Stereochemical Determination of Five-Membered Cyclic Ether Acetogenins Using a Spin-Spin Coupling Constant Approach and DFT Calculations


Abstract
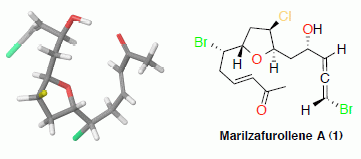
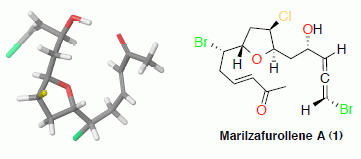
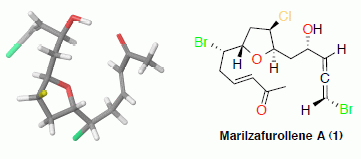
:
1. Introduction
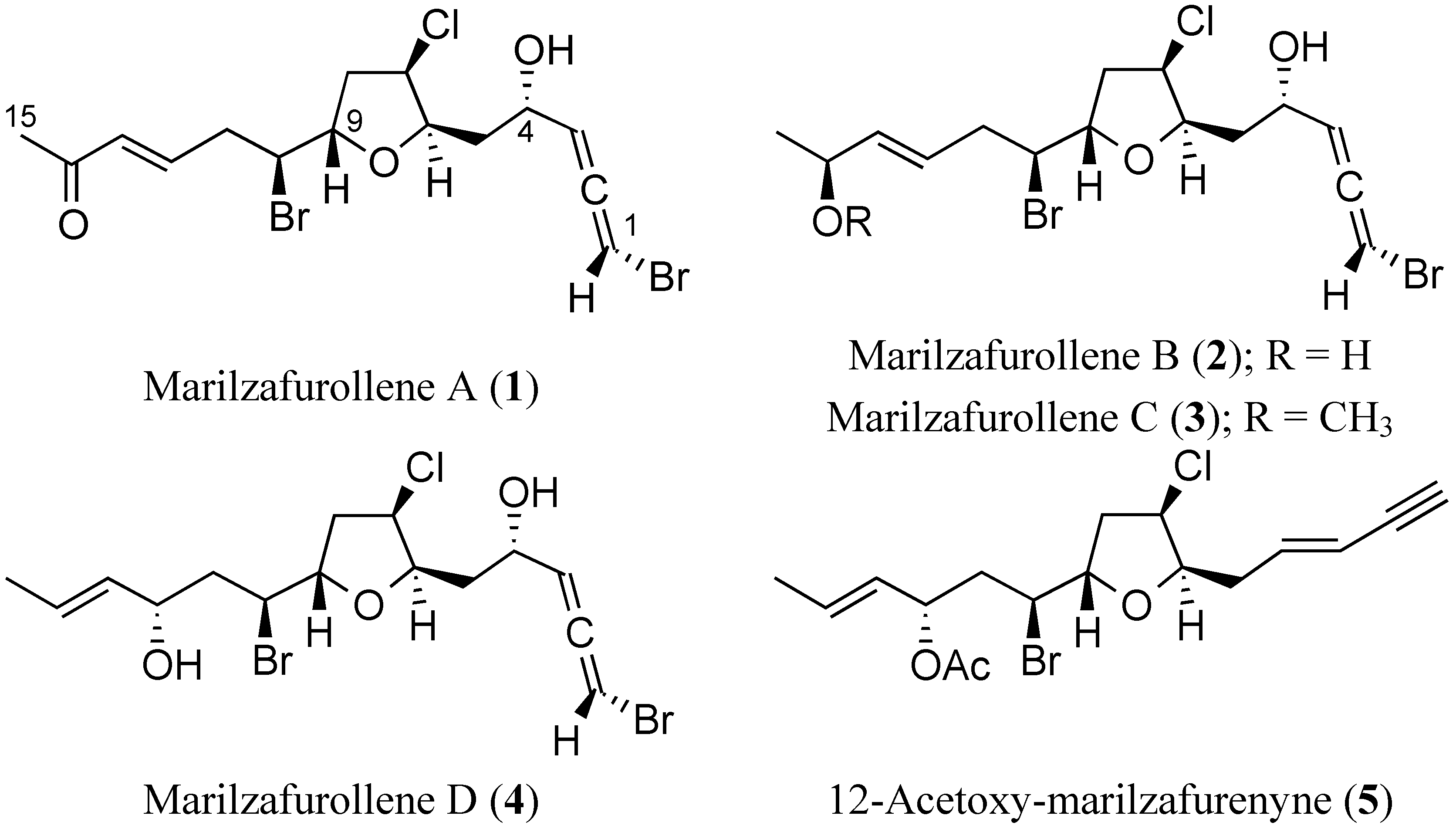
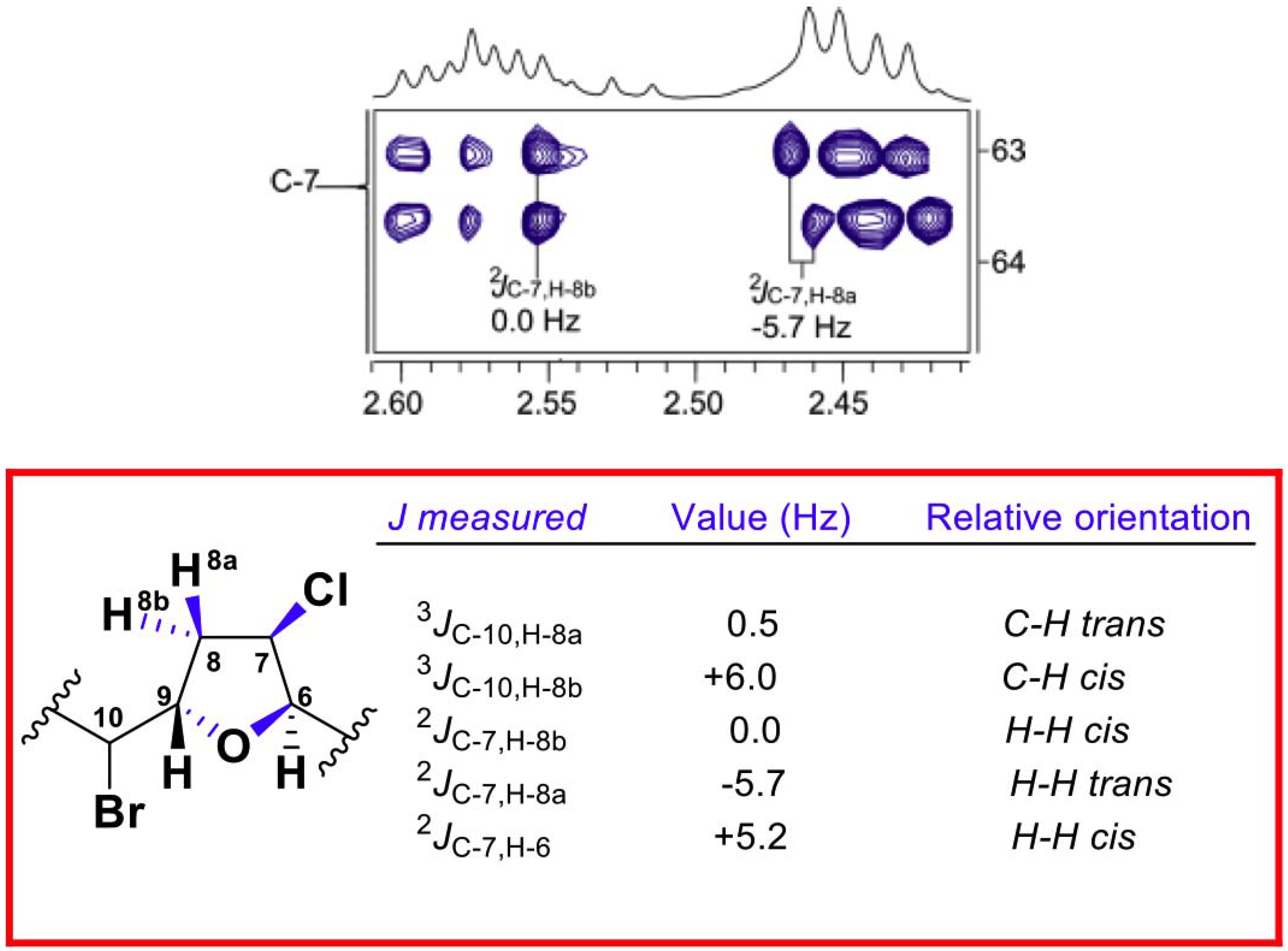
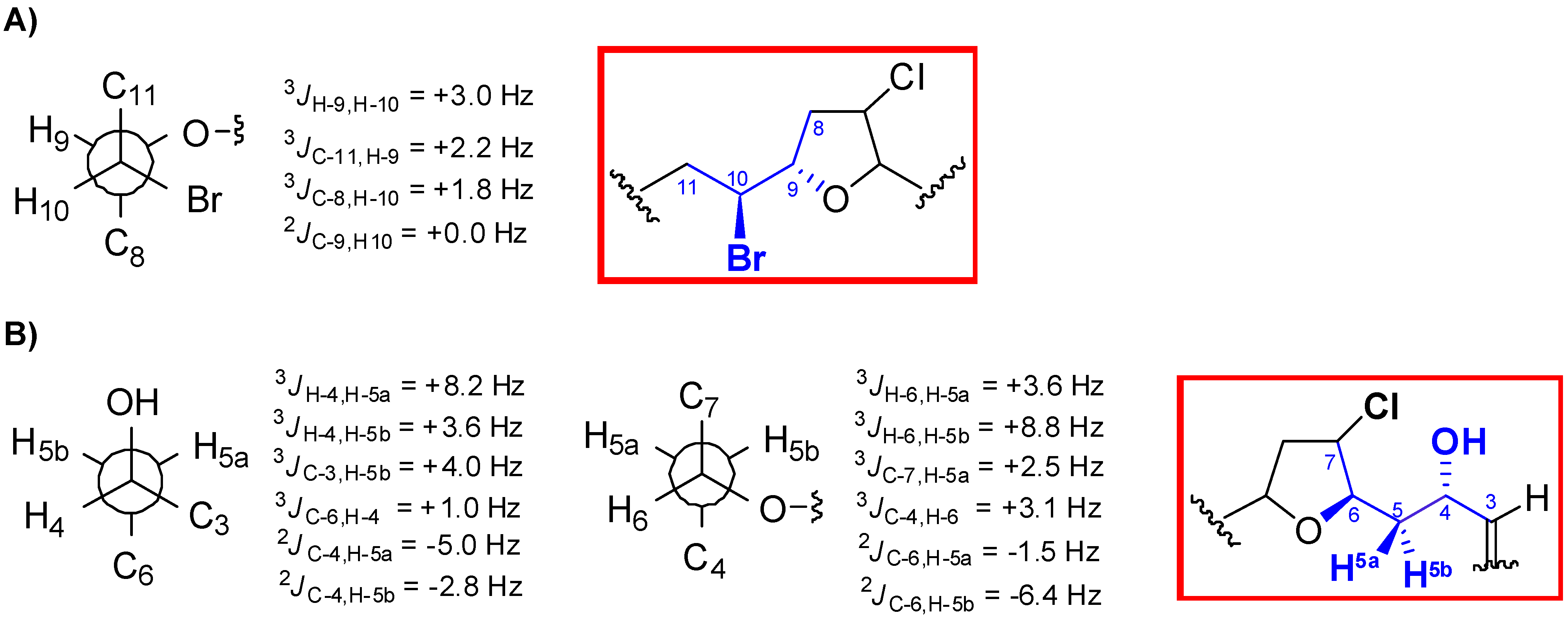
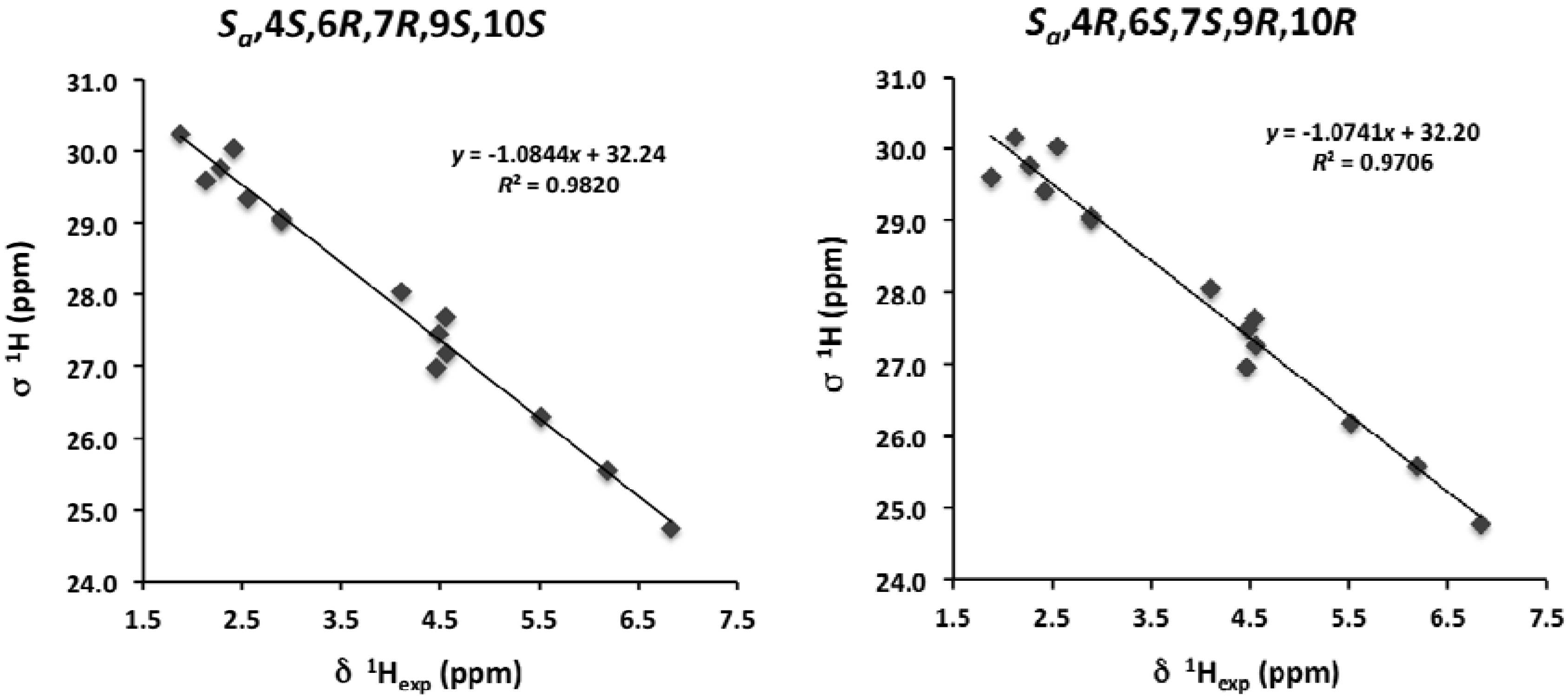
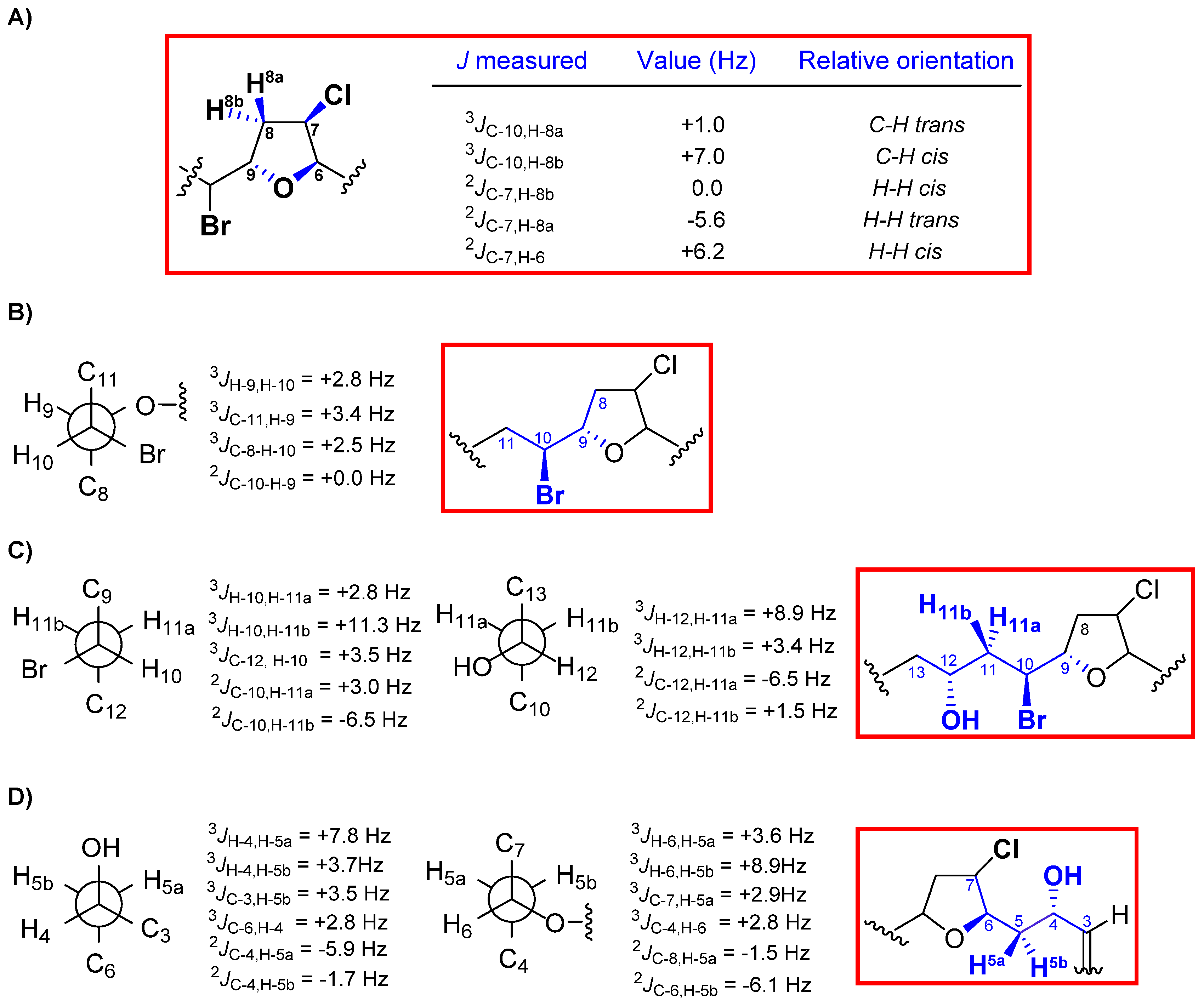
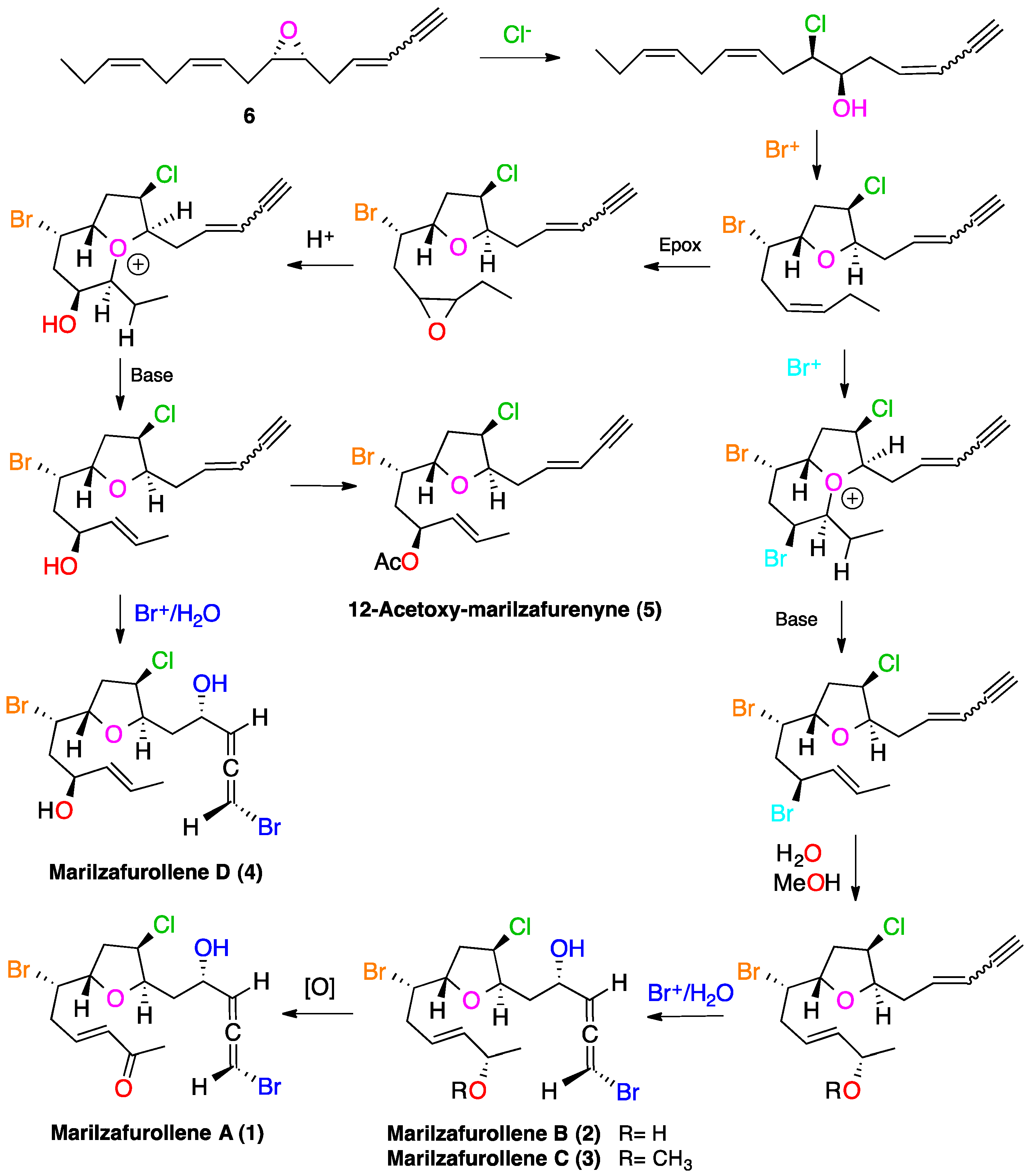
2. Results and Discussion

{kind=link}
{kind=link}
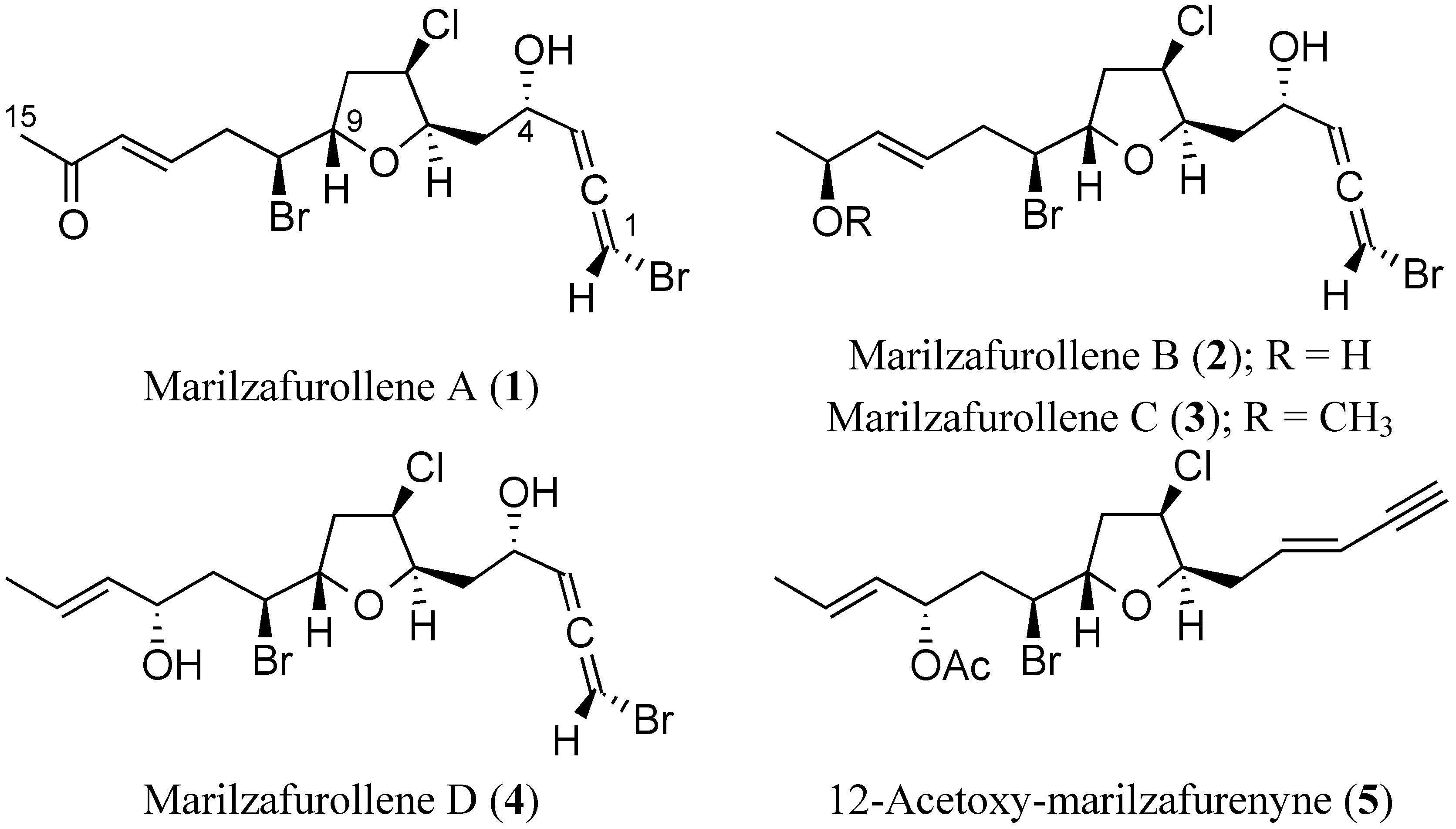{kind=link}
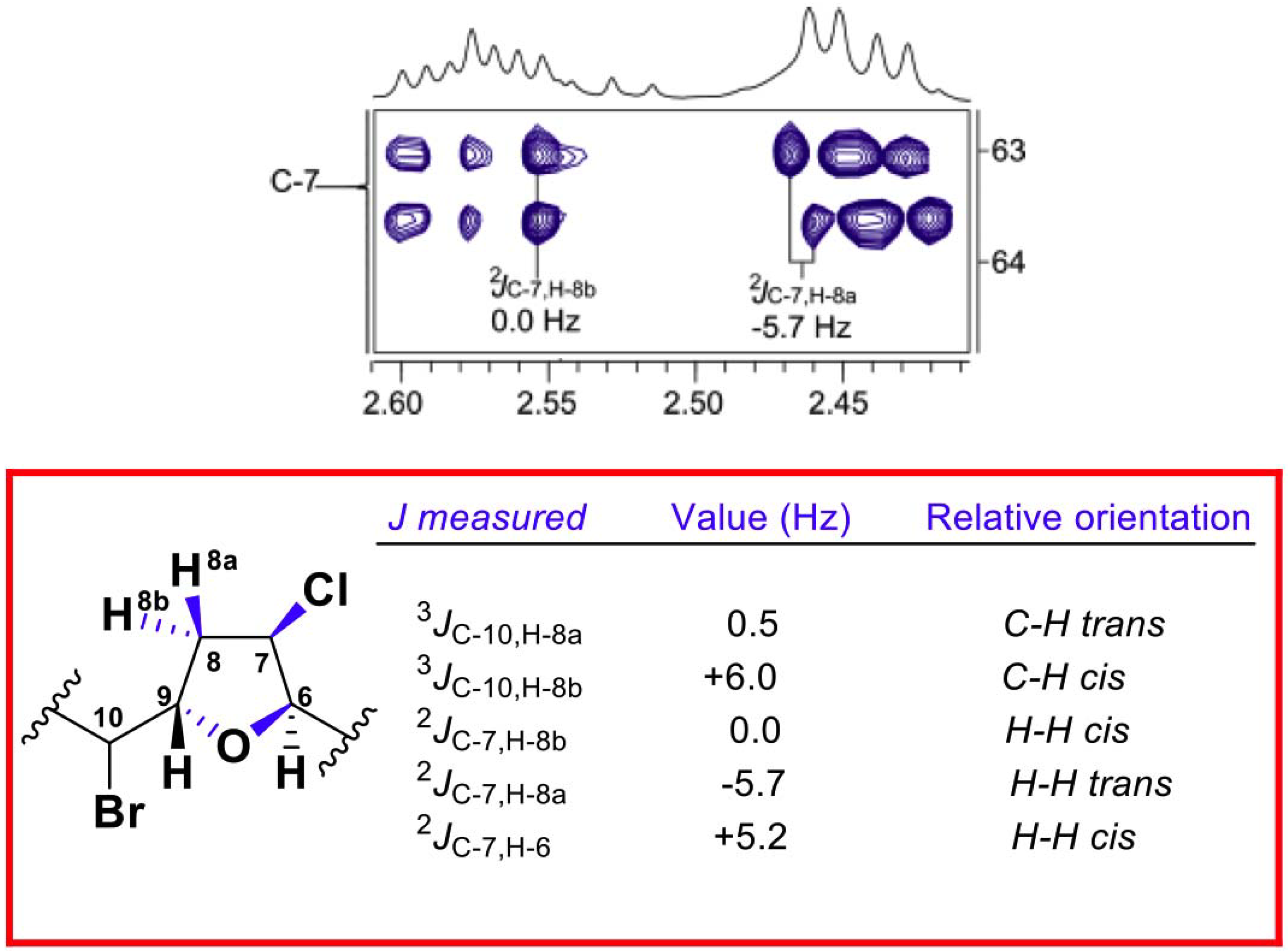{kind=link}
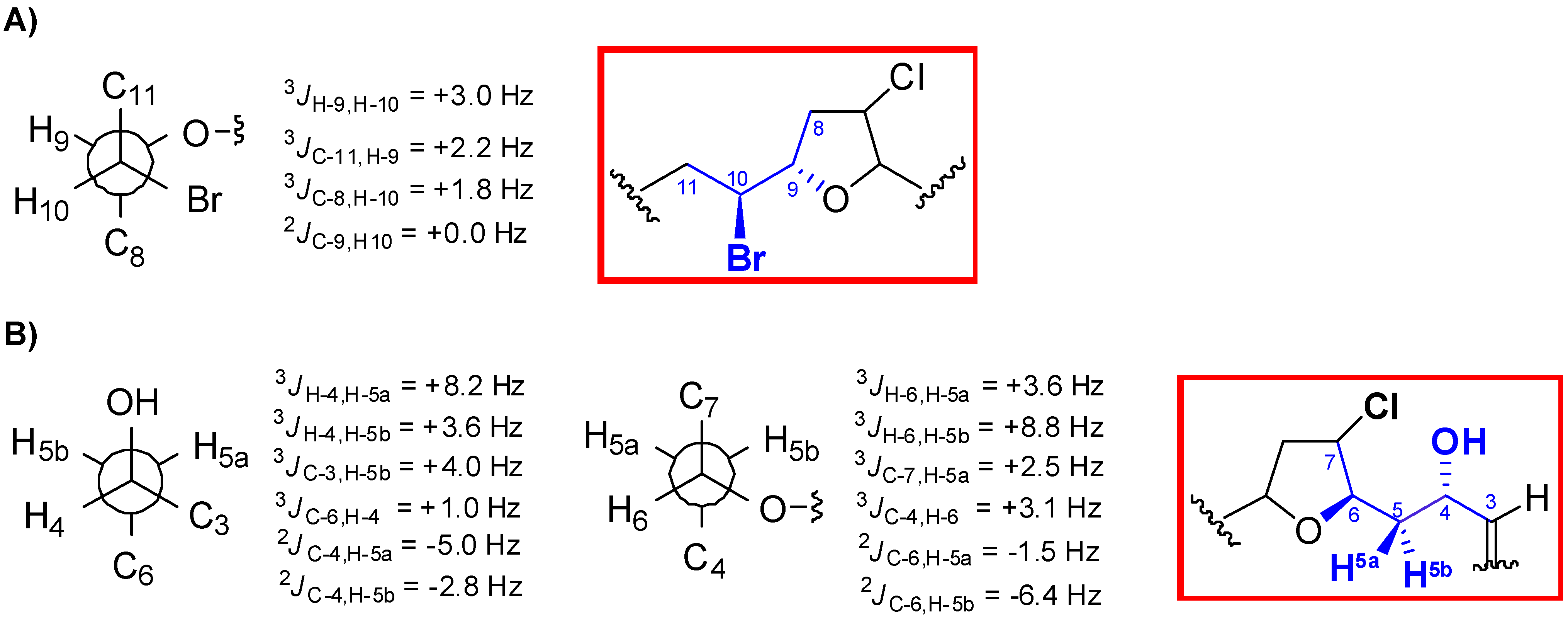{kind=link}
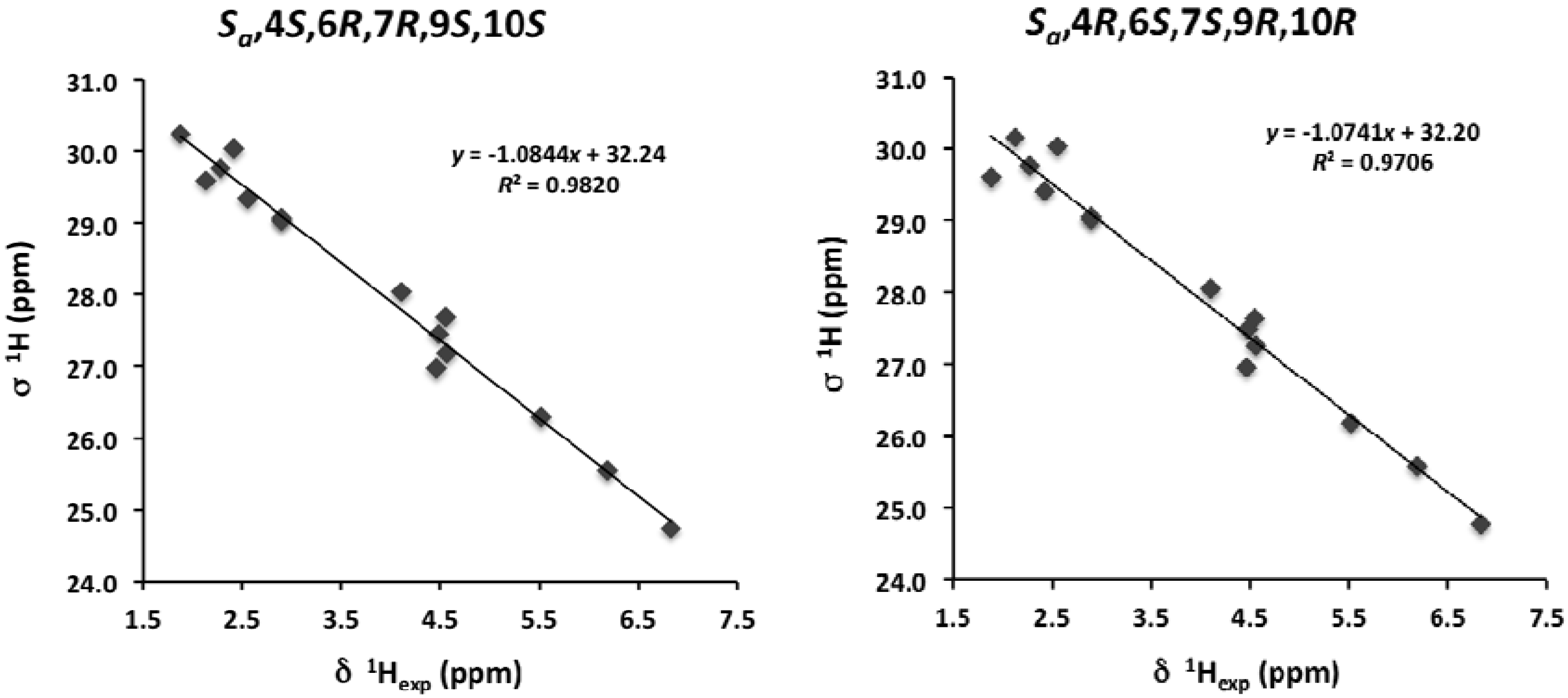{kind=link}
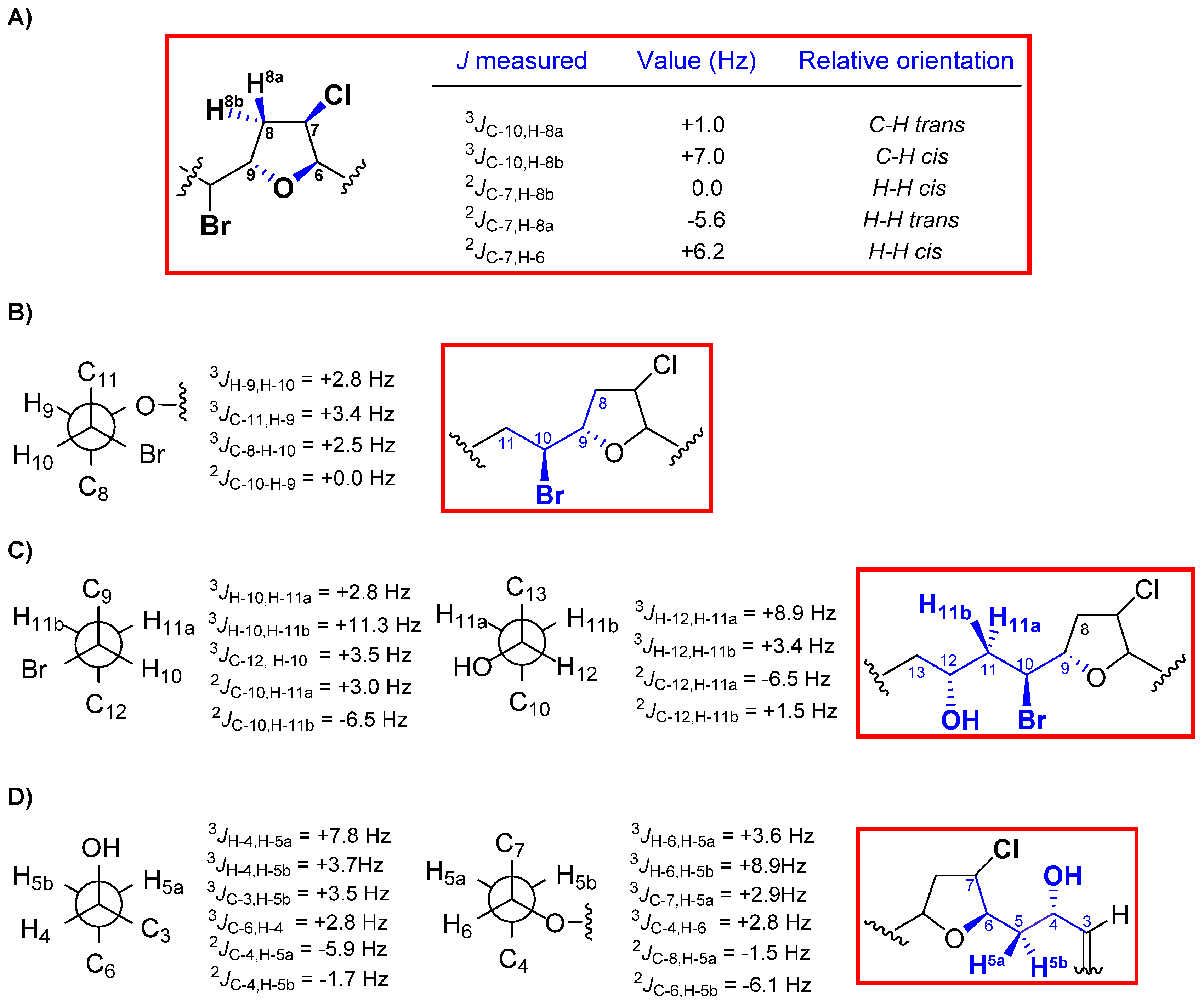{kind=link}
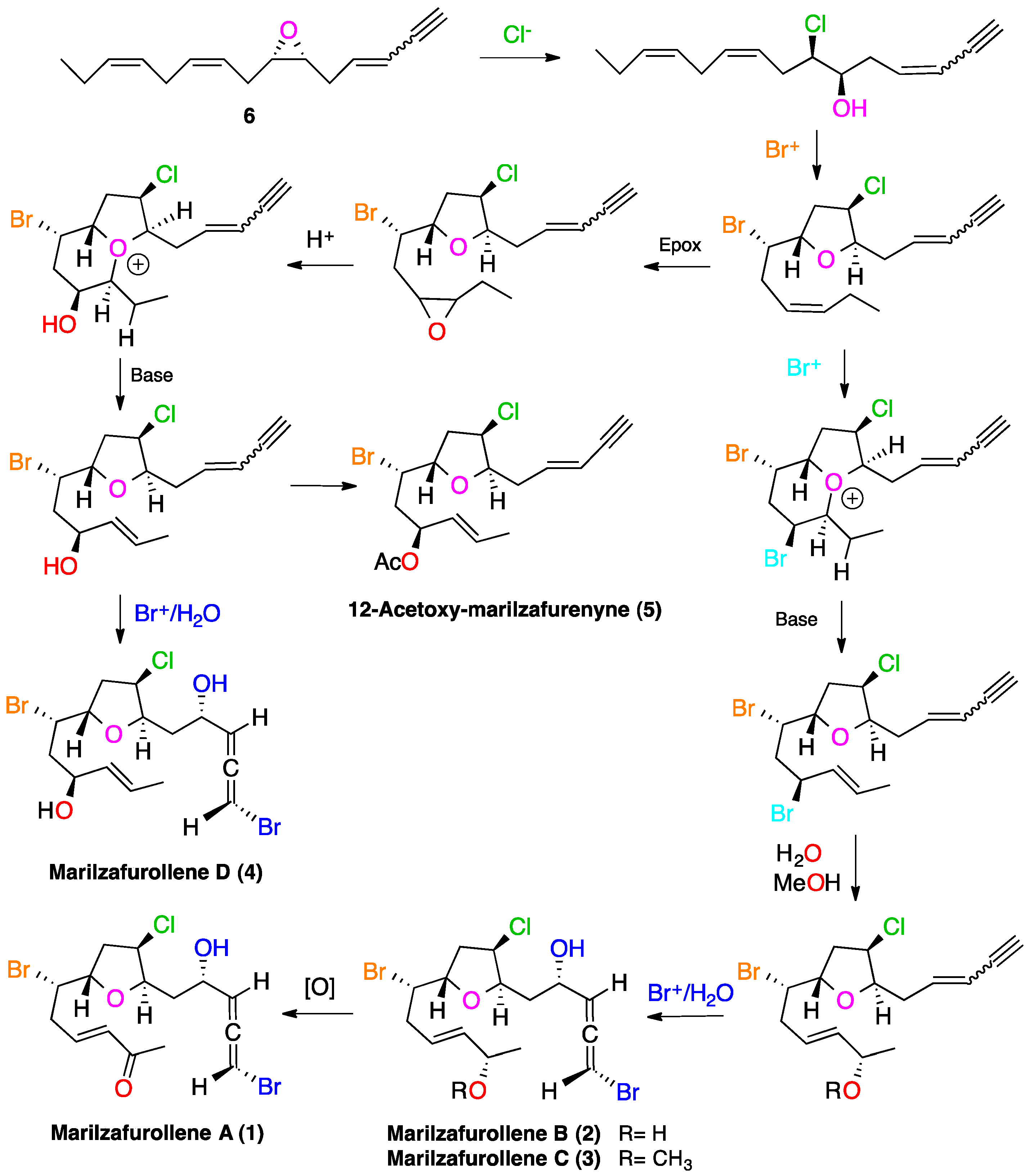{kind=link}
| C | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 1 | 74.9, CH | 74.8, CH | 74.8, CH | 74.8, CH | 77.0, CH |
| 2 | 200.6, C | 200.6, C | 200.8, C | 200.6, C | 82.0, C |
| 3 | 104.1, CH | 104.1, CH | 104.2, CH | 104.1, CH | 111.9, CH |
| 4 | 66.4, CH | 66.5, CH | 66.6, CH | 66.6, CH | 140.7, CH |
| 5 | 38.3, CH2 | 38.1, CH2 | 38.2, CH2 | 38.2, CH2 | 35.0, CH2 |
| 6 | 80.0, CH | 79.7, CH | 79.8, CH | 79.8, CH | 82.1, CH |
| 7 | 63.2, CH | 63.3, CH | 63.4, CH | 63.4, CH | 62.3, CH |
| 8 | 40.9, CH2 | 40.7, CH2 | 40.9, CH2 | 41.1, CH2 | 41.0, CH2 |
| 9 | 79.1, CH | 79.1, CH | 79.1, CH | 80.0, CH | 79.6, CH |
| 10 | 55.6, CH | 57.3, CH | 57.9, CH | 56.2, CH | 54.8, CH |
| 11 | 38.5, CH2 | 38.5, CH2 | 38.4, CH2 | 43.1, CH2 | 40.8, CH2 |
| 12 | 143.2, CH | 126.2, CH | 128.5, CH | 70.4, CH | 72.8, CH |
| 13 | 133.4, CH | 137.6, CH | 135.8, CH | 133.3, CH | 128.7, CH |
| 14 | 198.5, C | 68.5, CH | 77.7, CH | 127.5, CH | 130.0, CH |
| 15 | 27.3, CH3 | 23.4, CH3 | 21.3, CH3 | 17.7, CH3 | 17.8, CH3 |
| OCH3 | 56.0, CH3 | ||||
| CO(Ac) | 170.2, C | ||||
| CH3(Ac) | 21.3, CH3 |
| C | Marilzafurollene A (1) | Marilzafurollene B (2) | Marilzafurollene C (3) |
|---|---|---|---|
| 1 | 6.13, dd (2.2, 5.6) | 6.13, dd (2.2, 5.7) | 6.13, ddd (1.5, 2.2, 5.6) |
| 3 | 5.52, dd (5.6, 5.6) | 5.52, dd (5.5, 5.7) | 5.53, dd (5.6, 5.6) |
| 4 | 4.56, ddd (3.6, 5.6, 8.2) | 4.57, ddd (3.6, 5.5, 7.7) | 4.57, ddd (3.4, 5.6, 8.0) |
| 5 | 2.13, ddd (3.6, 8.8, 14.4) | 2.15, ddd (3.6, 9.1, 14.5) | 2.14, ddd (3.4, 8.7, 14.0) |
| 1.88, ddd (3.6, 8.2, 14.4) | 1.86, ddd (3.5, 7.7, 14.5) | 1.87, ddd (3.1, 8.0, 14.0) | |
| 6 | 4.48, ddd (3.0, 3.6, 8.8) | 4.46, ddd (3.3, 3.5, 9.1) | 4.46, ddd (2.5, 3.1, 8.7) |
| 7 | 4.55, ddd (0.8, 3.0, 4.8) | 4.53, ddd (0.8, 3.3, 4.8) | 4.54, ddd (2.5, 3.4, 4.5) |
| 8 | α 2.55, ddd (4.8, 9.6, 13.9) | α 2.52, ddd (4.8, 9.6, 13.9) | α 2.52, ddd (4.5, 8.9, 14.1) |
| β 2.42, ddd (0.8, 6.2, 13.9) | β 2.39, ddd (0.8, 6.2, 13.9) | β 2.39, ddd (3.4, 6.3, 14.1) | |
| 9 | 4.46, ddd (3.0, 6.2, 9.6) | 4.47, ddd (3.1, 6.2, 9.6) | 4.47, ddd (3.4, 6.3, 8.3) |
| 10 | 4.10, ddd (3.0, 5.2, 8.5) | 4.06, ddd (3.1, 5.7, 7.9) | 4.03, ddd (3.4, 4.6, 8.6) |
| 11 | 2.90, m (2H) | 2.70, m (2H) | 2.71, m (2H) |
| 12 | 6.83, ddd (7.0, 7.0, 15.9) | 5.72, ddd (6.4, 7.0, 15.6) | 5.67, ddd (7.0, 7.2, 15.5) |
| 13 | 6.18, br d (15.9) | 5.65, br dd (6.1, 15.6) | 5.48, dddd (1.5, 1.5, 6.8, 15.5) |
| 14 | 4.30, dd (6.1, 6.3) | 3.72, dd (6.6, 6.8) | |
| 15 | 2.28, s (3H) | 1.28, d (6.3) (3H) | 1.24, d (6.5) (3H) |
| OCH3 | 3.28, s (3H) |
| C | Marilzafurollene D (4) | 12-Acetoxy-marilzafurenyne (5) |
|---|---|---|
| 1 | 6.12, dd (2.1, 5.6) | 2.84, br d (1.7) |
| 3 | 5.52, dd (5.6, 5.6) | 5.64, dd (1.7, 16.1) |
| 4 | 4.57, ddd (3.7, 5.6, 7.8) | 6.20, ddd (7.4, 7.4, 16.1) |
| 5 | 2.15, ddd (3.7, 8.9, 14.3) | 2.60, ddd (6.8, 7.4, 14.7) |
| 1.87, ddd (3.6, 7.8, 14.3) | 2.50, ddd (6.8, 7.4, 14.7) | |
| 6 | 4.47, ddd (3.4, 3.6, 8.9) | 4.19, ddd (2.8, 6.8, 6.8) |
| 7 | 4.54, dd (3.4, 4.5) | 4.49, dd (2.8, 4.8) |
| 8 | α 2.56, ddd (4.5, 9.5, 13.9) | α 2.56, ddd (4.8, 9.8, 13.9) |
| β 2.40, dd (6.2, 13.9) | β 2.38, dd (6.1, 13.9) | |
| 9 | 4.42, ddd (2.8, 6.2, 9.5) | 4.39 ddd (2.5, 6.1, 9.8) |
| 10 | 4.35, ddd (2.8, 2.8, 11.3) | 4.05 ddd (2.5, 3.1, 10.7) |
| 11 | 2.09, ddd (3.4, 11.3, 15.0) | 2.22, ddd (3.3, 10.7, 14.3) |
| 1.91, ddd (2.8, 8.9, 15.0) | 2.17, ddd (3.1, 9.8, 14.3) | |
| 12 | 4.41, ddd (3.4, 6.7, 8.9) | 5.48, ddd (3.3, 7.0, 9.8) |
| 13 | 5.54, br dd (6.7, 15.2) | 5.43, br dd (7.0, 15.1) |
| 14 | 5.74, dq (6.4, 15.2) | 5.80, dq (6.5, 15.1) |
| 15 | 1.70, d (6.4) (3H) | 1.69, br d (6.5) (3H) |
| CH3(Ac) | 2.05, s (3H) |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Computational Methods
3.3. Biological Material
3.4. Extraction and Isolation
4. Conclusions
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinset, M.R. Marine natural products. Nat. Prod. Rep. 2014, 31, 160–258. [Google Scholar] [CrossRef]
- Gerwick, W.H.; Moore, B.S. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol. 2012, 27, 85–98. [Google Scholar] [CrossRef]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–83. [Google Scholar]
- Bifulco, G.; Dambruoso, P.; Gomez-Paloma, L.; Riccio, R. Determination of relative configuration in organic compounds by NMR spectroscopy and computational methods. Chem. Rev. 2007, 107, 3744–3779. [Google Scholar] [CrossRef]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational prediction of 1H and 13C chemical shifts: A useful tool for natural product, mechanistic, and synthetic organic chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef]
- Saielli, G.; Nicolaou, K.C.; Ortiz, A.; Zhang, H.; Bagno, A. Addressing the stereochemistry of complex organic molecules by density functional theory-NMR: Vannusal B in retrospective. J. Am. Chem. Soc. 2011, 133, 6072–6077. [Google Scholar] [CrossRef]
- Napolitano, J.G.; Gavín, J.A.; García, C.; Norte, M.; Fernández, J.J.; Hernández Daranas, A. On the configuration of five-membered rings: A spin-spin coupling constant approach. Chem. Eur. J. 2011, 17, 6338–6347. [Google Scholar] [CrossRef]
- Napolitano, J.G.; Norte, M.; Fernández, J.J.; Hernández Daranas, A. Corozalic acid: A key biosynthetic precursor with phosphatase inhibition activity. Chem. Eur. J. 2010, 16, 11576–11579. [Google Scholar]
- Gutiérrez-Cepeda, A.; Fernández, J.J.; Gil, L.V.; López-Rodríguez, M.; Norte, M.; Souto, M.L. Nonterpenoid C15 acetogenins from Laurencia marilzae. J. Nat. Prod. 2011, 74, 441–448. [Google Scholar]
- Gutiérrez-Cepeda, A.; Fernández, J.J.; Norte, M.; Souto, M.L. New bicyclotridecane C15 nonterpenoid bromoallenes from Laurencia marilzae. Org. Lett. 2011, 13, 2690–2693. [Google Scholar]
- Gil-Rodríguez, M.C.; Sentíes, A.; Díaz-Larrea, J.; Cassano, V.; Fujii, M.T. Laurencia marilzae sp. nov. (Ceramiales, Rhodophyta) from the Canary Islands, Spain, based on morphological and molecular evidence. J. Phycol. 2009, 45, 264–271. [Google Scholar] [CrossRef]
- Wang, B.-G.; Gloer, J.B.; Ji, N.-Y.; Zhao, J.-C. Halogenated organic molecules of Rhodomelaceae origin: Chemistry and biology. Chem. Rev. 2013, 113, 3632–3685. [Google Scholar] [CrossRef]
- Holmes, M.T.; Briton, R. Total synthesis and structural revision of laurefurenynes A and B. Chem. Eur. J. 2013, 19, 12649–12652. [Google Scholar] [CrossRef]
- Ji, N.-Y.; Li, X.-M.; Li, K.; Wang, B.-G. Corrigendum: Laurendecumallenes A–B and laurendecumenynes A–B, halogenated nonterpenoid C(15)-acetogenins from the marine red alga Laurencia decumbens. J. Nat. Prod. 2010, 73, 1192. [Google Scholar]
- Cen-Pacheco, F.; Rodríguez, J.; Norte, M.; Fernández, J.J.; Hernández Daranas, A. Connecting discrete stereoclusters by using DFT and NMR spectroscopy: The case of nivariol. Chem. Eur. J. 2013, 19, 8525–8532. [Google Scholar] [CrossRef]
- Matsumori, N.; Kaneno, D.; Murata, M.; Nakamura, H.; Tachibana, K. Stereochemical determination of acyclic structures based on carbon-proton spin-coupling constants. A method of configuration analysis for natural products. J. Org. Chem. 1999, 64, 866–876. [Google Scholar]
- The absolute configuration of the bromoallene moiety was proposed according to the Lowe-Brewster’s rule, and confirmed by CD data through comparison with related compounds whose absolute configurations were assigned from X-ray diffraction data.
- Lowe, G. The absolute configuration of allenes. J. Chem. Soc. Chem. Commun. 1965, 17, 411–413. [Google Scholar]
- Elsevier, C.J.; Vermeer, P.; Gedanken, A.; Runge, W. Synthesis and absolute configurations of halogenoallenes. J. Org. Chem. 1985, 50, 364–367. [Google Scholar]
- Guella, G.; Chiasera, G.; Mancini, I.; Öztunç, A.; Pietra, F. Twelve-membered O-bridged cyclic ethers of red seaweeds in the genus Laurencia exist in solution as slowly interconverting conformers. Chem. Eur. J. 1997, 3, 1223–1231. [Google Scholar] [CrossRef]
- Domínguez, H.J.; Crespín, G.D.; Santiago Benítez, A.J.; Gavín, J.A.; Norte, M.; Fernández, J.J.; Hernández Daranas, A. Stereochemistry of complex marine natural products by quantum mechanical calculations of NMR chemical shifts: Solvent and conformational effects on okadaic acid. Mar. Drugs 2014, 12, 176–192. [Google Scholar] [CrossRef]
- Smith, S.G.; Channon, J.A.; Paterson, I.; Goodman, J.M. The stereochemical assignment of acyclic polyols: A computational study of the NMR data of a library of stereopentad sequences from polyketide natural products. Tetrahedron 2010, 66, 6437–6444. [Google Scholar]
- Rassolov, V.A.; Ratner, M.A.; Pople, J.A.; Redfern, P.C.; Curtiss, L.A. 6–31G* basis set for third-row atoms. J. Comp. Chem. 2001, 22, 976–984. [Google Scholar] [CrossRef]
- Smith, S.G.; Goodman, J.M. Assigning stereochemistry to single diastereoisomers by GIAO NMR calculation: The DP4 probability. J. Am. Chem. Soc. 2010, 132, 12946–12959. [Google Scholar] [CrossRef]
- Murai, A. Biosynthesis of cyclic bromoethers from red algae. In Comprehensive Natural Product Chemistry; Barton, D., Nakanishi, K., Meth-Cohn, O., Eds.; Pergamon: Elmsford, NY, USA, 1999; Volume 1, pp. 303–324. [Google Scholar]
- Braddock, D.C. A hypothesis concerning the biosynthesis of the obtusallene family of marine natural products via electrophilic bromination. Org. Lett. 2006, 8, 6055–6058. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gutiérrez-Cepeda, A.; Daranas, A.H.; Fernández, J.J.; Norte, M.; Souto, M.L. Stereochemical Determination of Five-Membered Cyclic Ether Acetogenins Using a Spin-Spin Coupling Constant Approach and DFT Calculations. Mar. Drugs 2014, 12, 4031-4044. https://doi.org/10.3390/md12074031
Gutiérrez-Cepeda A, Daranas AH, Fernández JJ, Norte M, Souto ML. Stereochemical Determination of Five-Membered Cyclic Ether Acetogenins Using a Spin-Spin Coupling Constant Approach and DFT Calculations. Marine Drugs. 2014; 12(7):4031-4044. https://doi.org/10.3390/md12074031
Chicago/Turabian StyleGutiérrez-Cepeda, Adrián, Antonio Hernández Daranas, José J. Fernández, Manuel Norte, and María L. Souto. 2014. "Stereochemical Determination of Five-Membered Cyclic Ether Acetogenins Using a Spin-Spin Coupling Constant Approach and DFT Calculations" Marine Drugs 12, no. 7: 4031-4044. https://doi.org/10.3390/md12074031
APA StyleGutiérrez-Cepeda, A., Daranas, A. H., Fernández, J. J., Norte, M., & Souto, M. L. (2014). Stereochemical Determination of Five-Membered Cyclic Ether Acetogenins Using a Spin-Spin Coupling Constant Approach and DFT Calculations. Marine Drugs, 12(7), 4031-4044. https://doi.org/10.3390/md12074031