3.2. Synthesis of Compounds 1–20
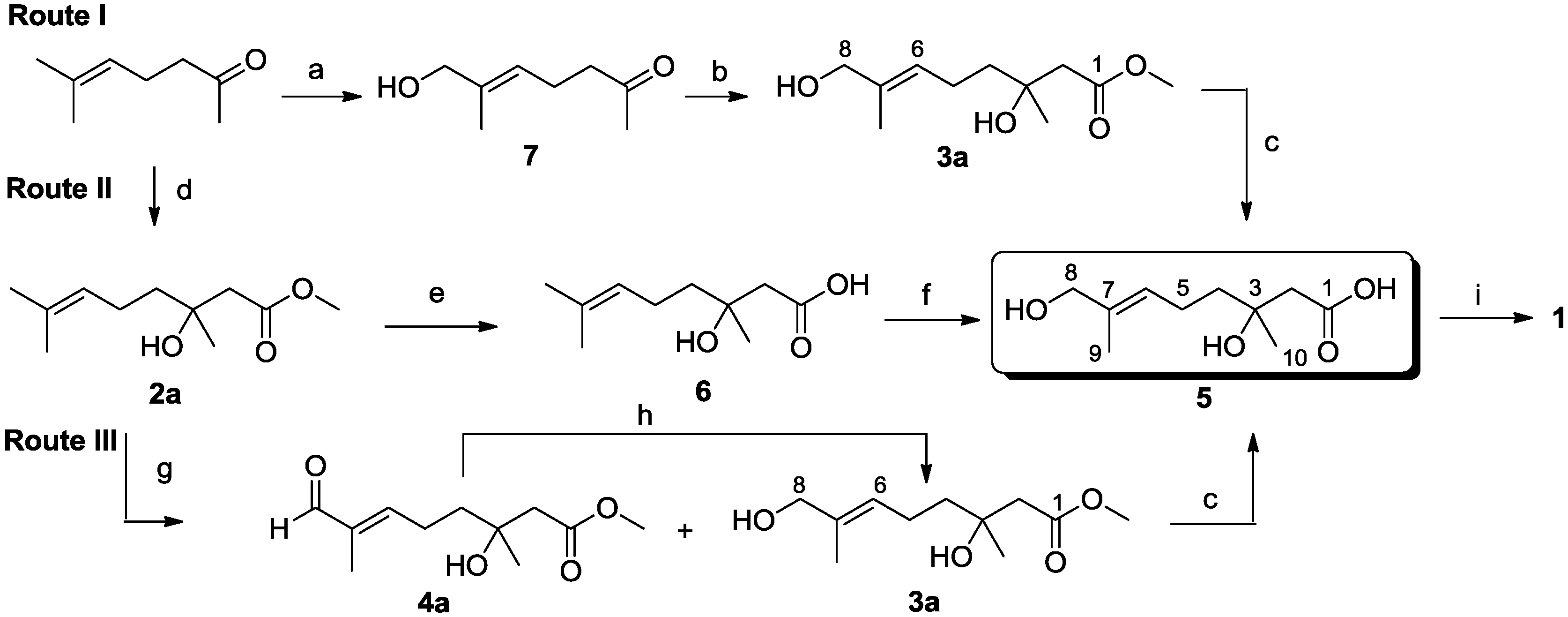
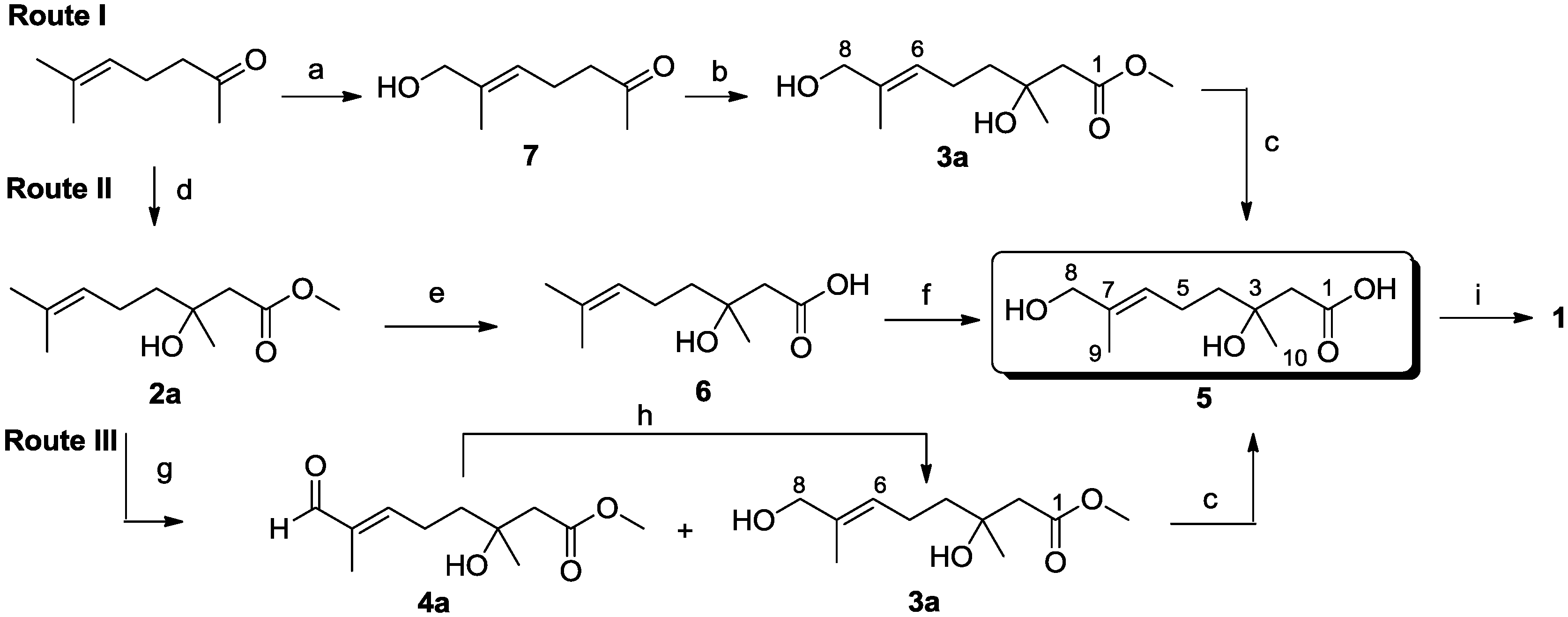
3.2.1. Synthesis of Methyl (±)-3-Hydroxy-3,7-dimethyloct-6-enoate (2a)
2-Methyl-2-hepten-6-one (3.16 g, 25.0 mol) and BrCH2COOCH3 (7.65 g, 50.0 mol) in 50 mL of anhydrous THF were added to a suspension of Zn (3.27 g, 50.0 mol) in anhydrous THF (50 mL) at reflux. After the reaction was initiated, the other portion of the solution was added dropwise to the reaction mixture. The mixture was allowed to stir at reflux until the color of the Zn changed from gray to brownish. After 2 h, the reaction mixture was cooled to room temperature, and the reaction was quenched with 100 mL of 10% AcOH. The reaction mixture was extracted with EtOAc (3 × 100 mL). The combined organic layers were washed with brine and dried with anhydrous Na2SO4. The solvent was evaporated in vacuo, and the crude product was purified by column chromatography (EtOAc–petroleum ether, 1:7) to yield 2a (4.25 g, 85%) as a pale yellow oil; Rf 0.32 (EtOAc–petroleum ether, 1:7); 1H NMR (500 MHz, CDCl3) δ 5.07 (t, J = 6.3 Hz, 1H, CH2CH=), 3.69 (s, 3H, OCH3), 3.08 (s, 1H, OH), 2.51 (d, J = 15.5 Hz, 1H, CH2COOCH3), 2.43 (d, J = 15.5 Hz, 1H, CH2COOCH3), 2.02 (m, 2H, =CHCH2), 1.66 (s, 3H, CH3), 1.59 (s, 3H, CH3), 1.52 (m, 2H, =CHCH2CH2), 1.22 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 173.4, 131.9, 124.2, 71.0, 51.7, 44.9, 41.9, 26.8, 25.7, 22.8, 17.7; HRESIMS m/z 201.1486 [M + H]+ (calcd. for C11H21O3, 201.1485).
3.2.2. Synthesis of Ethyl (±)-3-Hydroxy-3,7-dimethyloct-6-enoate (2b)
This compound was obtained from BrCH2COOCH2CH3 by a method analogous to that used for 2a. 2b (3.05 g, 95%); Rf 0.35 (EtOAc–petroleum ether, 1:7); 1H NMR (500 MHz, CDCl3) δ 5.06 (t, J = 6.3 Hz, 1H, CH2CH=), 4.14 (q, J = 7.1 Hz, 2H, OCH2CH3), 3.39 (br s, 1H, OH), 2.48 (d, J = 15.5 Hz, 1H, CH2COOCH2CH3), 2.40 (d, J = 15.5 Hz, 1H, CH2COOCH2CH3), 2.01 (m, 2H, =CHCH2), 1.64 (s, 3H, CH3), 1.57 (s, 3H, CH3), 1.50 (td, J = 7.3, 3.5 Hz, 2H, =CHCH2CH2), 1.24 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.21 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 173.1, 131.8, 124.2, 71.0, 60.7, 45.0, 41.9, 26.7, 25.7, 22.7, 17.6, 14.2; HRESIMS m/z 215.1640 [M + H]+ (calcd. for C12H23O3, 215.1642).
3.2.3. Syntheses of Methyl (E)-(±)-3,8-Dihydroxy-3,7-dimethyloct-6-enoate (3a) and Methyl (E)-(±)-3-Hydroxy-3,7-dimethyl-8-oxooct-6-enoate (4a)
Compound 2a (3.20 g, 15.9 mmol) was dissolved in CH2Cl2 (150 mL) and added dropwise to a solution of SeO2 (825 mg, 7.4 mmol) and t-BuOOH (70% in water, 3.2 mL, 22.3 mmol) in CH2Cl2 (50 mL) at room temperature. After stirring at room temperature for 19 h, the reaction mixture was quenched with aqueous 10% NaOH, H2O and brine; then, the organic layer was dried and concentrated. The resulting yellow oil was purified by flash column chromatography (EtOAc–petroleum ether, 1:7) on silica gel to yield 4a as a colorless oil (624 mg, 25%); Rf 0.33 (EtOAc–petroleum ether, 1:3); 1H NMR (500 MHz, CDCl3) δ 9.38 (s, 1H, CHO), 6.48 (t, J = 6.9 Hz, 1H, CH2CH=), 3.72 (s, 3H, OCH3), 2.55 (d, J = 15.7 Hz, 1H, CH2COOCH3), 2.49 (d, J = 15.7 Hz, 1H, CH2COOCH3), 2.46 (m, 2H, CH2CH2CH=), 1.75 (s, 3H, CH3), 1.67 (m, 2H, CH2CH2CH=), 1.28 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 195.3, 173.3, 154.3, 139.6, 70.7, 51.9, 44.9, 40.2, 26.7, 23.7, 9.3; HRESIMS m/z 237.1100 [M + Na]+ (calcd. for C11H18O4Na, 237.1097). Compound 3a was obtained in an analogous fashion; colorless oil (732 mg, 29%); Rf 0.22 (EtOAc–petroleum ether, 1:3); 1H NMR (500 MHz, CDCl3) δ 5.39 (t, J = 7.1 Hz, 1H, CH2CH=), 3.98 (s, 2H, CH2OH), 3.71 (s, 3H, OCH3), 2.53 (d, J = 15.6 Hz, 1H, CH2COOCH3), 2.46 (d, J = 15.6 Hz, 1H, CH2COOCH3), 2.12 (m, 2H, CH2CH=), 1.66 (s, 3H, CH3C=), 1.56 (m, 2H, CH2CH2CH=), 1.25 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 173.5, 135.3, 125.8, 71.0, 68.9, 51.8, 45.0, 41.6, 26.8, 22.4, 13.7; HRESIMS m/z 239.1252 [M + Na]+ (calcd. for C11H20O4Na, 239.1254). Additionally, 841 mg of material were recovered. The E-configuration of Compound 3a was verified by a NOESY correlation from CH2CH= (δ 5.39) to CH2OH (δ 3.98).
3.2.4. Reduction of Aldehyde 4a to Alcohol 3a
NaBH4 (117 mg, 3.1 mmol) was added to a stirred solution of aldehyde 4a (840 mg, 3.1 mmol) in MeOH (50 mL). The mixture was stirred for 1 h at room temperature, then concentrated under reduced pressure. The residue thus obtained was treated with EtOAc (20 mL) and saturated NH4Cl solution (50 mL); then, the separated aqueous phase was extracted with EtOAc (3 × 30 mL). The combined organic phases were dried (Na2SO4), filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography (EtOAc–petroleum ether, 1:5) on silica gel to yield a colorless oil, 3a (460 mg, 69%).
3.2.5. Syntheses of Ethyl (E)-(±)-3,8-Dihydroxy-3,7-dimethyloct-6-enoate (3b) and Ethyl (E)-(±)-3-Hydroxy-3,7-dimethyl-8-oxooct-6-enoate (4b)
These compounds were obtained from 2b in a manner similar to that described for the preparation of 3a and 4a. Colorless oil 4b (3.98 g, 16%); Rf 0.35 (EtOAc–petroleum ether, 1:3); 1H NMR (500 MHz, CDCl3) δ 9.39 (s, 1H, CHO), 6.49 (t, J = 7.1 Hz, 1H, CH2CH=), 4.20 (q, J = 7.1 Hz, 2H, OCH2CH3), 2.54 (d, J = 15.8 Hz, 1H, CH2COOCH2CH3), 2.48 (d, J = 15.5 Hz, 1H, CH2COOCH2CH3), 2.45 (m, 2H, CH2CH=), 1.76 (s, 3H, CH3), 1.67 (m, 2H, CH2CH2CH=), 1.29 (s, 3H, COHCH3), 1.29 (t, J = 7.0 Hz, 3H, OCH2CH3); 13C NMR (125 MHz, CDCl3) δ 195.3, 173.0, 154.3, 139.6, 70.7, 61.0, 45.1, 40.2, 26.7, 23.8, 14.3, 9.3; HRESIMS m/z 251.1253 [M + Na]+ (calcd. for C12H20O4Na, 251.1254). Colorless oil 3b (20.91 g, 81%); Rf 0.25 (EtOAc–petroleum ether, 1:3); 1H NMR (500 MHz, CDCl3) δ 5.38 (t, J = 6.7 Hz, 1H, CH2CH=), 4.16 (q, J = 7.1 Hz, 2H, OCH2CH3), 3.97 (s, 2H, CH2OH), 2.50 (d, J = 15.5 Hz, 1H, CH2COOCH2CH3), 2.43 (d, J = 15.6 Hz, 1H, CH2COOCH2CH3), 2.11 (m, 2H, =CHCH2), 1.65 (s, 3H, CH3), 1.54 (m, 2H, =CHCH2CH2), 1.26 (t, J = 7.2 Hz, 3H, OCH2CH3), 1.24 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 173.1, 135.2, 125.7, 71.0, 68.9, 60.8, 45.1, 41.6, 26.7, 22.3, 14.3, 13.7; HRESIMS m/z 253.1415 [M + Na]+ (calcd. for C12H22O4Na, 253.1410).
3.2.6. Synthesis of (E)-(±)-3,8-Dihydroxy-3,7-dimethyloct-6-enoic Acid (5)
To a solution of 3a (133 mg, 0.6 mmol) in MeOH (30 mL) and water (10 mL) was added KOH (76 mg, 1.4 mmol). The resulting reaction mixture was heated under reflux for 3 h, and the reaction was monitored by TLC. After completion of the reaction, the reaction mixture was acidified with 1 N HCl to pH 2~3, followed by extraction with CHCl3 (3 × 20 mL). The combined organic layers were dried (MgSO4) and concentrated in vacuo. The residue was chromatographed over silica gel (EtOAc–petroleum ether, 1:1) to afford the acid, 5 (105 mg, 85%), as a colorless oil; Rf 0.25 (EtOAc:CHCl3, 1:1); 1H NMR (500 MHz, CDCl3) δ 5.41 (t, J = 5.4 Hz, 1H, CH2CH=), 4.00 (s, 2H, CH2OH), 2.58 (d, J = 15.7 Hz, 1H, CH2COOH), 2.51 (d, J = 15.7 Hz, 1H, CH2COOH), 2.13 (m, 2H, CH2CH=), 1.67 (s, 3H, CH3C=), 1.63 (m, 2H, CH2CH2CH=), 1.31 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 175.5, 135.4, 125.7, 71.5, 68.9, 44.9, 41.5, 26.8, 22.4, 13.8; HRESIMS m/z 225.1076 [M + Na]+ (calcd. for C10H18O4Na, 225.1103).
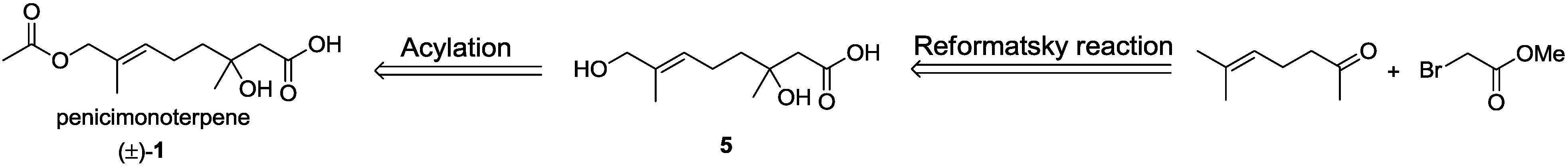
3.2.7. Syntheses of Acetic (E)-(±)-8-Acetoxy-3-hydroxy-3,7-dimethyloct-6-enoic Anhydride (10) and Penicimonoterpene (±)-1
Compound 5 (250 mg, 1.2 mmol) was dissolved in CH2Cl2 (20 mL) at room temperature. To this solution were added DMAP (3 mg, 0.03 mmol), Et3N (225 μL, 1.6 mmol) and, finally, Ac2O (152 μL, 1.6 mmol). The reaction was stirred overnight. After completion of the reaction as detected by TLC, the solvents were evaporated under reduced pressure, and the crude product was purified by column chromatography (EtOAc–petroleum ether, 1:1) to afford 10 and (±)-1.
10, colorless oil (65 mg, 18%); Rf 0.75 (EtOAc–petroleum ether, 1:3); 1H NMR (500 MHz, CDCl3) δ 5.44 (t, J = 6.5 Hz, 1H, CH2CH=), 4.44 (s, 2H, CH2OAc), 3.05 (d, J = 14.5 Hz, 1H, CH2COOAc), 2.90 (d, J = 14.5 Hz, 1H, CH2COOAc), 2.10 (m, 2H, CH2CH=), 2.07 (s, 3H, COCH3), 2.03 (m, 1H, CH2CH2CH=), 2.00 (s, 3H, CH3COOCOCH2), 1.81 (m, 1H, CH2CH2CH=), 1.65 (s, 3H, CH3C=), 1.55 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 175.4, 171.2, 170.7, 130.9, 128.4, 81.2, 70.1, 42.5, 38.3, 24.1, 22.3, 22.1, 21.1, 14.0; HRESIMS m/z 309.1307 [M + Na]+ (calcd. for C14H22O6Na, 309.1309).
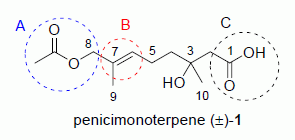
Penicimonoterpene (±)-1, colorless oil (165 mg, 55%); Rf 0.62 (EtOAc–petroleum ether, 2:1); 1H NMR (500 MHz, CDCl3) δ 5.39 (t, J = 6.7 Hz, 1H, CH2CH=), 4.38 (s, 2H, CH2O), 2.51 (d, J = 15.7 Hz, 1H, CH2COOH), 2.45 (d, J = 15.7 Hz, 1H, CH2COOH), 2.08 (m, 2H, CH2CH=), 2.01 (s, 3H, COCH3), 1.60 (s, 3H, CH3C=), 1.55 (m, 2H, CH2CH2CH=), 1.24 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 176.4, 171.5, 130.4, 129.0, 71.3, 70.3, 44.6, 41.0, 26.4, 22.3, 21.0, 13.8; HRESIMS m/z 267.1202 [M + Na]+ (calcd. for C12H20O5Na, 267.1203).
The spectral data (NMR, MS and HRMS) of (±)-
1 were in agreement with those of the reported natural penicimonoterpene [
15].
3.2.8. Synthesis of (±)-3-Hydroxy-3,7-dimethyloct-6-enoic Acid (6)
Compound 6 was obtained from 2a by a method similar to that described for the preparation of 5. Colorless oil 6 (443 mg, 94%); Rf 0.23 (EtOAc–CHCl3, 1:1); 1H NMR (500 MHz, CDCl3) δ 5.10 (t, J = 6.6 Hz, 1H, =CHCH2), 2.59 (d, J = 15.7 Hz, 1H, CH2COOH), 2.51 (d, J = 15.7 Hz, 1H, CH2COOH), 2.07 (m, 2H, =CHCH2), 1.68 (s, 3H, CH3), 1.61 (s, 3H, CH3), 1.58 (m, 2H, =CHCH2CH2), 1.30 (s, 3H, CH3COH); 13C NMR (125 MHz, CDCl3) δ 177.2, 132.3, 123.9, 71.5, 44.9, 41.8, 26.7, 25.8, 22.8, 17.8; HRESIMS m/z 187.1332 [M + H]+ (calcd. for C10H19O3, 187.1329).
3.2.9. Synthesis of (E)-7-Hydroxy-6-methylhept-5-en-2-one (7)
Compound 7 was obtained from 2-methyl-2-hepten-6-one in a manner similar to that described for the preparation of 3a. Yellow oil 7 (1.35 g, 38%); Rf 0.15 (EtOAc–petroleum ether, 1:5); 1H NMR (500 MHz, DMSO-d6) δ 5.26 (t, J = 7.2 Hz, 1H, =CHCH2), 4.64 (s, 1H, CH2OH), 3.75 (s, 2H, CH2OH), 2.46 (t, J = 7.4 Hz, 2H, CH2COCH3), 2.16 (q, J = 7.2 Hz, 2H, =CHCH2), 2.07 (s, 3H, COCH3), 1.54 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δ 208.0, 135.9, 122.1, 66.2, 42.6, 29.6, 21.4, 13.4.
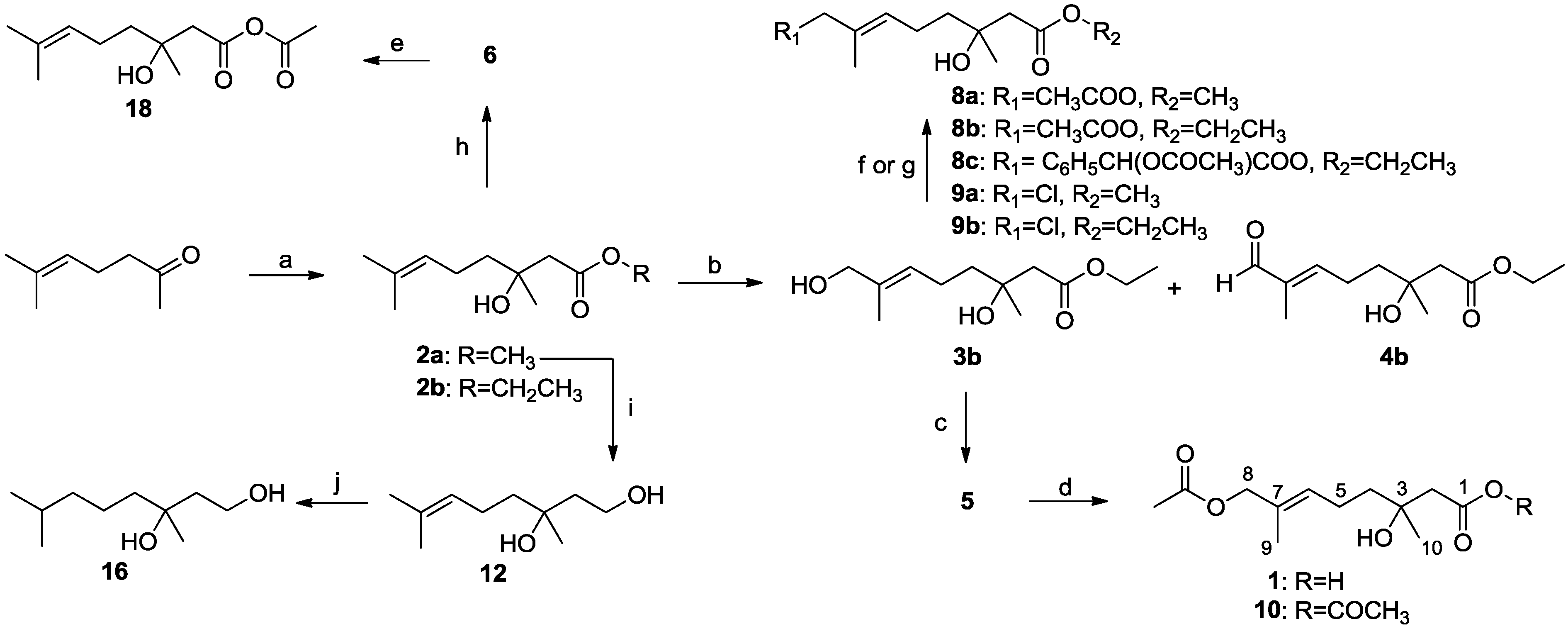
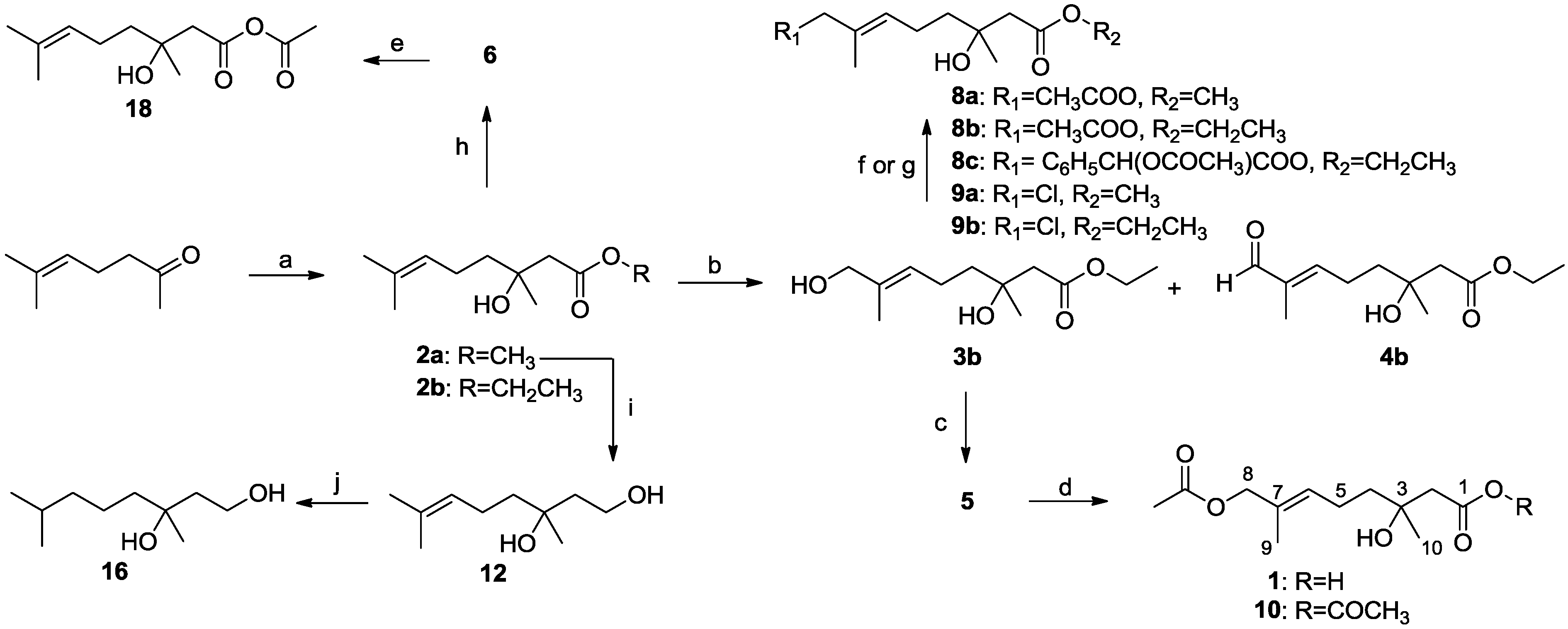
3.2.10. Synthesis of Methyl (E)-(±)-8-Acetoxy-3-hydroxy-3,7-dimethyloct-6-enoate (8a)
Compound 3a (130 mg, 0.6 mmol) was dissolved in CH2Cl2 (5 mL) at room temperature. To this solution were added DMAP (2 mg, 0.02 mmol), Et3N (125 μL, 0.9 mmol) and Ac2O (85 μL, 0.9 mmol). The reaction was stirred for 24 h. After completion of the reaction as detected by TLC, the solvents were evaporated under reduced pressure, and the crude products were purified by column chromatography (EtOAc–petroleum ether, 1:1) to afford 8a (141 mg, 91%) as a colorless oil; Rf 0.72 (EtOAc–petroleum ether, 1:3); 1H NMR (500 MHz, CDCl3) δ 5.42 (t, J = 7.1 Hz, 1H, CH2CH=), 4.41 (s, 2H, CH2O), 3.69 (s, 3H, OCH3), 2.51 (d, J = 15.6 Hz, 1H, CH2COOCH3), 2.44 (d, J = 15.6 Hz, 1H, CH2COOCH3), 2.12 (m, 2H, CH2CH=), 2.04 (s, 3H, COCH3), 1.63 (s, 3H, CH3C=), 1.53 (m, 2H, CH2CH2CH=), 1.23 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 172.7, 170.4, 129.8, 128.6, 70.2, 69.5, 51.1, 44.2, 40.7, 26.1, 21.7, 20.4, 13.3; HRESIMS m/z 281.1359 [M + Na]+ (calcd. for C13H22O5Na, 281.1359).
3.2.11. Synthesis of Ethyl (E)-(±)-8-Acetoxy-3-hydroxy-3,7-dimethyloct-6-enoate (8b)
Compound 8b was obtained from 3b by a method similar to that described for the preparation of 8a. Colorless oil 8b (250 mg, 95%); Rf 0.75 (EtOAc–petroleum ether, 1:3); 1H NMR (500 MHz, CDCl3) δ 5.43 (t, J = 7.0 Hz, 1H, CH2CH=), 4.43 (s, 2H, CH3COOCH2), 4.16 (q, J = 7.1 Hz, 2H, OCH2CH3), 3.24 (br s, 1H, COHCH3), 2.50 (d, J = 15.6 Hz, 1H, CH2COOCH2CH3), 2.43 (d, J = 15.6 Hz, 1H, CH2COOCH2CH3), 2.13 (m, 2H, CH2CH=), 2.05 (s, 3H, CH3COOCH2), 1.64 (s, 3H, CH3C=), 1.54 (m, 2H, CH2CH2CH=), 1.26 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.23 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 173.0, 171.0, 130.5, 129.3, 70.9, 70.2, 60.8, 45.1, 41.4, 26.7, 22.4, 21.1, 14.3, 14.0; HRESIMS m/z 295.1518 [M + Na]+ (calcd. for C14H24O5Na, 295.1516).
3.2.12. Synthesis of Ethyl (E)-(±)-8-((R)-2-Acetoxy-2-phenylacetoxy)-3-hydroxy-3,7-dimethyloct-6-enoate (8c)
DCC (563 mg, 2.7 mmol) was dissolved in CH2Cl2 (20 mL), and R-(−)-O-acyl mandelic acid (530 mg, 2.7 mmol) and DMAP (30 mg, 0.3 mmol) were added at room temperature. After stirring for 16 h at room temperature, the solvent volume was reduced in vacuo, and the remaining mixture was purified by column chromatography (EtOAc–petroleum ether, 1:8) to afford 8c (819 mg, 81%) as a colorless oil; Rf 0.76 (EtOAc–petroleum ether, 1:2); 1H NMR (500 MHz, CDCl3) δ 7.47 (dd, J = 6.4, 2.8 Hz, 2H, ArH), 7.38 (m, 3H, ArH), 5.92 (s, 1H, CHArH), 5.33 (t, J = 6.9 Hz, 1H, CH2CH=), 4.50 (dd, J = 12.2 Hz, 2H, COOCH2CCH3=), 4.18 (q, J = 7.1 Hz, 2H, OCH2CH3), 2.49 (d, J = 15.6 Hz, 1H, CH2COOCH2CH3), 2.42 (d, J = 15.6 Hz, 1H, CH2COOCH2CH3), 2.19 (s, 3H, CH3COO), 2.06 (m, 2H, CH2CH=), 1.50 (s, 3H, CH3C=), 1.48 (m, 2H, CH2CH2CH=), 1.28 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.22 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 173.1, 170.4, 168.8, 134.1, 129.9, 129.7, 129.3, 128.9 (two), 127.8 (two), 74.7, 71.1, 70.8, 60.8, 45.1, 41.3, 26.7, 22.4, 20.8, 14.3, 13.7; HRESIMS m/z 407.2065 [M + H]+ (calcd. for C22H31O7, 407.2064).
3.2.13. Synthesis of Methyl (E)-(±)-8-Chloro-3-hydroxy-3,7-dimethyloct-6-enoate (9a)
To a solution of 3a (76 mg, 0.3 mmol) and PPh3 (138 mg, 0.5 mmol) in 10 mL of dry CH2Cl2 was added NCS (70 mg, 0.5 mmol) at 0 °C under nitrogen. The reaction mixture was stirred at 0 °C for 1 h, then allowed to warm to room temperature and stirred for 2 h. The reaction was monitored by TLC. After completion of the reaction, the solvent volume was reduced in vacuo, and the remaining mixture was purified by column chromatography (EtOAc–petroleum ether, 1:5) to afford 9a (63 mg, 77%) as a colorless oil; Rf 0.63 (EtOAc–petroleum ether, 1:3); 1H NMR (500 MHz, CDCl3) δ 5.49 (t, J = 7.0 Hz, 1H, CH2CH=), 3.97 (s, 2H, CH2Cl), 3.69 (s, 3H, OCH3), 3.20 (br s, 1H, COHCH3), 2.50 (d, J = 15.6 Hz, 1H, CH2COOCH3), 2.43 (d, J = 15.6 Hz, 1H, CH2COOCH3), 2.11 (m, 2H, CH2CH=), 1.71 (s, 3H, CH3C=), 1.55 (m, 2H, CH2CH2CH=), 1.22 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 173.3, 132.1, 130.4, 70.8, 52.3, 51.7, 44.9, 41.1, 26.7, 22.7, 14.1; HRESIMS m/z 257.0892 [M + Na]+ (calcd. for C11H19ClO3Na, 257.0920).
3.2.14. Synthesis of Ethyl (E)-(±)-8-Chloro-3-hydroxy-3,7-dimethyloct-6-enoate (9b)
This compound was obtained from 3b by a method similar to that used for 9a. Colorless oil 9b (157 mg, 69%); Rf 0.82 (EtOAc–petroleum ether, 1:2); 1H NMR (500 MHz, CDCl3) δ 5.51 (t, J = 7.0 Hz, 1H, CH2CH=), 4.17 (q, J = 7.1 Hz, 2H, OCH2CH3), 3.99 (s, 2H, CH2Cl), 3.17 (br s, 1H, COHCH3), 2.50 (d, J = 15.6 Hz, 1H, CH2COOCH2CH3), 2.43 (d, J = 15.6 Hz, 1H, CH2COOCH2CH3), 2.13 (m, 2H, CH2CH=), 1.73 (s, 3H, CH3C=), 1.55 (m, 2H, CH2CH2CH=), 1.27 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.24 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 173.0, 132.1, 130.5, 70.8, 60.8, 52.4, 45.1, 41.2, 26.8, 22.8, 14.3, 14.2; HRESIMS m/z 249.1253 [M + H]+ (calcd. for C12H22ClO3, 249.1252).
3.2.15. Synthesis of (±)-3,7-Dimethyloct-6-ene-1,3-diol (12)
Compound 2a (100 mg, 0.5 mmol) in THF (10 mL) was added dropwise to a stirred slurry of LiAlH4 (57 mg, 1.5 mmol) in THF (10 mL) in an ice bath. After stirring in an ice bath for 2 h, the solution was heated to 65 °C about 10 h. The reaction was monitored by TLC. After completion of the reaction, the reaction mixture was successively treated with water (10 mL), 20% aqueous NaOH (10 mL) and then again with water (10 mL). The resulting white granular suspension was filtered, and the filter cake was washed with CHCl3 (3 × 10 mL). The organic layers were combined, dried (Na2SO4), and then, the solvent was evaporated under reduced pressure and the residue purified by chromatography (EtOAc–petroleum ether, 1:4) to afford 12 (83 mg, 97%) as a colorless oil; Rf 0.34 (EtOAc–petroleum ether, 1:2); 1H NMR (500 MHz, CDCl3) δ 5.09 (t, 1H, CH2CH=), 3.84 (m, 2H, CH2OH), 3.30 (s, 2H, 2 × OH), 2.01 (m, 2H, CH2CH=), 1.76 (m, 1H, CH2CH2OH), 1.66 (s, 3H, CH3),1.63 (m, 1H, CH2CH2OH), 1.59 (s, 3H, CH3), 1.51 (m, 2H, CH2CH2CH=), 1.21 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 131.9, 124.4, 73.9, 59.7, 42.5, 41.7, 26.7, 25.7, 22.8, 17.7; HRESIMS m/z 195.1335 [M + Na]+ (calcd. for C10H20O2Na, 195.1361).
3.2.16. Synthesis of Acetic (±)-3-Hydroxy-3,7-dimethyloct-6-enoic Anhydride (18)
Compound 6 (345 mg, 1.8 mmol) was dissolved in CH2Cl2 (20 mL) at room temperature. To this solution were added DMAP (23 mg, 0.18 mmol), Et3N (517 μL, 3.7 mmol) and, finally, Ac2O (350 μL, 3.7 mmol). The reaction was stirred overnight. After completion of the reaction as detected by TLC, the solvents were evaporated under reduced pressure, and the crude product was purified by column chromatography (EtOAc–petroleum ether, 1:10) to afford 18 as a colorless oil (360 mg, 85%); Rf 0.75 (EtOAc–petroleum ether, 1:3); 1H NMR (500 MHz, CDCl3) δ 5.07 (t, J = 6.5 Hz, 1H, CH2CH=), 3.04 (d, J = 14.3 Hz, 1H, CH2COOCOCH3), 2.89 (d, J = 14.3 Hz, 1H, CH2COOCOCH3), 2.00 (s, 3H, COCH3), 1.99 (m, 2H, CH2CH=), 1.99 (m, 1H, CH2CH2CH=), 1.76 (m, 1H, CH2CH2CH=), 1.67 (s, 3H, CH3C=), 1.59 (s, 3H, CH3C=), 1.54 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3)δ 176.1, 170.8, 132.2, 123.5, 81.4, 42.5, 38.9, 25.8, 24.1, 22.4, 22.3, 17.7; HRESIMS m/z 251.1260 [M + Na]+ (calcd. for C12H20O4Na, 251.1254).
3.2.17. Syntheses of Compounds 11, 13–17, 19 and 20
General procedure: A mixture of the starting material (i.e., a double-bond-containing monoterpene derivative) with 10% Pd/C in EtOAc was stirred at room temperature for an appropriate time (12 to 36 h). After the catalyst was removed by filtration, the filtrate was concentrated in vacuo to give a residue, which was purified by column chromatography on silica gel.
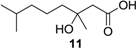
(±)-3-Hydroxy-3,7-dimethyloctanoic acid (11): Colorless oil (47 mg, 92%); Rf 0.52 (EtOAc–petroleum ether, 1:1); 1H NMR (500 MHz, CDCl3) δ 2.57 (d, J = 15.7 Hz, 1H, CH2COOH), 2.49 (d, J = 15.7 Hz, 1H, CH2COOH), 1.55 (m, 1H, CH2CH(CH3)2), 1.51 (m, 2H, CH2COH), 1.34 (m, 2H, CH2CH2CH2), 1.28 (s, 3H, COHCH3), 1.16 (m, 2H, CH2CH2CH2), 0.87 (s, 3H, CH(CH3)2), 0.86 (s, 3H, CH(CH3)2); 13C NMR (125 MHz, CDCl3) δ 177.2, 71.7, 44.8, 42.4, 39.4, 28.0, 26.7, 22.7, 22.6, 21.8; HRESIMS m/z 187.1353 [M − H]− (calcd. for C10H19O3, 187.1340).
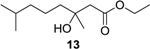
Ethyl (±)-3-hydroxy-3,7-dimethyloctanoate (13): Colorless oil (61 mg, 96%); Rf 0.73 (EtOAc–petroleum ether, 1:5); 1H NMR (500 MHz, CDCl3) δ 4.17 (q, J = 7.1 Hz, 2H, OCH2CH3), 2.50 (d, J = 15.5 Hz, 1H, CH2COOCH2CH3), 2.42 (d, J = 15.5 Hz, 1H, CH2COOCH2CH3), 1.53 (m, 1H, CH(CH3)2), 1.47 (m, 2H, CH2CH2CH2COHCH3), 1.33 (m, 2H, CH2CH2CH2COHCH3), 1.27 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.22 (s, 3H, COHCH3), 1.15 (m, 2H, CH2CH2CH2COHCH3), 0.87 (s, 3H, CH(CH3)2), 0.86 (s, 3H, CH(CH3)2); 13C NMR (125 MHz, CDCl3) δ 173.2, 71.2, 60.7, 45.0, 42.5, 39.5, 28.0, 26.8, 22.7, 22.6, 21.8, 14.3; HRESIMS m/z 239.1624 [M + Na]+ (calcd. for C12H24O3Na, 239.1618).
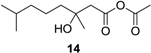
Acetic (±)-3-hydroxy-3,7-dimethyloctanoic anhydride (14): Colorless oil (45 mg, 93%); Rf 0.75 (EtOAc–petroleum ether, 1:1); 1H NMR (500 MHz, CDCl3) δ 3.02 (d, J = 14.4 Hz, 1H, CH2COOCOCH3), 2.88 (d, J = 14.4 Hz, 1H, CH2COOCOCH3), 1.99 (s, 3H, COCH3), 1.93 (m, 1H, CH2CH2CH2COHCH3), 1.73 (m, 1H, CH2CH2CH2COHCH3), 1.53 (m, 1H, CH(CH3)2), 1.52 (s, 3H, COHCH3), 1.31 (m, 2H, CH2CH2CH2COHCH3), 1.17 (m, 2H, CH2CH2CH2COHCH3), 0.87 (s, 3H, CH(CH3)2), 0.86 (s, 3H, CH(CH3)2); 13C NMR (125 MHz, CDCl3) δ 176.2, 170.8, 81.6, 42.6, 39.2, 39.1, 27.9, 24.2, 22.7, 22.4, 21.3; HRESIMS m/z 253.1409 [M + Na]+ (calcd. for C12H22O4Na, 253.1410).
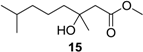
Methyl (±)-3-hydroxy-3,7-dimethyloctanoate (15): Colorless oil (45 mg, 93%); Rf 0.72 (EtOAc–petroleum ether, 1:3); 1H NMR (500 MHz, CDCl3) δ 3.71 (s, 3H, OCH3), 3.41 (br s, 1H, COHCH3), 2.52 (d, J = 15.1 Hz, 1H, CH2COOCH3), 2.45 (d, J = 15.6 Hz, 1H, CH2COOCH3), 1.54 (m, 1H, CH(CH3)2), 1.46 (m, 2H, CH2CH2CH2COHCH3), 1.33 (m, 2H, CH2CH2CH2COHCH3), 1.23 (s, 3H, COHCH3), 1.16 (m, 2H, CH2CH2CH2COHCH3), 0.87 (s, 3H, CH(CH3)2), 0.86 (s, 3H, CH(CH3)2); 13C NMR (125 MHz, CDCl3) δ 173.6, 71.2, 51.7, 44.9, 42.5, 39.5, 28.1, 26.9, 22.7, 22.6, 21.8; HRESIMS m/z 225.1461 [M + Na]+ (calcd. for C11H22O3Na, 225.1461).
(±)-3,7-Dimethyloctane-1,3-diol (16): Colorless oil (26 mg, 89%); Rf 0.52 (EtOAc–petroleum ether, 1:3); 1H NMR (500 MHz, CDCl3) δ 3.88 (m, 2H, CH2OH), 2.49 (s, 2H, 2 × OH), 1.79 (m, 1H, CH2CH2OH), 1.66 (m, 1H, CH2CH2OH), 1.55 (m, 1H, CH(CH3)2), 1.50 (m, 2H, CH2CH2CH2COHCH3), 1.31 (m, 2H, CH2CH2CH2COHCH3), 1.24 (s, 3H, COHCH3), 1.18 (m, 2H, CH2CH2CH2COHCH3), 0.88 (s, 3H, CH(CH3)2), 0.87 (s, 3H, CH(CH3)2); 13C NMR (125 MHz, CDCl3) δ 74.2, 60.1, 43.2, 41.7, 39.6, 28.1, 26.9, 22.7, 22.7, 21.9; HRESIMS m/z 197.1518 [M + Na]+ (calcd. for C10H22O2Na, 197.1512).
(±)-3-Hydroxy-3,7-dimethyl-8-oxooctanoic acid (17): Colorless oil (90 mg, 45%); Rf 0.33 (EtOAc–petroleum ether, 2:1); 1H NMR (500 MHz, CDCl3) δ 9.60 (s, 1H, CHO), 2.55 (d, J = 15.7 Hz, 1H, CH2COOH), 2.48 (d, J = 15.7 Hz, 1H, CH2COOH), 2.35 (m, 1H, CH3CHCHO), 1.70 (m, 1H, CH2CH2CH2COHCH3), 1.54 (m, 2H, CH2CH2CH2COHCH3), 1.41 (m, 2H, CH2CH2CH2COHCH3), 1.38 (m, 1H, CH2CH2CH2COHCH3), 1.26 (s, 3H, COHCH3), 1.09 (d, J = 7.0 Hz, 3H, CH3CHCHO); 13C NMR (125 MHz, CDCl3) δ 205.5, 177.0, 71.3, 46.3, 44.8, 41.9, 30.8, 26.7, 21.4, 13.5; HRESIMS m/z 225.1101 [M + Na]+ (calcd. for C10H18O4Na, 225.1097).
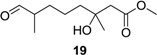
Methyl (±)-3-hydroxy-3,7-dimethyl-8-oxooctanoate (19): Colorless oil (94 mg, 27%); Rf 0.53 (EtOAc–petroleum ether, 1:1); 1H NMR (500 MHz, CDCl3) δ 9.61 (s, 1H, CHO), 3.71 (s, 3H, OCH3), 2.50 (d, J = 15.6 Hz, 1H, CH2COOCH3), 2.43 (d, J = 15.6 Hz, 1H, CH2COOCH3), 2.34 (m, 1H, CHCH3), 1.70 (m, 1H, CHCH2CH2CH2), 1.50 (m, 2H, CHCH2CH2CH2), 1.39 (m, 1H, CHCH2CH2CH2), 1.38 (m, 2H, CHCH2CH2CH2), 1.22 (s, 3H, COHCH3 ), 1.09 (d, J = 7.0 Hz, 3H, CHCH3); 13C NMR (125 MHz, CDCl3) δ 205.1, 173.5, 70.9, 51.8, 46.4, 44.8, 42.0, 31.0, 26.8, 21.4, 13.5; HRESIMS m/z 239.1260 [M + Na]+ (calcd. for C11H20O4Na, 239.1254).
Ethyl (±)-3-hydroxy-3,7-dimethyl-8-oxooctanoate (20): Colorless oil (15 mg, 25%); Rf 0.52 (EtOAc–petroleum ether, 1:2); 1H NMR (500 MHz, CDCl3) δ 9.56 (s, 1H, CHO), 4.12 (q, J = 7.1 Hz, 2H, OCH2CH3), 2.44 (d, J = 15.5 Hz, 1H, CH2COOCH2CH3), 2.37 (d, J = 15.5 Hz, 1H, CH2COOCH2CH3), 2.30 (m, 1H, CHCH3), 1.64 (m, 1H, CHCH2CH2CH2), 1.46 (m, 2H, CHCH2CH2CH2), 1.35 (m, 1H, CHCH2CH2CH2), 1.34 (m, 2H, CHCH2CH2CH2), 1.22 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.17 (s, 3H, COHCH3), 1.05 (d, J = 7.0 Hz, 3H, CHCH3); 13C NMR (125 MHz, CDCl3) δ 205.0, 172.9, 70.9, 60.7, 46.3, 45.0, 41.9, 30.9, 26.7, 21.4, 14.2, 13.4; HRESIMS m/z 231.1595 [M + H]+ (calcd. for C12H23O4, 231.1591).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}