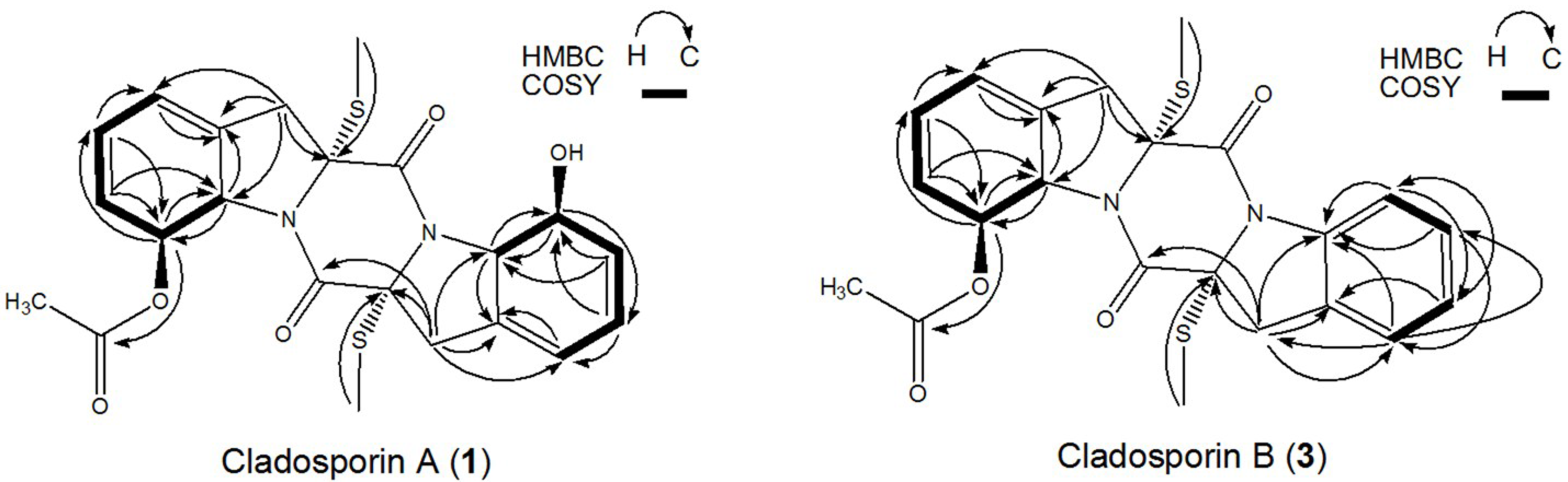
2.5. Structural Identification and Cytotoxic Activity
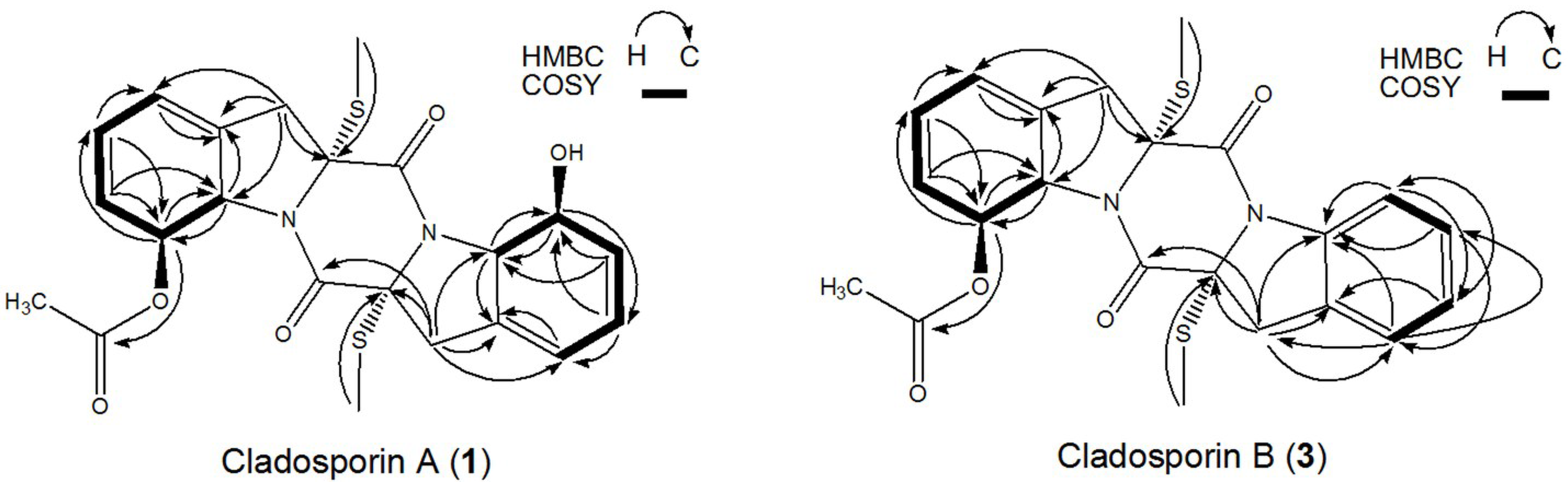
3. Experimental Section
3.1. Reagents and Materials
3.2. Apparatus
3.3. Preparation of Crude Sample
3.4. Measurement of Partition Coefficients (KD) and Preparation of Two-Phase Solvent and Sample Solution
3.5. Separation Procedure
3.6. HPLC Analysis and Identification of the Peaks
3.7. Cell Proliferation Assay
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kong, D.-X.; Jiang, Y.-Y.; Zhang, H.-Y. Marine natural products as sources of novel scaffolds: Achievement and concern. Drug Discov. Today 2010, 15, 884–886. [Google Scholar] [CrossRef] [PubMed]
- Montaser, R.; Luesch, H. Marine natural products: A new wave of drugs? Future Med. Chem. 2011, 3, 1475–1489. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E. Untersuchungen über aminosäuren, polypeptide und proteine. Ber. Dtsch. Chem. Ges. 1906, 39, 530–610. [Google Scholar] [CrossRef]
- Gu, B.; He, S.; Yan, X.; Zhang, L. Tentative biosynthetic pathways of some microbial diketopiperazines. Appl. Microbiol. Biotechnol. 2013, 97, 8439–8453. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.B.; Carvalho, I. Diketopiperazines: Biological activity and synthesis. Tetrahedron 2007, 63, 9923–9932. [Google Scholar] [CrossRef]
- Huang, R.; Zhou, X.; Xu, T.; Yang, X.; Liu, Y. Diketopiperazines from marine organisms. Chem. Biodivers. 2010, 7, 2809–2829. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Takahashi, H.; Esumi, Y.; Arie, T.; Morita, T.; Koshino, H.; Uzawa, J.; Uramoto, M.; Yamaguchi, I. Haematocin, a new antifungal diketopiperazine produced by Nectria haematococca Berk. et Br. (880701a-1) causing nectria blight disease on ornamental plants. J. Antibiot (Tokyo) 2000, 53, 45–49. [Google Scholar] [CrossRef]
- Ito, Y. Golden rules and pitfalls in selecting optimum conditions for high-speed counter-current chromatography. J. Chromatogr. A 2005, 1065, 145–168. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Dong, H.; Liu, Y.; Yang, B.; Wang, X.; Huang, L. Application of high-speed counter-current chromatography for preparative separation of cyclic peptides from Vaccaria segetalis. J. Chromatogr. B 2011, 879, 811–814. [Google Scholar] [CrossRef]
- Sutherland, I.A.; Fisher, D. Role of counter-current chromatography in the modernisation of Chinese herbal medicines. J. Chromatogr. A 2009, 1216, 740–753. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, H.; Yan, X.; Zhu, P.; Chen, J.; Yang, R. Preparative isolation and purification of macrolactin antibiotics from marine bacterium Bacillus amyloliquefaciens using high-speed counter-current chromatography in stepwise elution mode. J. Chromatogr. A 2013, 1272, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; He, S.; Yan, X. Efficient preparation of pseudoalteromone A from marine Pseudoalteromonas rubra QD1–2 by combination of response surface methodology and high-speed counter-current chromatography: A comparison with high-performance liquid chromatography. Appl. Microbiol. Biotechnol. 2014, 98, 4369–4377. [Google Scholar] [CrossRef] [PubMed]
- Oka, F.; Oka, H.; Ito, Y. Systematic search for suitable two-phase solvent systems for high-speed counter-current chromatography. J. Chromatogr. A 1991, 538, 99–108. [Google Scholar] [CrossRef]
- Wu, H.; Su, Z.; Yang, Y.; Ba, H.; Aisa, H.A. Isolation of three sesquiterpene lactones from the roots of Cichorium glandulosum Boiss. et Huet. by high-speed counter-current chromatography. J. Chromatogr. A 2007, 1176, 217–222. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Lu, Y.; Jiang, L.; Wu, B.; Zhang, F.; Pan, Y. Preparative isolation and purification of antioxidative stilbene oligomers from Vitis chunganeniss using high-speed counter-current chromatography in stepwise elution mode. J. Sep. Sci. 2009, 32, 2339–2345. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Ye, P.; Chen, C.-T.A.; Wang, K.; Liu, P.; He, S.; Wu, X.; Gan, L.; Ye, Y.; Wu, B. Two Novel Hepatocellular Carcinoma Cycle Inhibitory clodepsipeptides from a Hydrothermal Vent Crab-Associated Fungus Aspergillus clavatus C2WU. Mar. Drugs 2013, 11, 4761–4772. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, B.; Zhang, Y.; Ding, L.; He, S.; Wu, B.; Dong, J.; Zhu, P.; Chen, J.; Zhang, J.; Yan, X. Preparative Separation of Sulfur-Containing Diketopiperazines from Marine Fungus Cladosporium sp. Using High-Speed Counter-Current Chromatography in Stepwise Elution Mode. Mar. Drugs 2015, 13, 354-365. https://doi.org/10.3390/md13010354
Gu B, Zhang Y, Ding L, He S, Wu B, Dong J, Zhu P, Chen J, Zhang J, Yan X. Preparative Separation of Sulfur-Containing Diketopiperazines from Marine Fungus Cladosporium sp. Using High-Speed Counter-Current Chromatography in Stepwise Elution Mode. Marine Drugs. 2015; 13(1):354-365. https://doi.org/10.3390/md13010354
Chicago/Turabian StyleGu, Binbin, Yanying Zhang, Lijian Ding, Shan He, Bin Wu, Junde Dong, Peng Zhu, Juanjuan Chen, Jinrong Zhang, and Xiaojun Yan. 2015. "Preparative Separation of Sulfur-Containing Diketopiperazines from Marine Fungus Cladosporium sp. Using High-Speed Counter-Current Chromatography in Stepwise Elution Mode" Marine Drugs 13, no. 1: 354-365. https://doi.org/10.3390/md13010354
APA StyleGu, B., Zhang, Y., Ding, L., He, S., Wu, B., Dong, J., Zhu, P., Chen, J., Zhang, J., & Yan, X. (2015). Preparative Separation of Sulfur-Containing Diketopiperazines from Marine Fungus Cladosporium sp. Using High-Speed Counter-Current Chromatography in Stepwise Elution Mode. Marine Drugs, 13(1), 354-365. https://doi.org/10.3390/md13010354