Comparative Analysis of the Cytotoxic Effects of Okadaic Acid-Group Toxins on Human Intestinal Cell Lines


Abstract
:1. Introduction
2. Results and Discussion
2.1. Neutral Red Uptake Assay
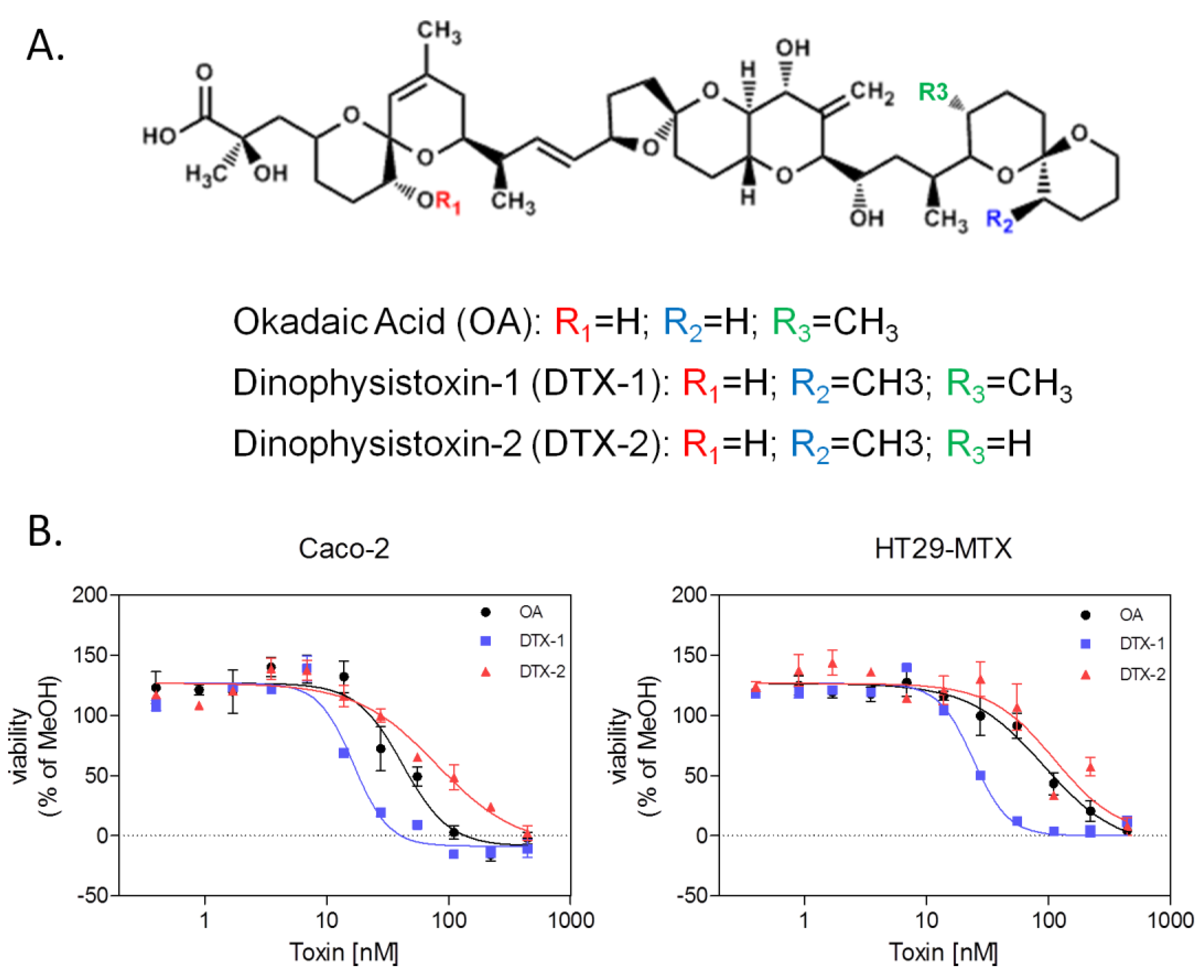

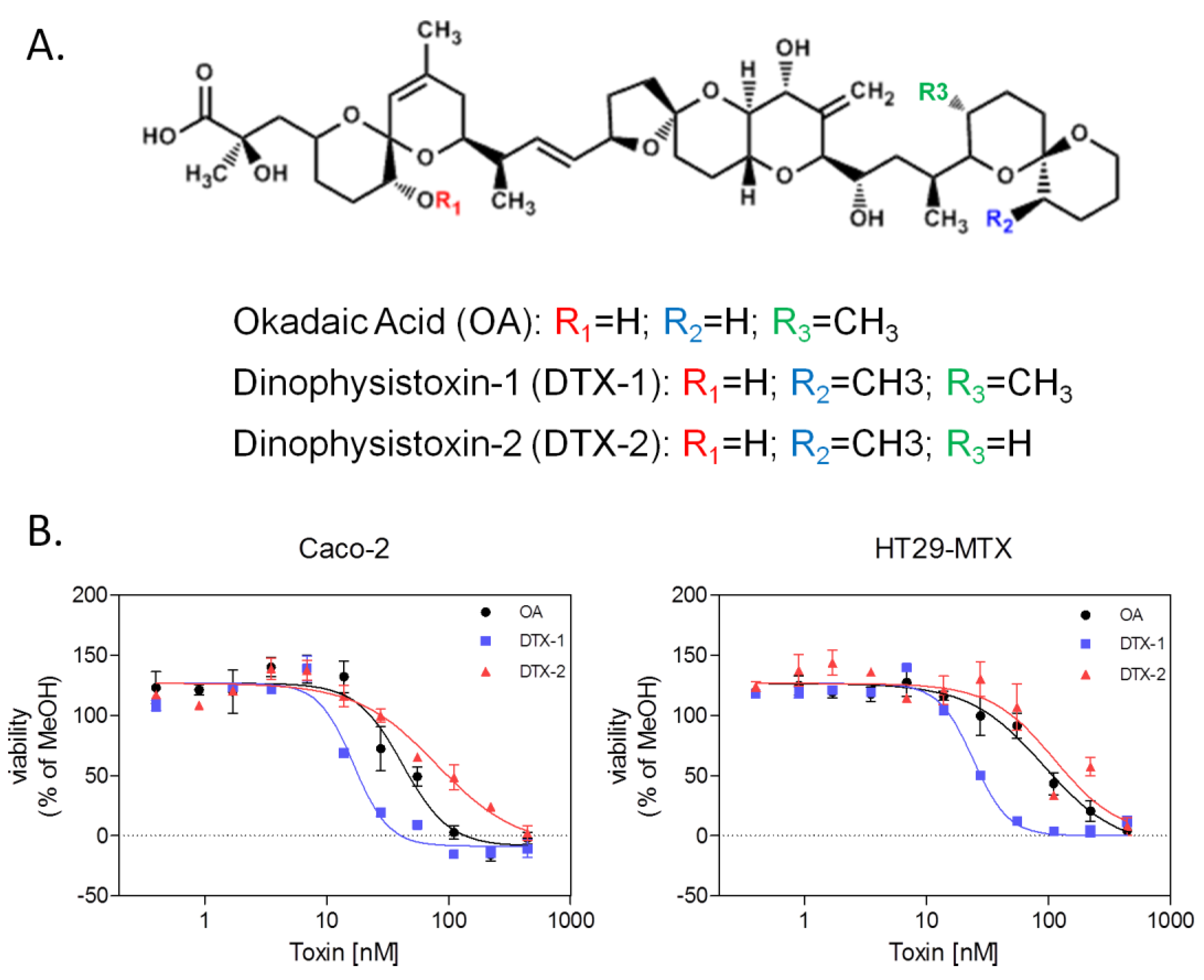{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Toxins | Caco-2 | HT29-MTX | ||
|---|---|---|---|---|
| IC50 [nM] | 95% CI | IC50 [nM] | 95% CI | |
| OA | 49.67 | 16.59–82.75 | 75.33 | 48.54–102.1 |
| DTX-1 | 22.50 | 8.83–36.16 | 22.00 | 10.62–33.38 |
| DTX-2 | 106.00 | 21.54–190.5 | 213.3 | 49.28–377.4 |
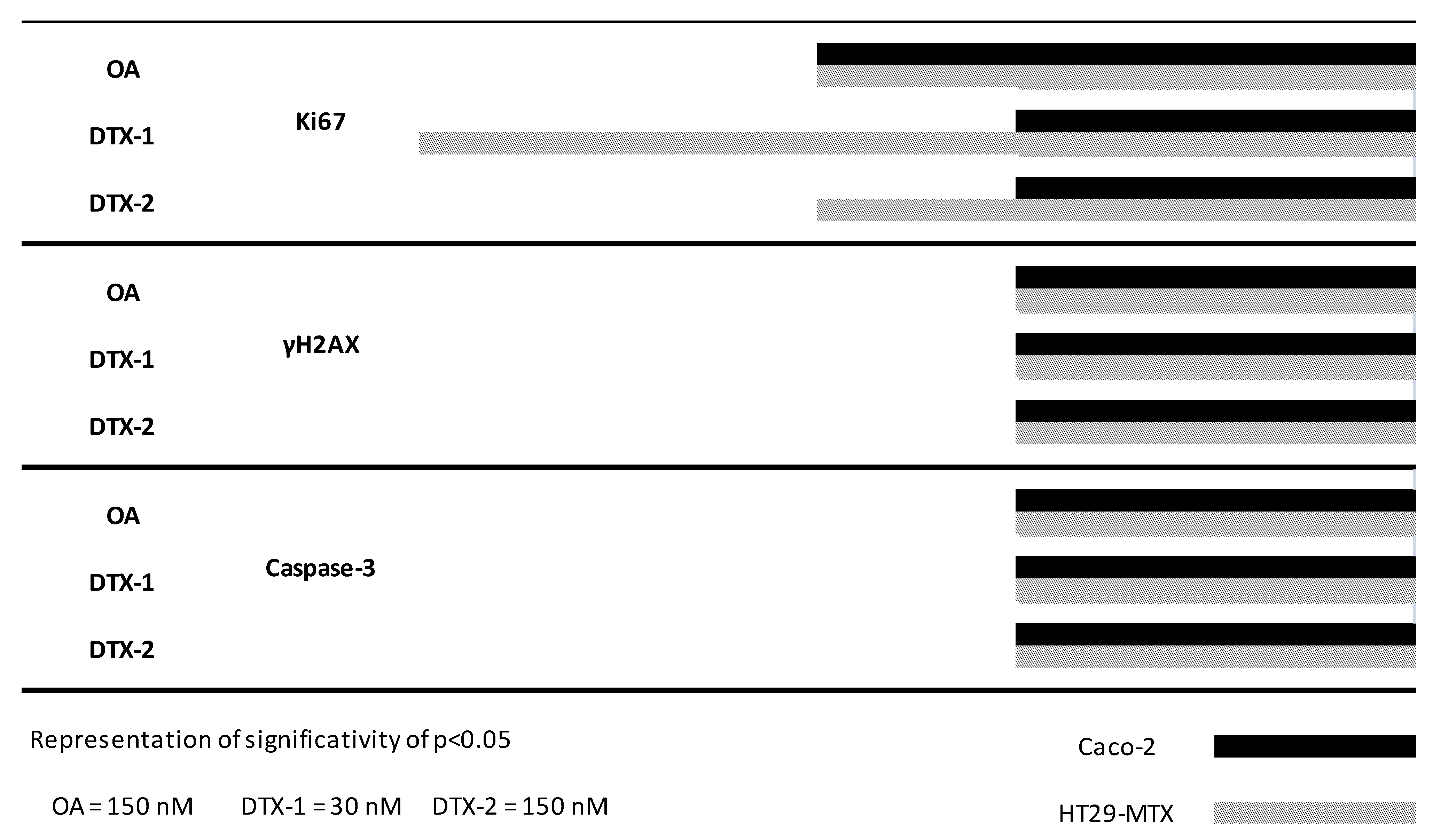
2.2. Cell Cycle Disruption

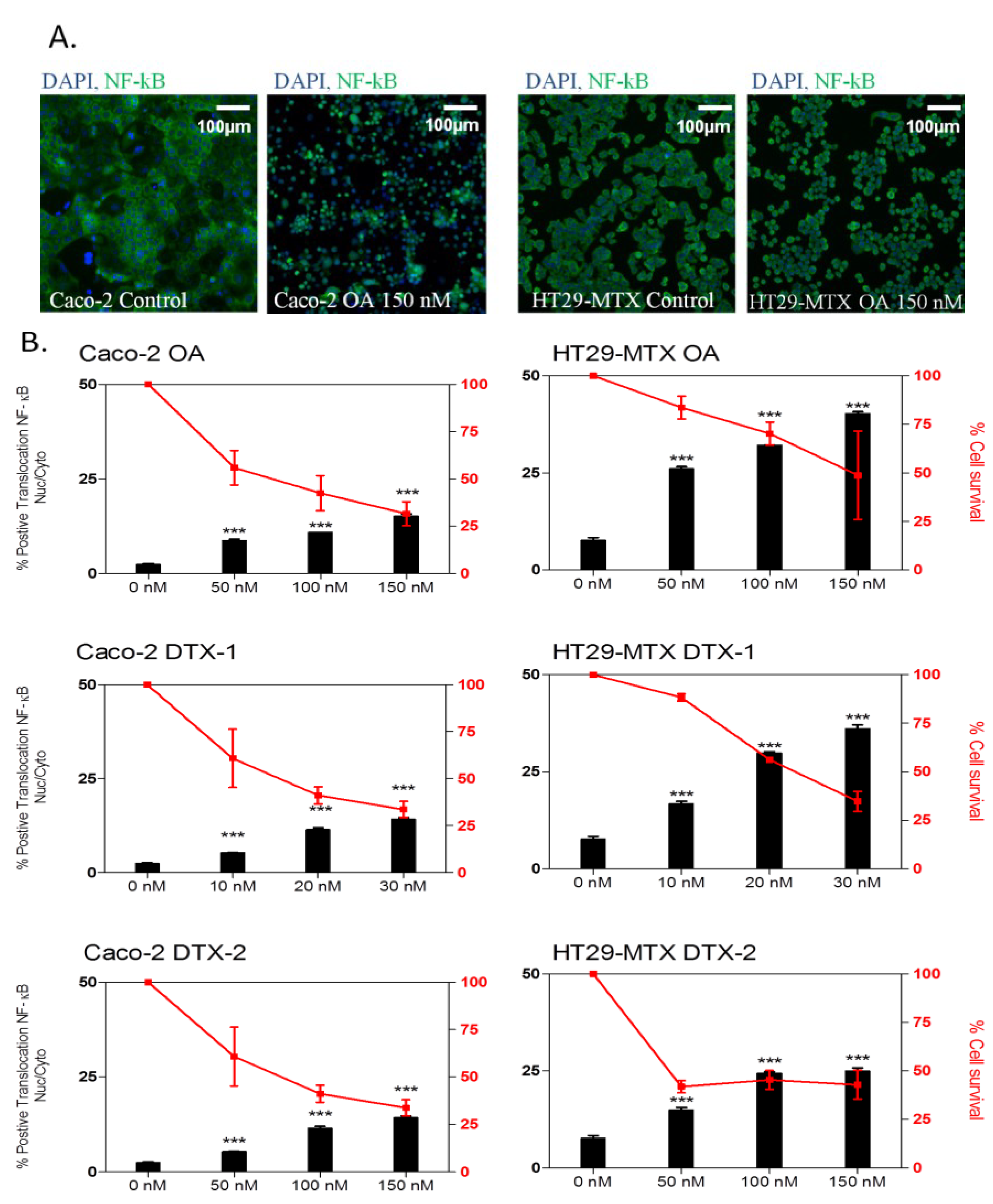
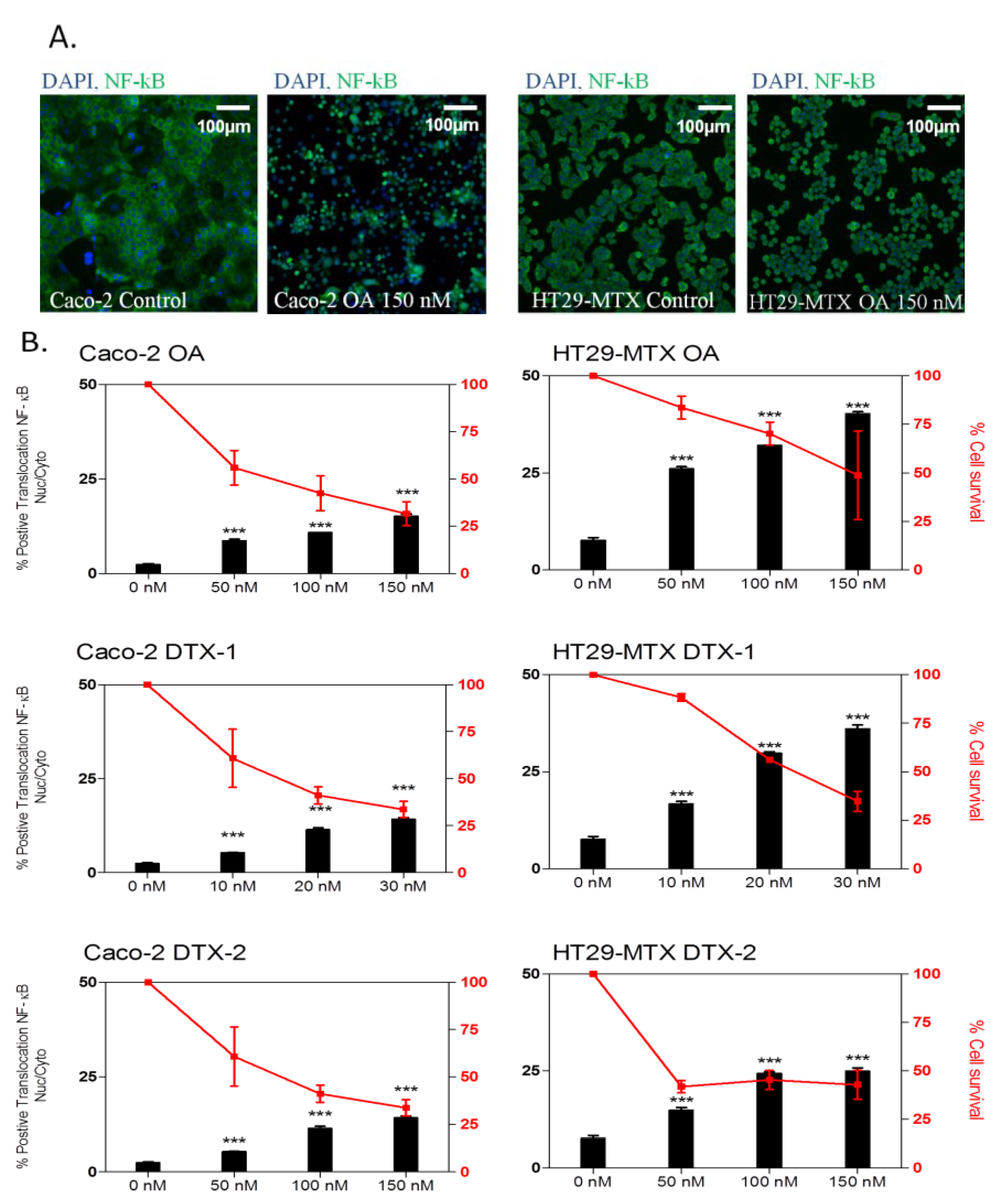
2.3. Inflammatory Response
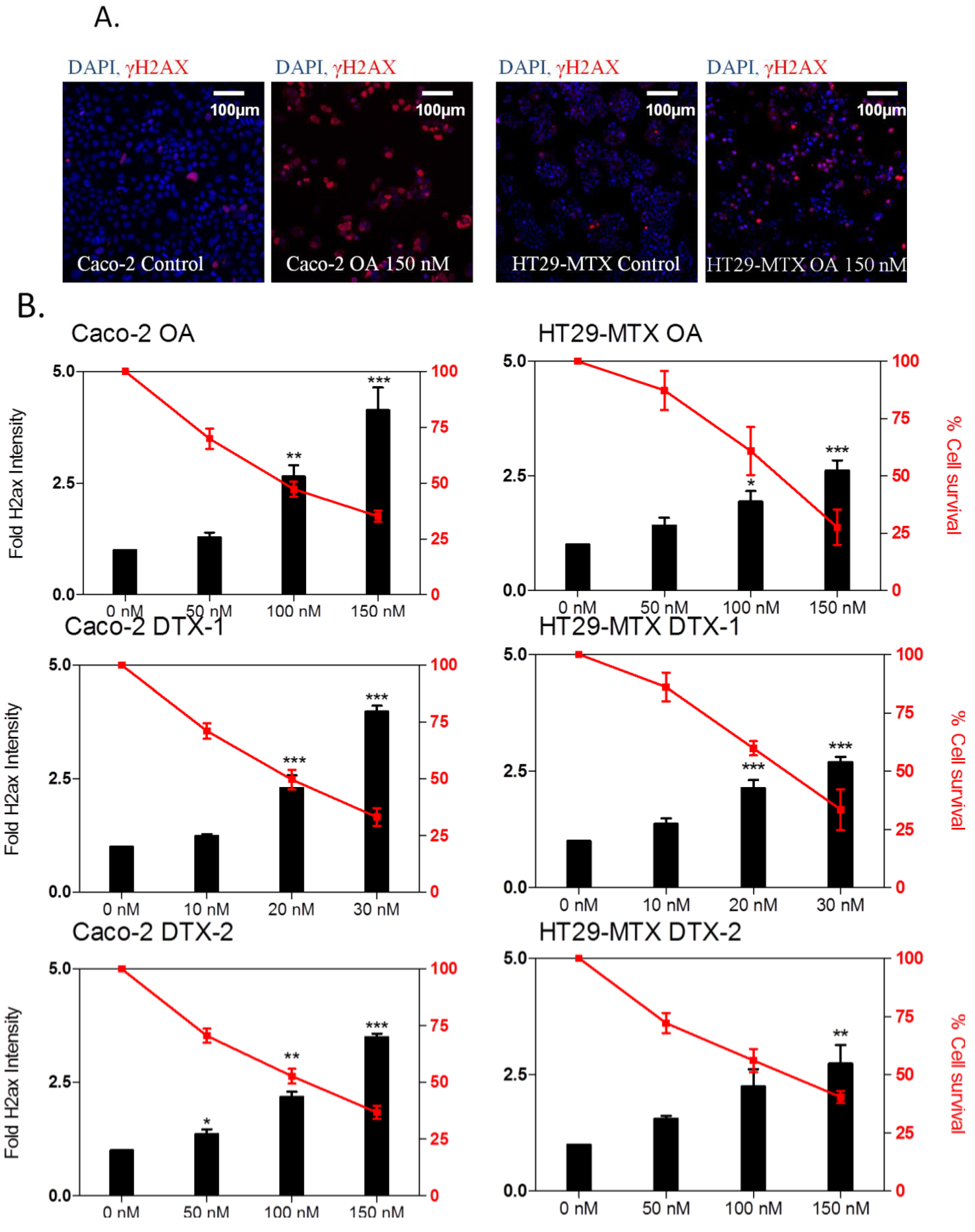
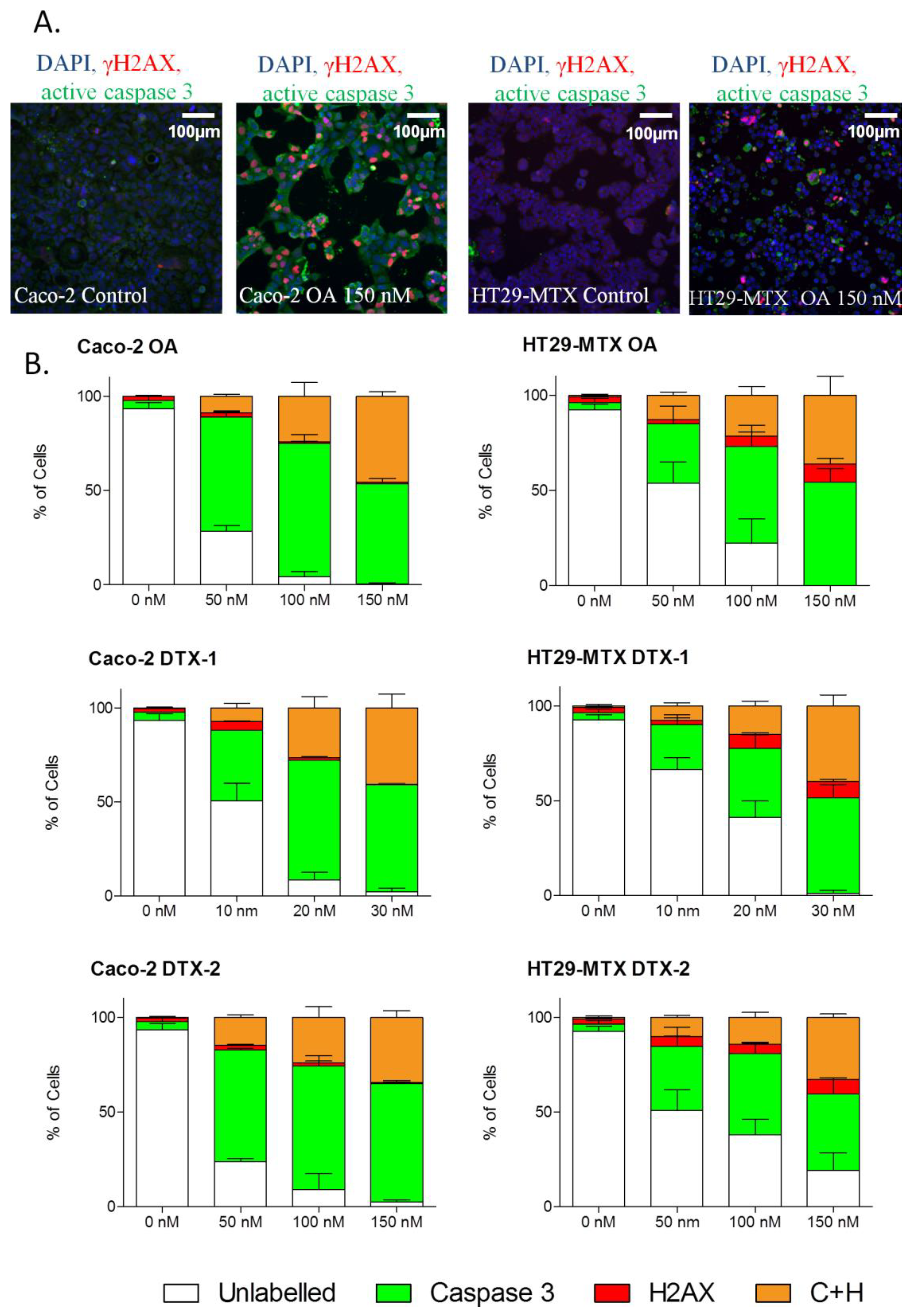
2.4. DNA Damage
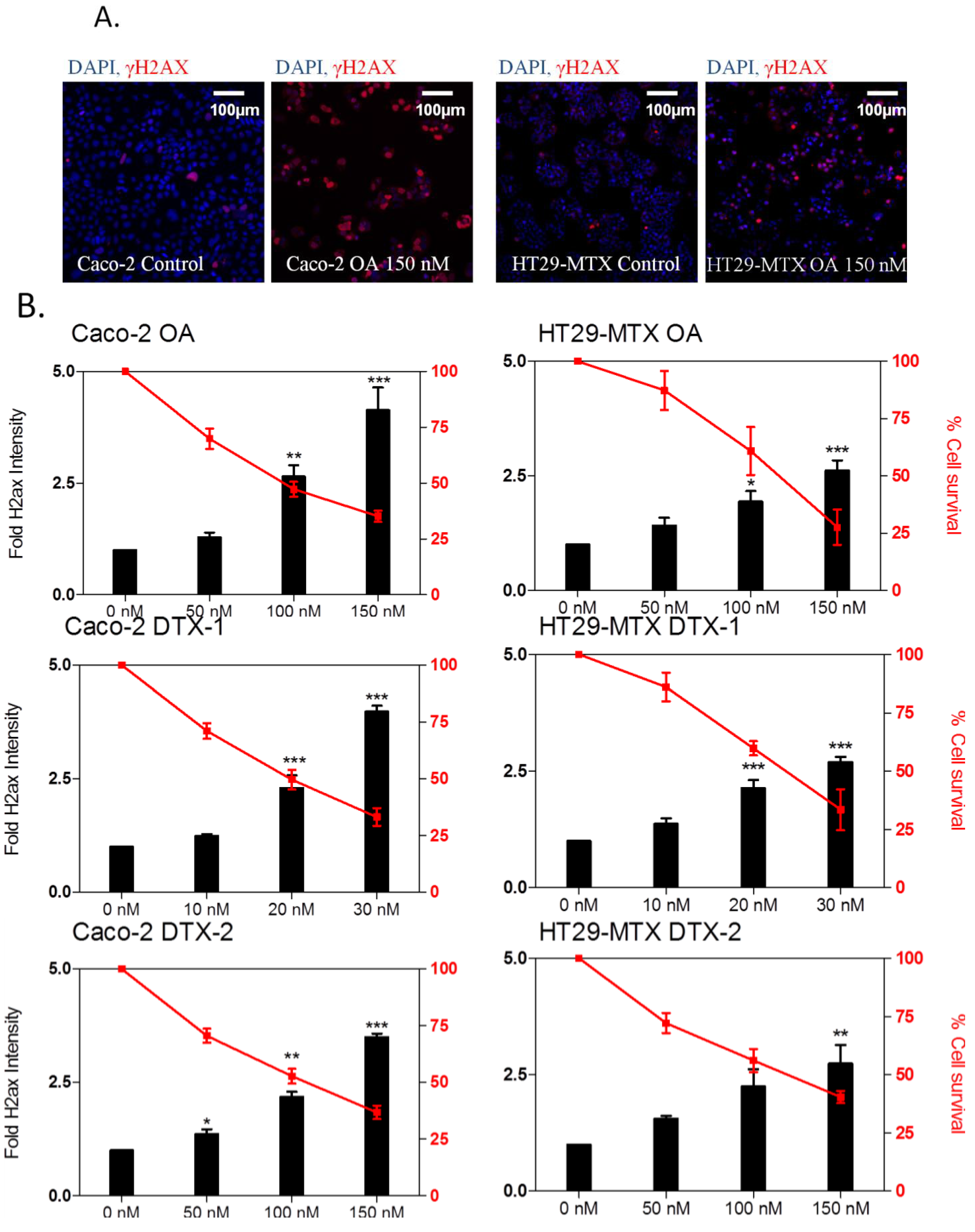
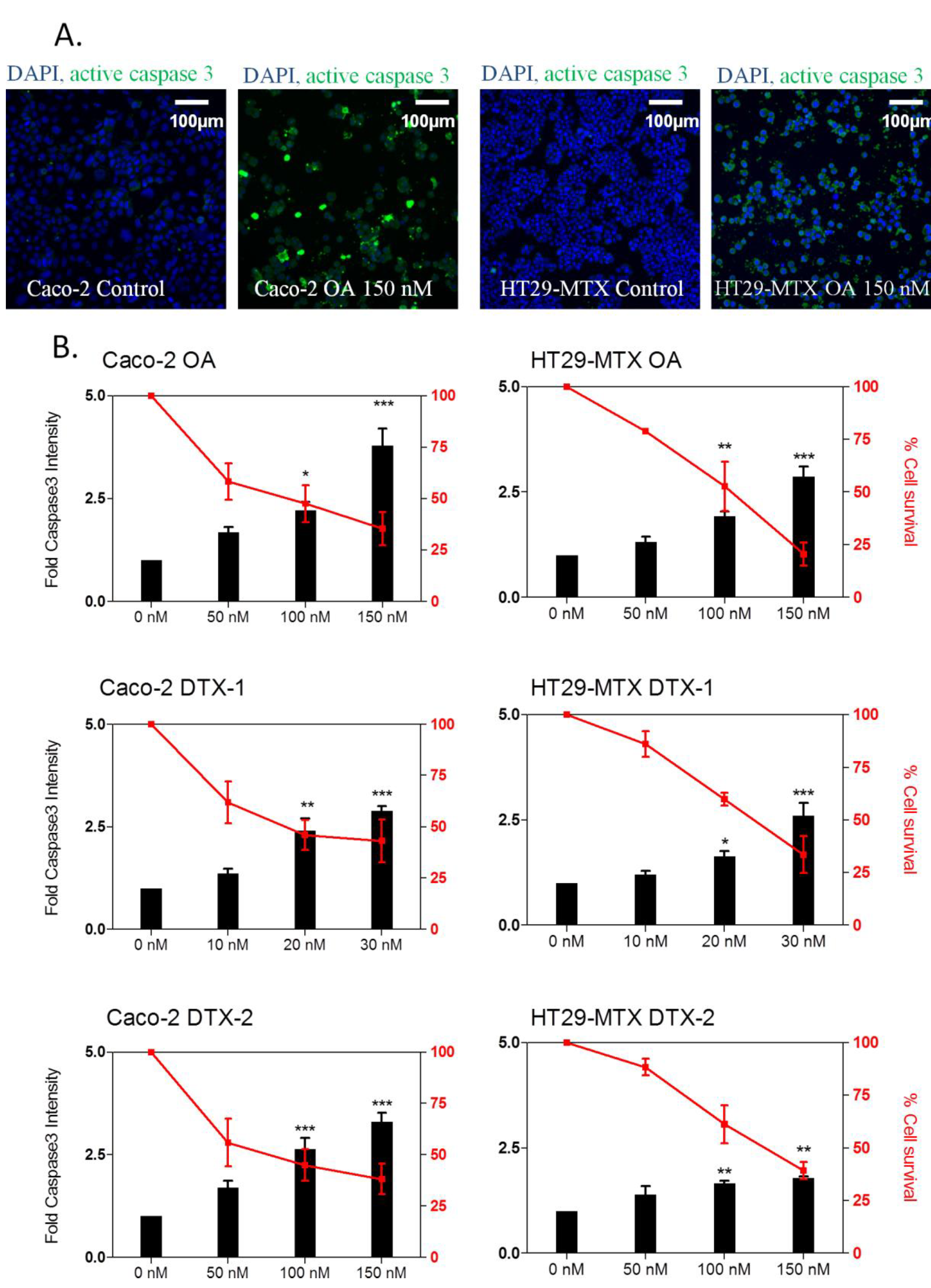
2.5. Apoptosis Induction
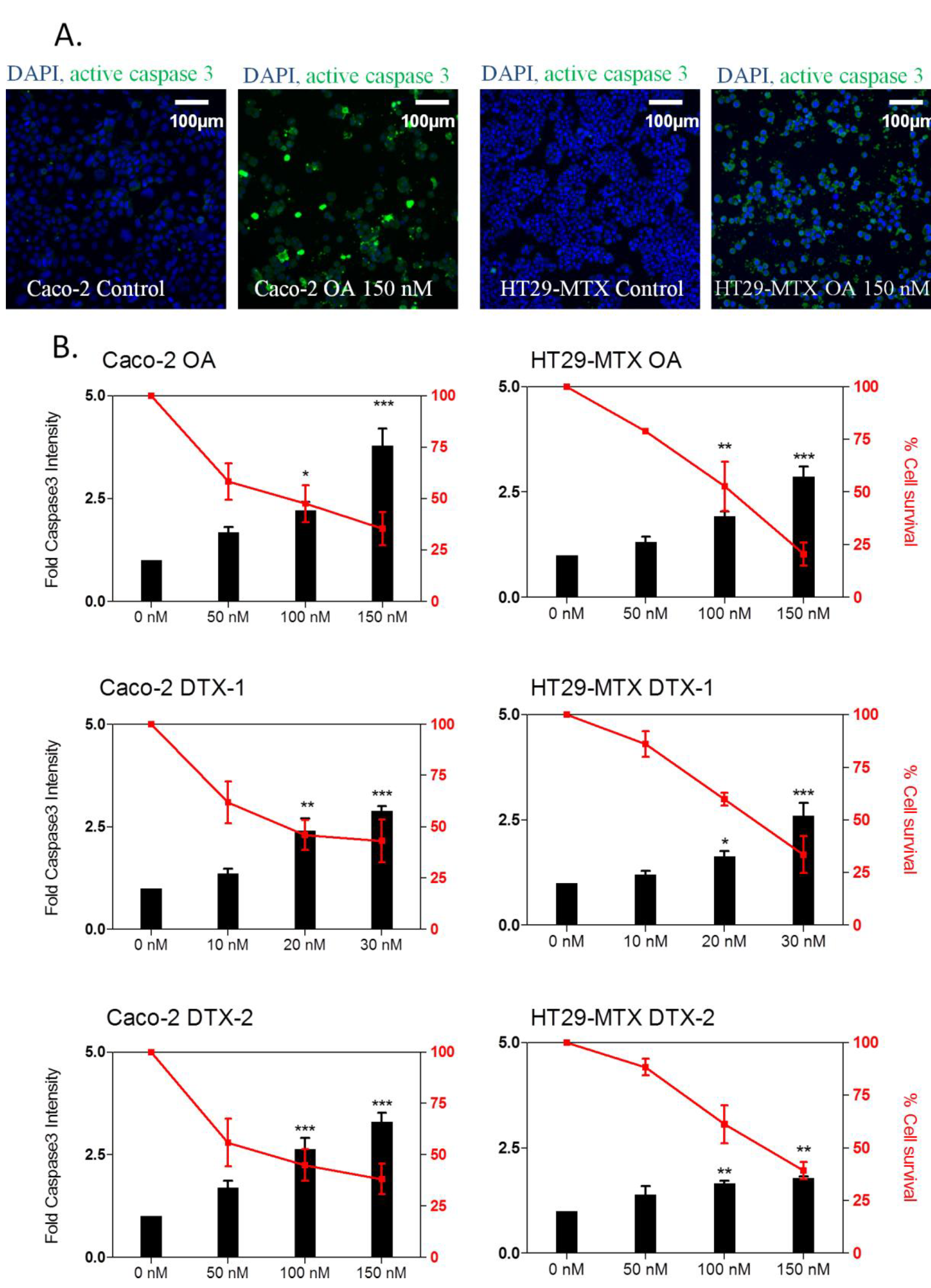
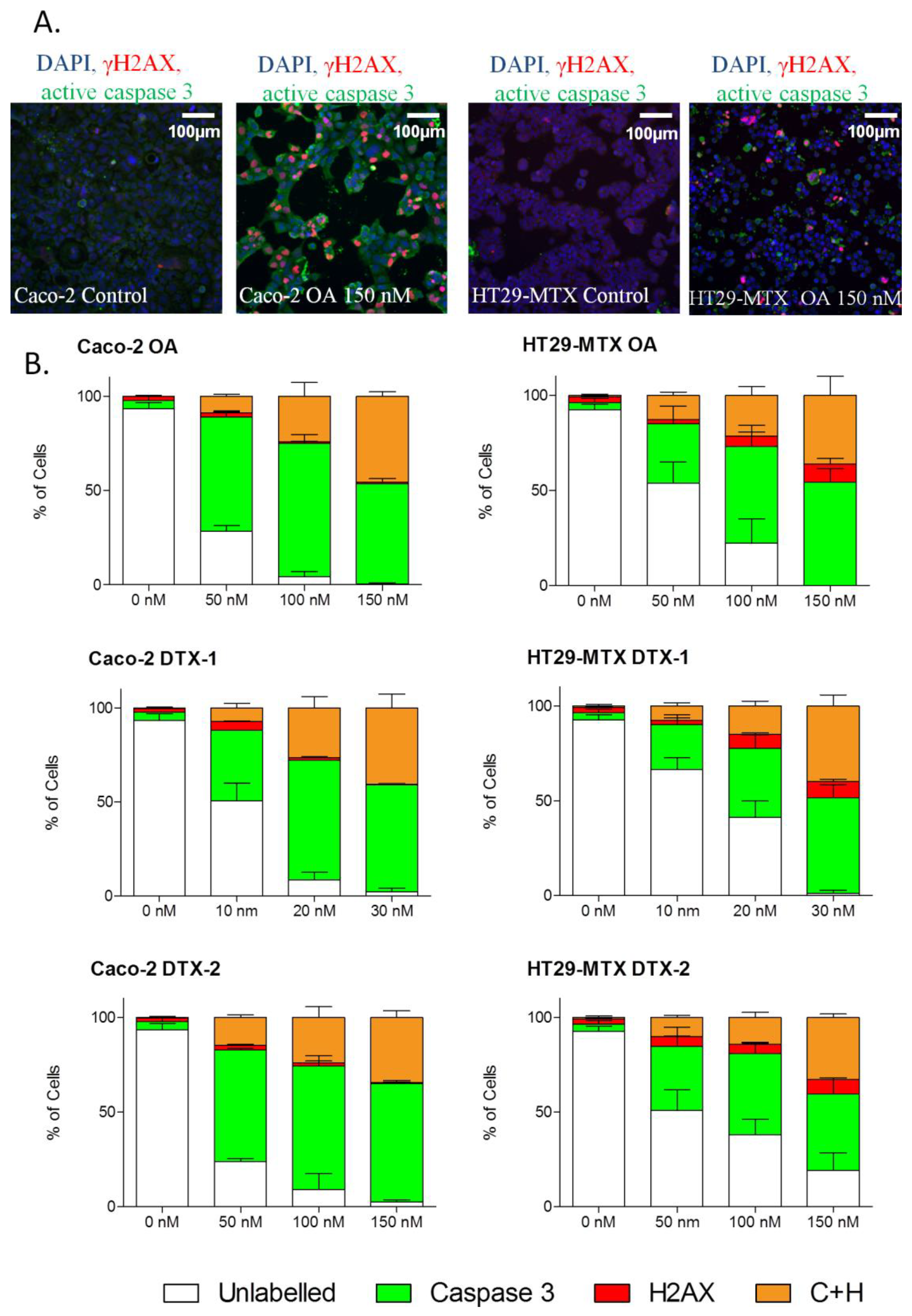
2.6. γH2AX Related to Apoptosis
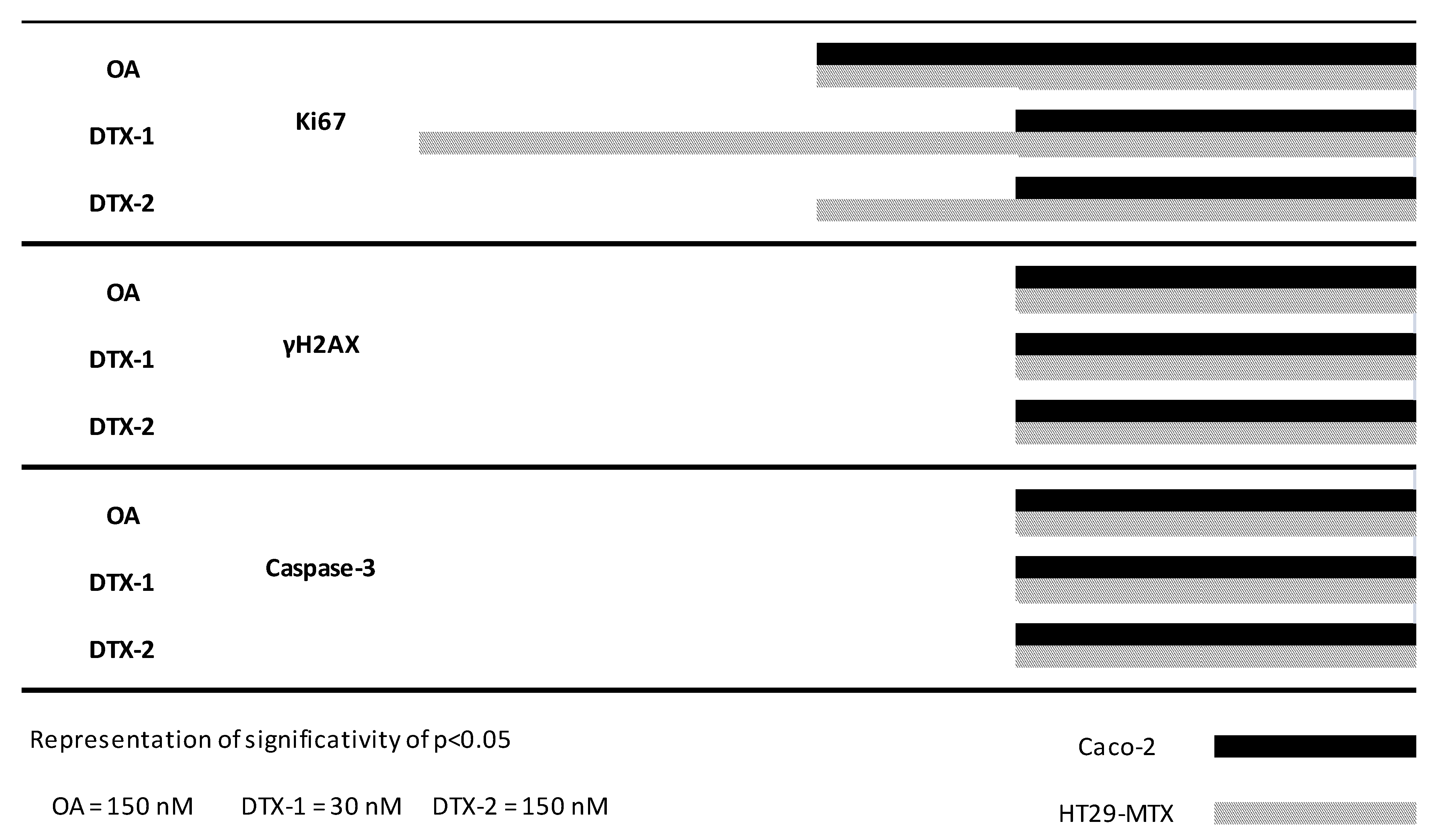
2.7. Temporal Sequence in Cytotoxicity Responses
3. Experimental Section
3.1. Cell Cultures
3.2. Toxins
3.3. Neutral Red Uptake Assay
3.4. Immunofluorescence
3.5. High Content Analysis
3.6. Statistics
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Fujiki, H.; Suganuma, M.; Suguri, H.; Yoshizawa, S.; Takagi, K.; Uda, N.; Wakamatsu, K.; Yamada, K.; Murata, M.; Yasumoto, T. Diarrhetic shellfish toxin, dinophysistoxin-1, is a potent tumor promoter on mouse skin. Jpn. J. Cancer Res. Gann. 1988, 79, 1089–1093. [Google Scholar] [CrossRef]
- Murata, M.; Shimatani, M.; Sugitani, H.; Oshima, Y.; Yasumoto, T. Isolation and structural elucidation of the causative toxin of the diarrhetic shellfish poisoning. Bull. Jpn. Soc. Sci. Fish. 1982, 48, 549–552. [Google Scholar] [CrossRef]
- Vale, P.; Antónia, M.; Sampayo, M. Esters of okadaic acid and dinophysistoxin-2 in Portuguese bivalves related to human poisonings. Toxicon 1999, 37, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yasumoto, T. Liquid chromatography–electrospray ionization mass spectrometry of the diarrhetic shellfish-poisoning toxins okadaic acid, dinophysistoxin-1 and pectenotoxin-6 in bivalves. J. Chromatogr. A 2000, 874, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Carmody, E.P.; James, K.J.; Kelly, S.S. Dinophysistoxin-2: The predominant diarrhoetic shellfish toxin in Ireland. Toxicon 1996, 34, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Moroño, A.; Arévalo, F.; Fernández, M.; Maneiro, J.; Pazos, Y.; Salgado, C.; Blanco, J. Accumulation and transformation of DSP toxins in mussels Mytilus galloprovincialis during a toxic episode caused by Dinophysis acuminata. Aquat. Toxicol. 2003, 62, 269–280. [Google Scholar]
- Terao, K.; Ito, E.; Yanagi, T.; Yasumoto, T. Histopathological studies on experimental marine toxin poisoning. I. Ultrastructural changes in the small intestine and liver of suckling mice induced by dinophysistoxin-1 and pectenotoxin-1. Toxicon Off. J. Int. Soc. Toxinol. 1986, 24, 1141–1151. [Google Scholar]
- García, C.; Truan, D.; Lagos, M.; Santelices, J.P.; Díaz, J.C.; Lagos, N. Metabolic transformation of dinophysistoxin-3 into dinophysistoxin-1 causes human intoxication by consumption of O-acyl-derivatives dinophysistoxins contaminated shellfish. J. Toxicol. Sci. 2005, 30, 287–296. [Google Scholar]
- Tubaro, A.; Giangaspero, A.; Ardizzone, M.; Soranzo, M.R.; Vita, F.; Yasumoto, T.; Maucher, J.M.; Ramsdell, J.S.; Sosa, S. Ultrastructural damage to heart tissue from repeated oral exposure to yessotoxin resolves in 3 months. Toxicon 2008, 51, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Marine biotoxins in shellfish—Summary on regulated marine biotoxins. EFSA J. 2009, 1306, 1–23.
- Fujiki, H.; Suganuma, M. Tumor promotion by inhibitors of protein phosphatases 1 and 2A: The okadaic acid class of compounds. Adv. Cancer Res. 1993, 61, 143–194. [Google Scholar] [PubMed]
- Takai, A.; Murata, M.; Torigoe, K.; Isobe, M.; Mieskes, G.; Yasumoto, T. Inhibitory effect of okadaic acid derivatives on protein phosphatases. A study on structure-affinity relationship. Biochem. J. 1992, 284, 539–544. [Google Scholar]
- Cohen, P.T. Novel protein serine/threonine phosphatases: Variety is the spice of life. Trends Biochem. Sci. 1997, 22, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Huhn, J.; Jeffrey, P.D.; Larsen, K.; Rundberget, T.; Rise, F.; Cox, N.R.; Arcus, V.; Shi, Y.; Miles, C.O. A structural basis for the reduced toxicity of dinophysistoxin-2. Chem. Res. Toxicol. 2009, 22, 1782–1786. [Google Scholar] [CrossRef] [PubMed]
- Larsen, K.; Petersen, D.; Wilkins, A.L.; Samdal, I.A.; Sandvik, M.; Rundberget, T.; Goldstone, D.; Arcus, V.; Hovgaard, P.; Rise, F.; et al. Clarification of the C-35 stereochemistries of dinophysistoxin-1 and dinophysistoxin-2 and its consequences for binding to protein phosphatase. Chem. Res. Toxicol. 2007, 20, 868–875. [Google Scholar] [PubMed]
- Munday, R. Is Protein phosphatase inhibition responsible for the toxic effects of okadaic acid in animals? Toxins 2013, 5, 267–285. [Google Scholar] [PubMed]
- Messner, D.J.; Ao, P.; Jagdale, A.B.; Boynton, A.L. Abbreviated cell cycle progression induced by the serine/threonine protein phosphatase inhibitor okadaic acid at concentrations that promote neoplastic transformation. Carcinogenesis 2001, 22, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Kurimchak, A.; Graña, X. PP2A holoenzymes negatively and positively regulate cell cycle progression by dephosphorylating pocket proteins and multiple CDK substrates. Gene 2012, 499, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yuasa, H.; Yoshida, K.; Iwata, H.; Nakanishi, H.; Suganuma, M.; Tatematsu, M. Increase of labeling indices in gastrointestinal mucosae of mice and rats by compounds of the okadaic acid type. J. Cancer Res. Clin. Oncol. 1994, 120, 208–212. [Google Scholar] [PubMed]
- Fessard, V.; Grosse, Y.; Pfohl-Leszkowicz, A.; Puiseux-Dao, S. Okadaic acid treatment induces DNA adduct formation in BHK21 C13 fibroblasts and HESV keratinocytes. Mutat. Res. 1996, 361, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Le Hégarat, L.; Jacquin, A.-G.; Bazin, E.; Fessard, V. Genotoxicity of the marine toxin okadaic acid, in human Caco-2 cells and in mice gut cells. Environ. Toxicol. 2006, 21, 55–64. [Google Scholar]
- Abolhassani, M.; Wertz, X.; Pooya, M.; Chaumet-Riffaud, P.; Guais, A.; Schwartz, L. Hyperosmolarity causes inflammation through the methylation of protein phosphatase 2A. Inflamm. Res. 2008, 57, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Rubiolo, J.A.; López-Alonso, H.; Vega, F.V.; Vieytes, M.R.; Botana, L.M. Comparative study of toxicological and cell cycle effects of okadaic acid and dinophysistoxin-2 in primary rat hepatocytes. Life Sci. 2012, 90, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Fernández, D.; Louzao, M.; Fraga, M.; Vilariño, N.; Vieytes, M.; Botana, L. Experimental basis for the high oral toxicity of dinophysistoxin 1: A comparative study of DSP. Toxins 2014, 6, 211–228. [Google Scholar]
- Del Campo, M.; Toledo, H.; Lagos, N. Okadaic acid toxin at sublethal dose produced cell proliferation in gastric and colon epithelial cell lines. Mar. Drugs 2013, 11, 4751–4760. [Google Scholar]
- Sérandour, A.-L.; Ledreux, A.; Morin, B.; Derick, S.; Augier, E.; Lanceleur, R.; Hamlaoui, S.; Moukha, S.; Furger, C.; Biré, R.; et al. Collaborative study for the detection of toxic compounds in shellfish extracts using cell-based assays. Part I: Screening strategy and pre-validation study with lipophilic marine toxins. Anal. Bioanal. Chem. 2012, 403, 1983–1993. [Google Scholar]
- Leteurtre, E.; Gouyer, V.; Rousseau, K.; Moreau, O.; Barbat, A.; Swallow, D.; Huet, G.; Lesuffleur, T. Differential mucin expression in colon carcinoma HT-29 clones with variable resistance to 5-fluorouracil and methotrexate. Biol. Cell 2004, 96, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Wikman-Larhed, A.; Artursson, P. Co-cultures of human intestinal goblet (HT29-H) and absorptive (Caco-2) cells for studies of drug and peptide absorption. Eur. J. Pharm. Sci. 1995, 3, 171–183. [Google Scholar]
- Haney, S.A.; LaPan, P.; Pan, J.; Zhang, J. High-content screening moves to the front of the line. Drug Discov. Today 2006, 11, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.M.; Fedorov, Y.; Brown, D.D.; Suh, M.; Proctor, D.M.; Kuriakose, L.; Haws, L.C.; Harris, M.A. Assessment of Cr(VI)-induced cytotoxicity and genotoxicity using High Content Analysis. PLoS One 2012, 7, e42720. [Google Scholar] [CrossRef] [PubMed]
- Scholzen, T.; Gerdes, J. The Ki-67 protein: From the known and the unknown. J. Cell. Physiol. 2000, 182, 311–322. [Google Scholar] [PubMed]
- Porter, A.G.; Jänicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Yang, Y.; Tirtha, D.; Yen, Y.; Zhou, B.-S.; Zhou, M.-M.; Ohlmeyer, M.; Ko, E.C.; Cagan, R.; Rosenstein, B.S.; et al. γ-H2AX kinetics as a novel approach to High Content Screening for small molecule radiosensitizers. PLoS One 2012, 7, e38465. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, M. Role of NF-κB in epithelial biology. Immunol. Rev. 2012, 246, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Rubiolo, J.A.; López-Alonso, H.; Vega, F.V.; Vieytes, M.R.; Botana, L.M. Okadaic acid and dinophysis toxin 2 have differential toxicological effects in hepatic cell lines inducing cell cycle arrest, at G0/G1 or G2/M with aberrant mitosis depending on the cell line. Arch. Toxicol. 2011, 85, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.H.A.; Held, M.; Janssens, V.; Hutchins, J.R.A.; Hudecz, O.; Ivanova, E.; Goris, J.; Trinkle-Mulcahy, L.; Lamond, A.I.; Poser, I.; et al. Live-cell imaging RNAi screen identifies PP2A-B55alpha and importin-beta1 as key mitotic exit regulators in human cells. Nat. Cell Biol. 2010, 12, 886–893. [Google Scholar] [PubMed]
- Liu, Y.; Bodmer, W.F. Analysis of P53 mutations and their expression in 56 colorectal cancer cell lines. Proc. Natl. Acad. Sci. USA 2006, 103, 976–981. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458, 1127–1130. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, A.; Scholz, J.; These, A.; Hessel, S.; Preiss-Weigert, A.; Lampen, A. Analysis of the passage of the marine biotoxin okadaic acid through an in vitro human gut barrier. Toxicology 2011, 279, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Kittler, K.; Preiss-Weigert, A.; These, A. Identification strategy using combined mass spectrometric techniques for elucidation of phase I and phase II in vitro metabolites of lipophilic marine biotoxins. Anal. Chem. 2010, 82, 9329–9335. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; An, T.; Rein, K.S. The algal hepatoxoxin okadaic acid is a substrate for human cytochromes CYP3A4 and CYP3A5. Toxicon 2010, 55, 325–332. [Google Scholar] [PubMed]
- Feng, G.; Ohmori, Y.; Chang, P.-L. Production of chemokine CXCL1/KC by okadaic acid through the nuclear factor-κB pathway. Carcinogenesis 2006, 27, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Lin, A. NF-kappaB at the crossroads of life and death. Nat. Immunol. 2002, 3, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Tinel, A.; Janssens, S.; Lippens, S.; Cuenin, S.; Logette, E.; Jaccard, B.; Quadroni, M.; Tschopp, J. Autoproteolysis of PIDD marks the bifurcation between pro-death caspase-2 and pro-survival NF-kappaB pathway. EMBO J. 2007, 26, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Tubaro, A.; Sosa, S.; Altinier, G.; Soranzo, M.R.; Satake, M.; Della Loggia, R.; Yasumoto, T. Short-term oral toxicity of homoyessotoxins, yessotoxin and okadaic acid in mice. Toxicon 2004, 43, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Souid-Mensi, G.; Moukha, S.; Mobio, T.A.; Maaroufi, K.; Creppy, E.E. The cytotoxicity and genotoxicity of okadaic acid are cell-line dependent. Toxicon Off. J. Int. Soc. Toxinol. 2008, 51, 1338–1344. [Google Scholar]
- Le Hégarat, L.; Fessard, V.; Poul, J.M.; Dragacci, S.; Sanders, P. Marine toxin okadaic acid induces aneuploidy in CHO-K1 cells in presence of rat liver postmitochondrial fraction, revealed by cytokinesis-block micronucleus assay coupled to FISH. Environ. Toxicol. 2004, 19, 123–128. [Google Scholar]
- Traoré, A.; Baudrimont, I.; Ambaliou, S.; Dano, S.D.; Creppy, E.E. DNA breaks and cell cycle arrest induced by okadaic acid in Caco-2 cells, a human colonic epithelial cell line. Arch. Toxicol. 2001, 75, 110–117. [Google Scholar]
- Lu, C.; Zhu, F.; Cho, Y.-Y.; Tang, F.; Zykova, T.; Ma, W.; Bode, A.M.; Dong, Z. Cell apoptosis: Requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol. Cell 2006, 23, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, J.; Gupta, N.; Agrawal, M.; Bala Bhaskar, A.S.; Lakshmana Rao, P.V. Modulation of ROS/MAPK signaling pathways by okadaic acid leads to cell death via, mitochondrial mediated caspase-dependent mechanism. Apoptosis 2011, 16, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Valdiglesias, V.; Laffon, B.; Pásaro, E.; Cemeli, E.; Anderson, D.; Méndez, J. Induction of oxidative DNA damage by the marine toxin okadaic acid depends on human cell type. Toxicon Off. J. Int. Soc. Toxinol. 2011, 57, 882–888. [Google Scholar] [CrossRef]
- Haneji, T.; Hirashima, K.; Teramachi, J.; Morimoto, H. Okadaic acid activates the PKR pathway and induces apoptosis through PKR stimulation in MG63 osteoblast-like cells. Int. J. Oncol. 2013, 42, 1904–1910. [Google Scholar] [PubMed]
- Fujita, M.; Goto, K.; Yoshida, K.; Okamura, H.; Morimoto, H.; Kito, S.; Fukuda, J.; Haneji, T. Okadaic acid stimulates expression of Fas receptor and Fas ligand by activation of nuclear factor kappa-B in human oral squamous carcinoma cells. Oral Oncol. 2004, 40, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Kitazumi, I.; Maseki, Y.; Nomura, Y.; Shimanuki, A.; Sugita, Y.; Tsukahara, M. Okadaic acid induces DNA fragmentation via caspase-3-dependent and caspase-3-independent pathways in Chinese hamster ovary (CHO)-K1 cells. FEBS J. 2010, 277, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Millward, T.A.; Zolnierowicz, S.; Hemmings, B.A. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem. Sci. 1999, 24, 186–191. [Google Scholar] [PubMed]
- Chang, F.; Steelman, L.S.; Shelton, J.G.; Lee, J.T.; Navolanic, P.M.; Blalock, W.L.; Franklin, R.; McCubrey, J.A. Regulation of cell cycle progression and apoptosis by the Ras/Raf/MEK/ERK pathway (Review). Int. J. Oncol. 2003, 22, 469–480. [Google Scholar] [PubMed]
- Li, H.-H.; Cai, X.; Shouse, G.P.; Piluso, L.G.; Liu, X. A specific PP2A regulatory subunit, B56γ, mediates DNA damage-induced dephosphorylation of p53 at Thr55. EMBO J. 2007, 26, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Kepp, O.; Galluzzi, L.; Kroemer, G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat. Immunol. 2012, 13, 343–351. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ferron, P.-J.; Hogeveen, K.; Fessard, V.; Hégarat, L.L. Comparative Analysis of the Cytotoxic Effects of Okadaic Acid-Group Toxins on Human Intestinal Cell Lines. Mar. Drugs 2014, 12, 4616-4634. https://doi.org/10.3390/md12084616
Ferron P-J, Hogeveen K, Fessard V, Hégarat LL. Comparative Analysis of the Cytotoxic Effects of Okadaic Acid-Group Toxins on Human Intestinal Cell Lines. Marine Drugs. 2014; 12(8):4616-4634. https://doi.org/10.3390/md12084616
Chicago/Turabian StyleFerron, Pierre-Jean, Kevin Hogeveen, Valérie Fessard, and Ludovic Le Hégarat. 2014. "Comparative Analysis of the Cytotoxic Effects of Okadaic Acid-Group Toxins on Human Intestinal Cell Lines" Marine Drugs 12, no. 8: 4616-4634. https://doi.org/10.3390/md12084616
APA StyleFerron, P.-J., Hogeveen, K., Fessard, V., & Hégarat, L. L. (2014). Comparative Analysis of the Cytotoxic Effects of Okadaic Acid-Group Toxins on Human Intestinal Cell Lines. Marine Drugs, 12(8), 4616-4634. https://doi.org/10.3390/md12084616