Two New Lyngbyatoxin Derivatives from the Cyanobacterium, Moorea producens

 ,
, 
Abstract
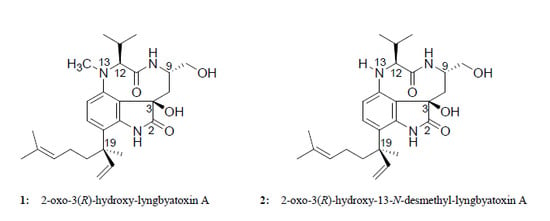
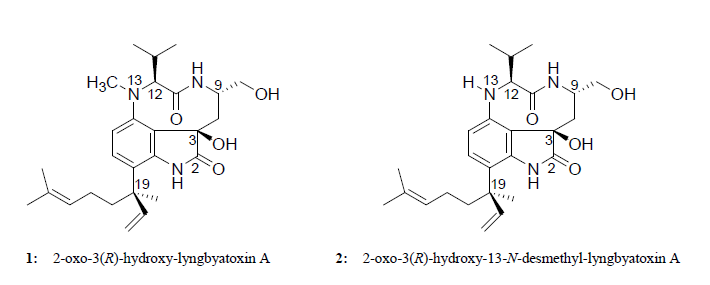
:
1. Introduction
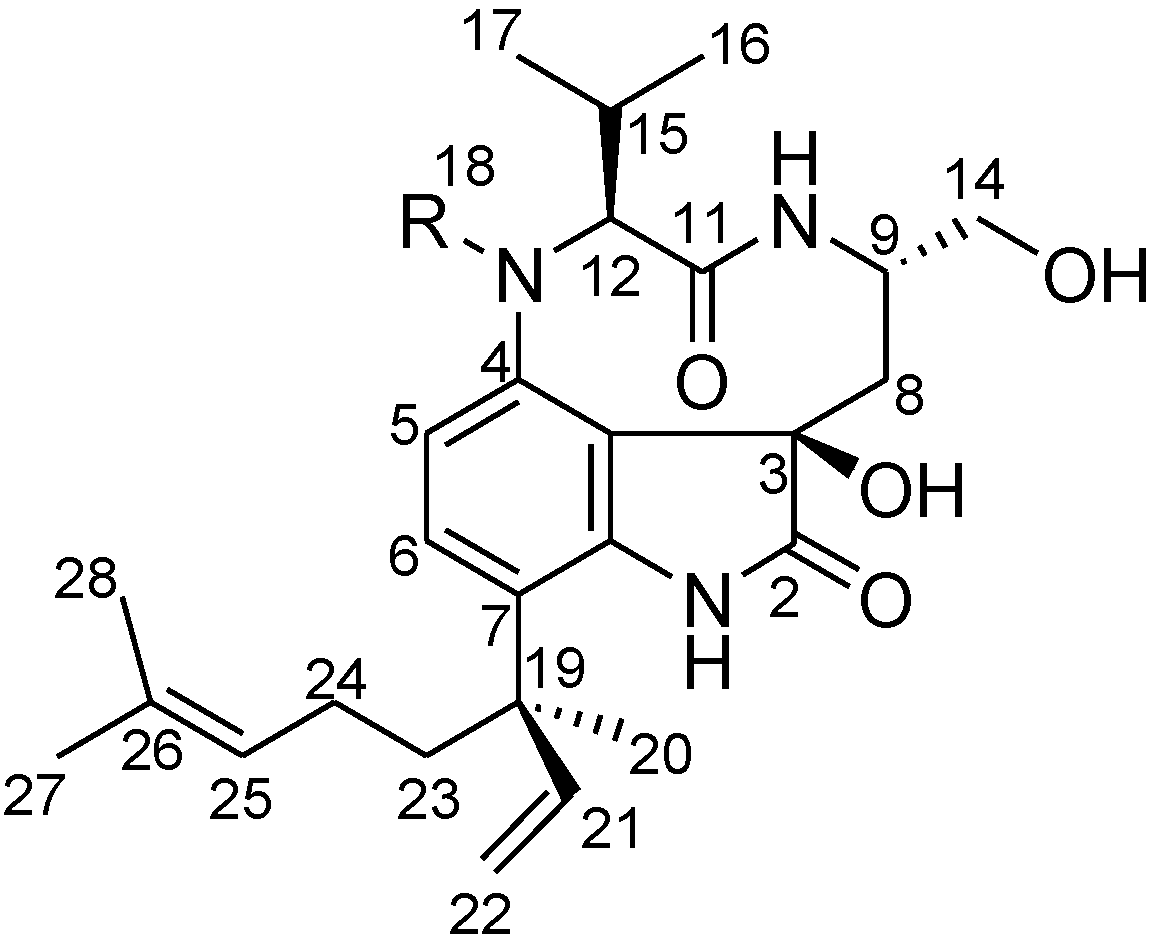
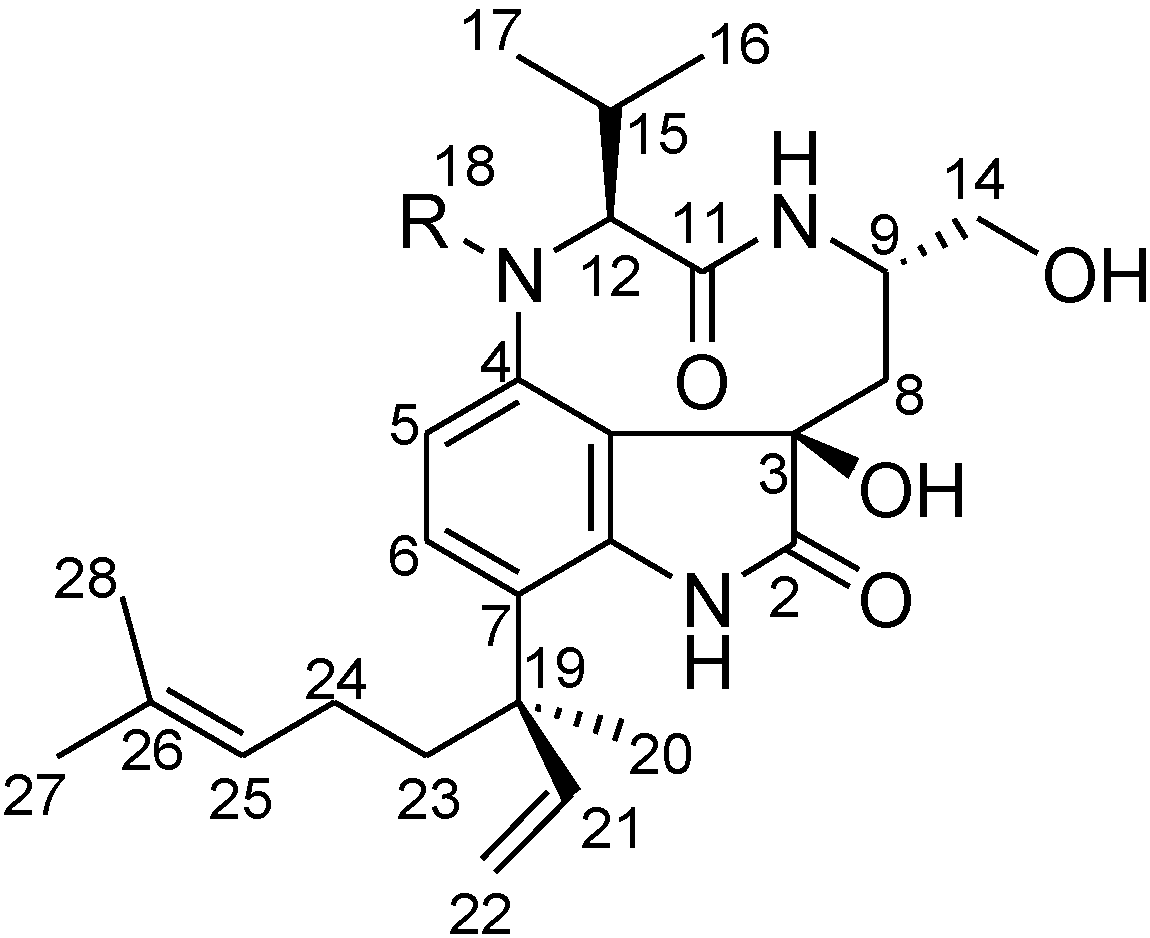
2. Results and Discussion
2.1. Isolation of 1 and 2
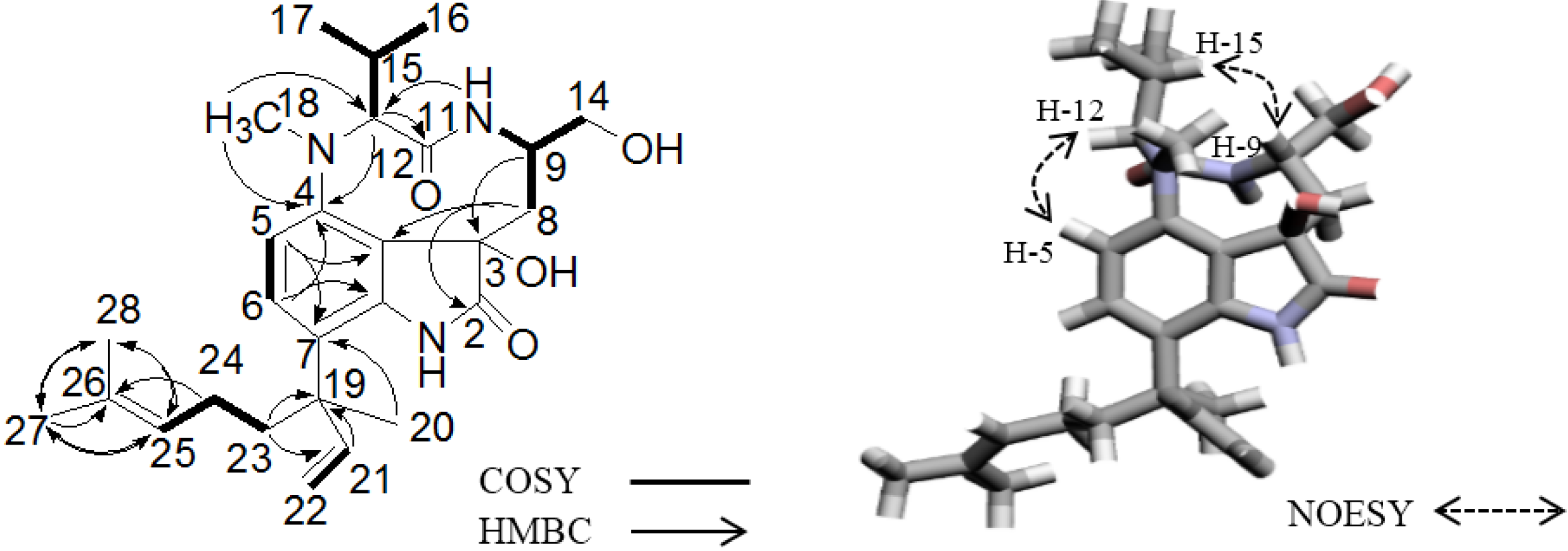
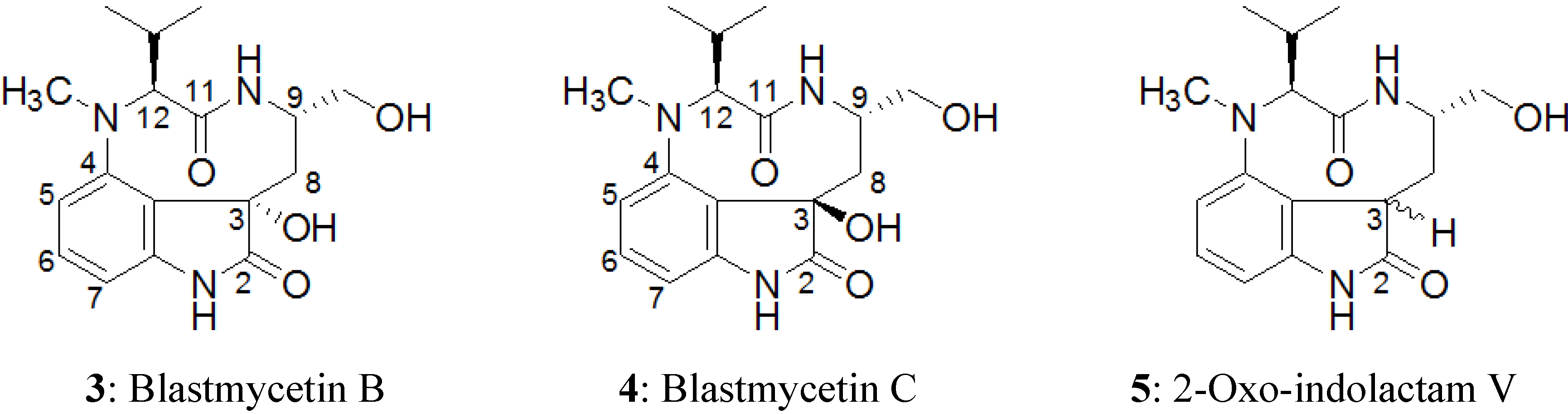
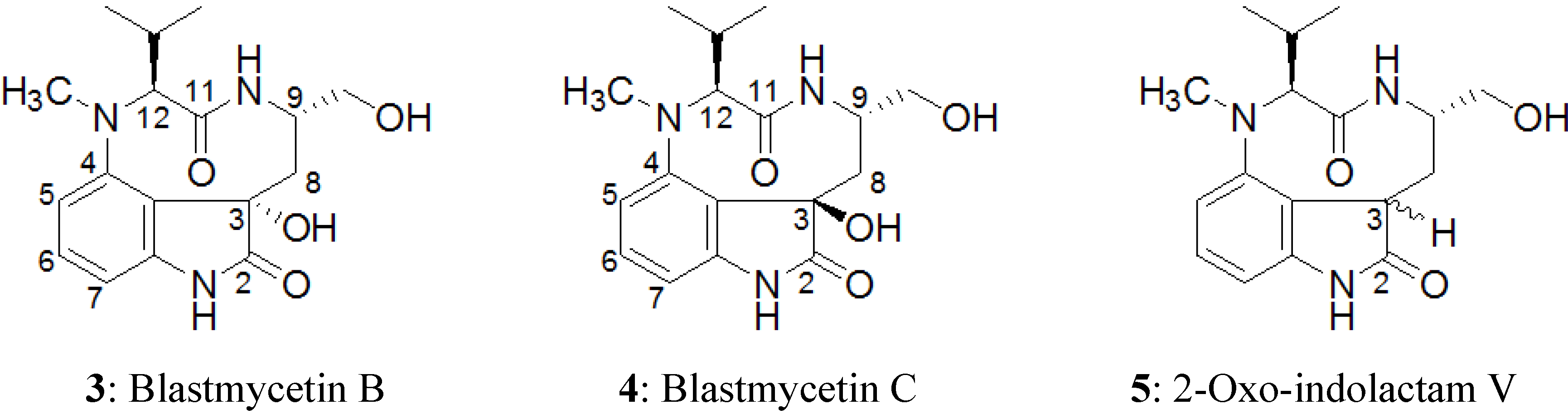
2.2. Elucidation of the Planar Structures of 1 and 2

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 | 2 | ||
|---|---|---|---|---|
| 13C | 1H (mult, J in Hz) | 13C | 1H (mult, J in Hz) | |
| 2 | 182.1 | 181.5 | ||
| 3 | 76.0 | 75.9 | ||
| 3a | 128.6 | 122.5 | ||
| 4 | 151.0 | 145.6 | ||
| 5 | 120.0 | 7.15, 1H (d, 8.6) | 116.9 | 6.69, 1H (d, 8.6) |
| 6 | 130.2 | 7.29, 1H (d, 8.6) | 129.8 | 7.15, 1H (d, 8.6) |
| 7 | 128.3 | 123.7 | ||
| 7a | 139.7 | 138.8 | ||
| 8 | 42.4 | 2.24, 1H (d, 15.1) | 42.9 | 2.15, 1H (m) |
| 1.46, 1H (dd, 9.0, 15.1) | 1.50, 1H (dd, 9.4, 14.7) | |||
| 9 | 51.1 | 5.63, 1H (m) | 51.6 | 4.49, 1H (m) |
| 11 | 175.1 | 176.4 | ||
| 12 | 78.1 | 3.81, 1H (d, 5.5) | 74.3 | 3.78, 1H (d, 10.5) |
| 14 | 67.5 | 3.60, 1H (dd, 4.7, 10.9) | 67.1 | 3.54, 1H (dd, 5.0, 11.1) |
| 3.54, 1H (dd, 5.6, 10.9) | 3.49, 1H (dd, 7.2, 11.1) | |||
| 15 | 30.0 | 2.43, 1H (m) | 32.6 | 2.15, 1H (m) |
| 16 | 22.7 | 1.22, 3H (d, 6.9) | 21.3 | 1.29, 3H (d, 6.5) |
| 17 | 19.4 | 1.20, 3H (d, 6.9) | 20.6 | 1.13, 3H (d, 6.5) |
| 18 | 42.3 | 2.67, 3H (s) | ||
| 19 | 44.6 | 44.4 | ||
| 20 | 25.4 | 1.41, 3H (s) | 25.6 | 1.38, 3H (s) |
| 21 | 146.8 | 6.09, 1H (dd, 10.7, 17.6) | 147.2 | 6.08, 1H (dd, 10.7, 17.6) |
| 22 | 114.0 | 5.22, 1H (d, 10.7) | 113.6 | 5.19, 1H (d, 10.7) |
| 5.06, 1H (d, 17.7) | 5.04, 1H (d, 17.7) | |||
| 23 | 40.2 | 1.86, 1H (m) | 40.5 | 1.83, 1H (m) |
| 1.76, 1H (m) | 1.72, 1H (m) | |||
| 24 | 24.2 | 1.97, 1H (m) | 24.3 | 1.94, 1H (m) |
| 1.76, 1H (m) | 1.74, 1H (m) | |||
| 25 | 125.3 | 5.10, 1H (t, 5.8) | 125.4 | 5.11, 1H (m) |
| 26 | 132.5 | 132.4 | ||
| 27 | 25.8 | 1.68, 3H (s) | 25.8 | 1.67, 3H (s) |
| 28 | 17.7 | 1.55, 3H (s) | 17.7 | 1.55, 3H (s) |
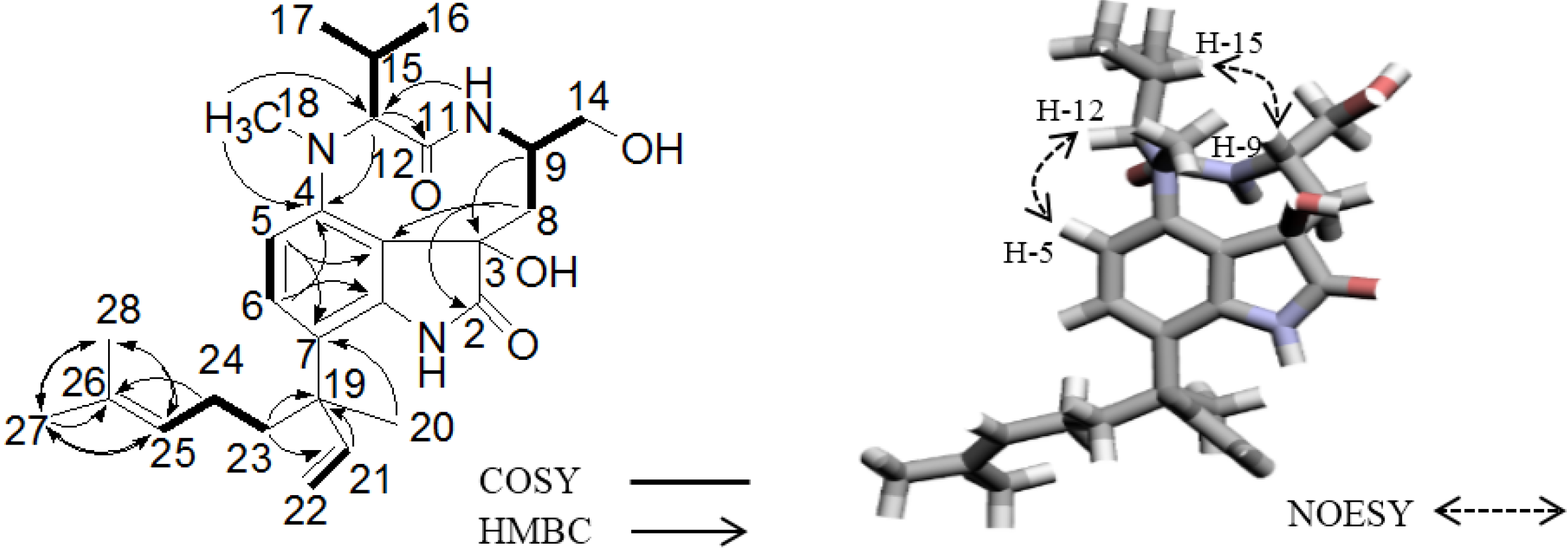
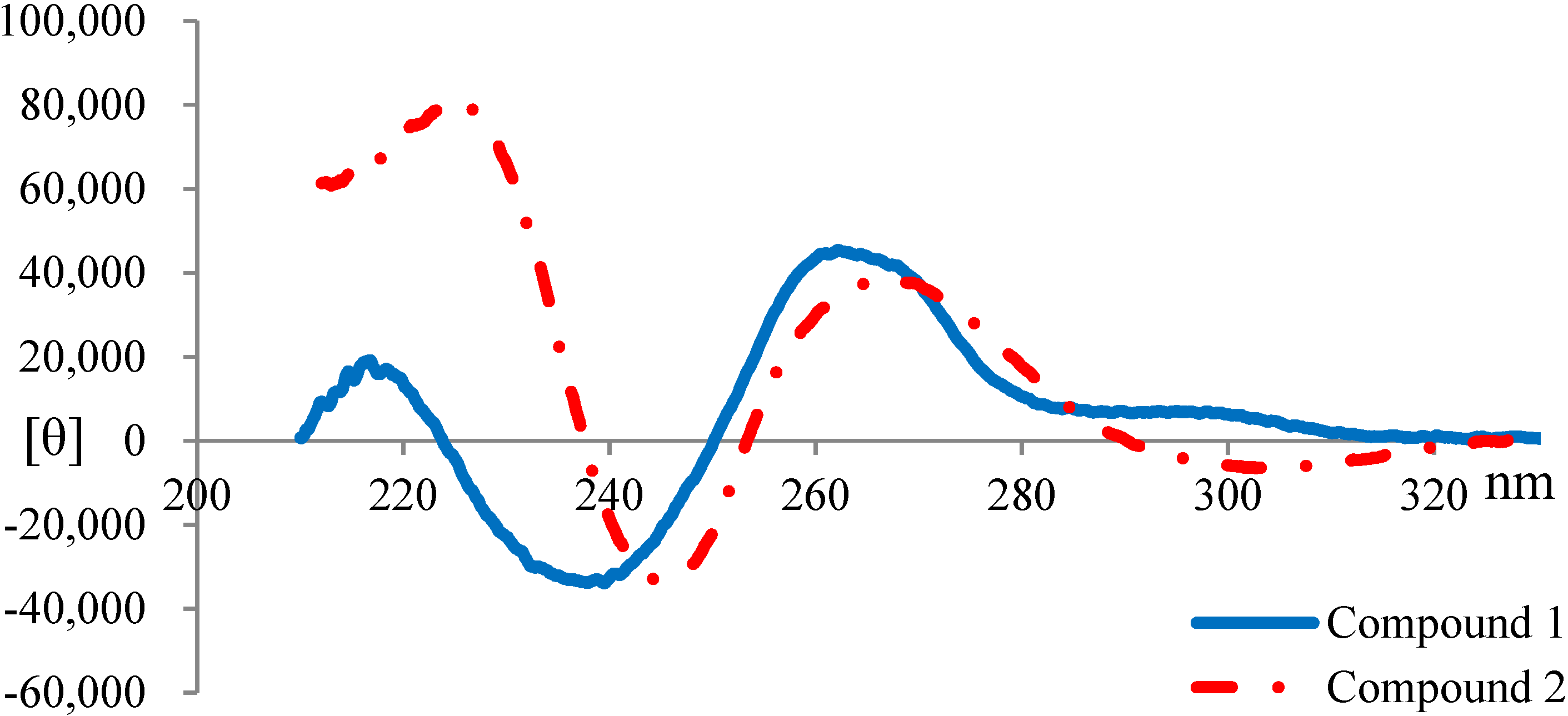
2.3. Elucidation of Absolute Stereochemistry of 1 and 2
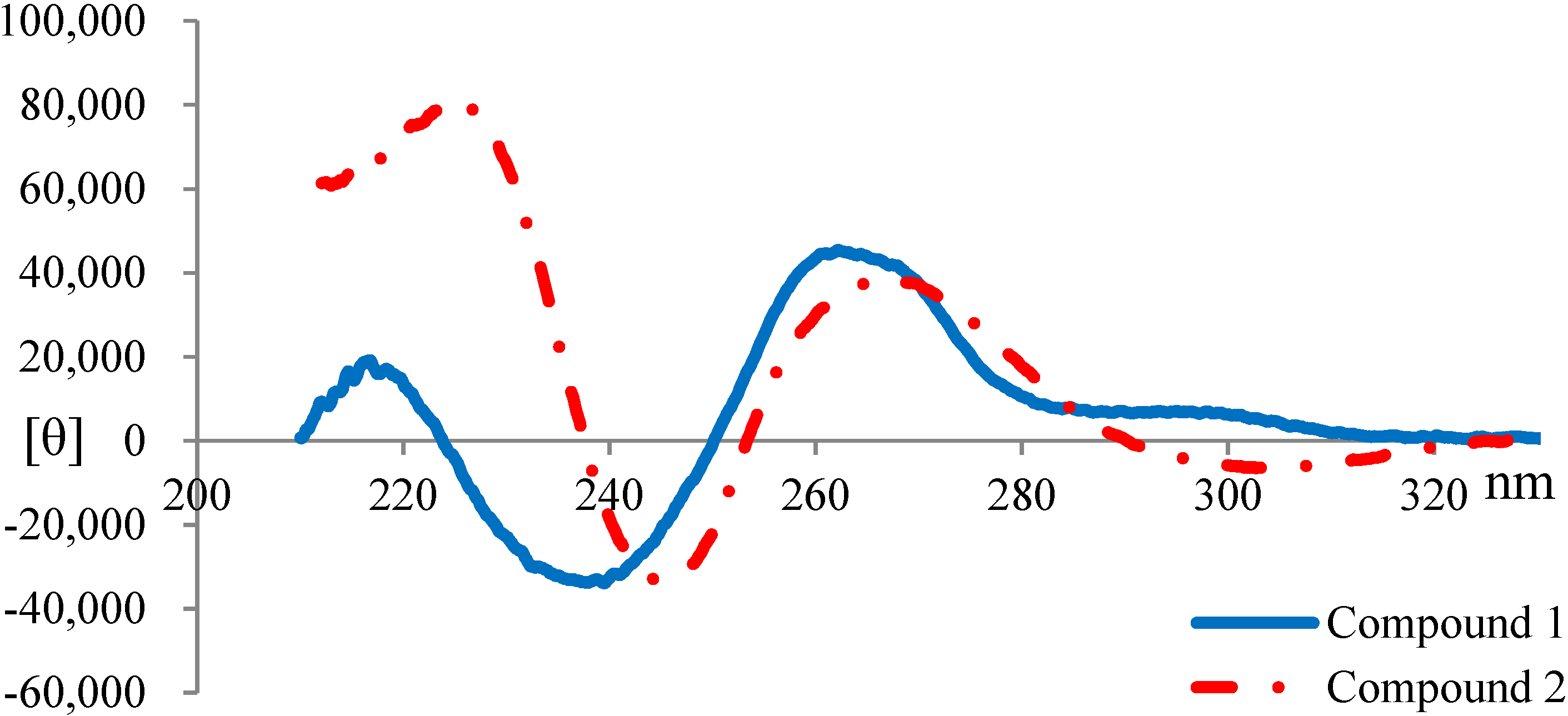
2.4. Biological Activity Tests
3. Experimental Section
3.1. Instrumentation and General Methods
3.2. Sample Collection
3.3. Extraction and Purification
3.4. HR-ESI-MS and LC-MS/MS Analyses
3.5. Nuclear Magnetic Resonance Experiments
3.6. Circular Dichroism (CD) Spectroscopy
3.7. Cytotoxicity Assay
3.8. Crustacean Lethality Test
3.9. Binding Assay of PKC Ligands Using the PKCδ-C1B Peptide
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hoffman, L. Marine cyanobacteria in tropical regions: Diversity and ecology. Eur. J. Phycol. 1999, 34, 371–379. [Google Scholar] [CrossRef]
- Carmichael, W.W. Cyanobacteria secondary metabolites-the cyanotoxins. J. Appl. Microbiol. 1992, 72, 445–459. [Google Scholar]
- Engene, N.; Rottacker, E.C.; Kaštovský, J.; Byrum, T.; Choi, H.; Ellisman, M.H.; Komárek, J.; Gerwick, W.H. Moorea producens gen. nov., sp. nov. and Moorea bouillonii comb. nov., tropical marine cyanobacteria rich in bioactive secondary metabolites. Int. J. Syst. Evol. Microbiol. 2012, 62, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Nunnery, J.K.; Mevers, E.; Gerwick, W.H. Biologically active secondary metabolites from marine cyanobacteria. Curr. Opin. Biotechnol. 2010, 21, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Tiwari, S.P.; Rai, A.K.; Mohapatra, T.M. Cyanobacteria: An emerging source for drug discovery. J. Antibiot. 2011, 64, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.T. Marine cyanobacteria: A prolific source of bioactive natural products as drug leads. In Marine Microbiology: Bioactive Compounds and Biotechnological Applications, 1st ed.; Kim, S.K., Ed.; Wiley-VCH: Weinheim, Germany, 2013; Volume 5, pp. 59–81. [Google Scholar]
- Mynderse, J.S.; Moore, R.E.; Kashiwagi, M.; Norton, T.R. Antileukemia activity in the osillatoriaceae: Isolation of debromoaplysiatoxin from Lyngbya. Science 1977, 196, 538–540. [Google Scholar] [PubMed]
- Cardellina, J.H.; Marner, F.J.; Moore, R.E. Seaweed dermatitis: Structure of lyngbyatoxin A. Science 1979, 204, 193–195. [Google Scholar] [CrossRef]
- Moore, R.E.; Blackman, A.J.; Cheuk, C.E.; Mynderse, J.S.; Matsumoto, G.K.; Clardy, J.; Woodard, R.W.; Craig, J.C. Absolute stereochemistries of the aplysiatoxins and oscillatoxin A. J. Org. Chem. 1984, 49, 2484–2489. [Google Scholar] [CrossRef]
- Banner, A.H. A dermatitis-producing alga in Hawaii. Preliminary report. Hawaii Med. J. 1959, 19, 35–36. [Google Scholar] [PubMed]
- Grauer, F.H. Dermatitis escharotica caused by a marine alga. Hawaii Med. J. 1959, 19, 32–34. [Google Scholar] [PubMed]
- Osborne, N.J.; Webb, P.M.; Shaw, G.R. The toxins of Lyngbya majuscule and their human and ecological health effects. Environ. Int. 2001, 27, 381–392. [Google Scholar] [CrossRef]
- Champetier, R.G.; Ranaivoson, G.; Ravaonindrina, N.; Rakotonjanabelo, A.L.; Rasolofonirina, N.; Roux, J.F.; Yasumoto, T. Un problème de santé publique réémergent à madagascar: Les intoxications collectives par consommation d’animaux marins. Aspects épidémiologiques, cliniques et toxicologiques des épisodes notifiés de janvier 1993 à janvier 1998. Arch. Inst. Pasteur Madag. 1998, 64, 71–76. [Google Scholar]
- Ito, E.; Satake, M.; Yasumoto, T. Pathological effects of lyngbyatoxin A upon mice. Toxicon 2002, 40, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Yasumoto, T. Fish poisoning due to toxins of microalgal origins in the Pacific. Toxicon 1998, 36, 1515–1518. [Google Scholar] [CrossRef] [PubMed]
- Hanne, M.; Matsubayashi, H.; Vogt, R.; Wakida, C.; Hau, S.; Nagai, H.; Hokama, Y.; Solorzano, L. Outbreak of gastrointestinal illness associated with consumption of seaweed-Hawaii, 1994. Morb. Mortal. Wkly. Rep. 1995, 44, 724–727. [Google Scholar]
- Nagai, H.; Yasumoto, T.; Hokama, Y. Aplysiatoxin and debromoaplysiatoxin as the causative agents of a red alga Gracilaria coronopifolia poisoning in Hawaii. Toxicon 1996, 34, 753–761. [Google Scholar] [PubMed]
- Marshall, K.L.E.; Vogt, R.L. Illness associated with eating seaweed, Hawaii, 1994. West. J. Med. 1998, 169, 293–295. [Google Scholar] [PubMed]
- Nagai, H.; Yasumoto, T.; Hokama, Y. Manauealides, some of the causative agents of a red alga Gracilaria coronopifolia poisoning in Hawaii. J. Nat. Prod. 1997, 60, 925–928. [Google Scholar] [CrossRef] [PubMed]
- Nagai, H.; Kan, Y.; Fujita, T.; Sakamoto, B.; Hokama, Y. Manuauealide C and anhydrodebromoaplysiatoxin, toxic constituents of the Hawaiian red alga, Gracilaria coronopifolia. Biosci. Biotech. Biochem. 1998, 62, 1011–1013. [Google Scholar] [CrossRef]
- Ito, E.; Nagai, H. Morphological observations of diarrhea in mice caused by aplysiatoxin, the causative agent of the red alga Gracilaria coronopifolia poisoning in Hawaii. Toxicon 1998, 36, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Fujiki, H.; Mori, M.; Nakayasu, M.; Terada, M.; Sugimura, T.; Moore, R.E. Indole alkaloids: Dihydroteleocidin B, teleocidin, and lyngbyatoxin A as members of a new class of tumor promoters. Proc. Natl. Acad. Sci. USA 1981, 78, 3872–3876. [Google Scholar] [CrossRef] [PubMed]
- Fujiki, H.; Tanaka, Y.; Miyake, R.; Kikkawa, U.; Nishizuka, Y.; Sugimura, T. Activation of calcium-activated, phospholipid-dependent protein kinase (protein kinase C) by new classes of tumor promoters: Teleocidin and debromoaplysiatoxin. Biochem. Biophys. Res. Commun. 1984, 120, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Fujiki, H.; Sugimura, T. New classes of tumor promoters: Teleocidin, aplysiatoxin, and palytoxin. Adv. Cancer Res. 1987, 49, 223–264. [Google Scholar] [PubMed]
- Hagiwara, N.; Irie, K.; Funaki, A.; Hayashi, H.; Arai, M.; Koshimizu, K. Structure and tumor-promoting activity of new teleocidin-related metabolites (blastmycetins) from Streptoverticillium blastmyceticum. Agric. Biol. Chem. 1988, 52, 641–648. [Google Scholar] [CrossRef]
- Izumikawa, M.; Khan, S.T.; Komaki, H.; Takagi, M.; Shin-ya, K. JBIR-31, a new teleocidin analog, produced by salt-requiring Streptomyces sp. NBRC 105896 isolated from a marine sponge. J. Antibiot. 2010, 63, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Irie, K.; Hagiwara, N.; Kurome, T.; Hayashi, H.; Arai, M.; Koshimizu, K. New teleochidin-related mtabolites from Streptoverticillium blastmyceticum producing tumor-promoting indole alkaloids. Agric. Biol. Chem. 1987, 51, 285–287. [Google Scholar] [CrossRef]
- Aimi, N.; Odaka, H.; Sakai, S.I.; Fujiki, H.; Suganuma, M.; Moore, R.E.; Patterson, G.M.L. Lyngbyatoxins B and C, two new irritants from Lyngbya majuscula. J. Nat. Prod. 1990, 53, 1593–1596. [Google Scholar] [CrossRef] [PubMed]
- Muratake, H.; Okabe, K.; Natsume, M. Synthesis of teleocidins A, B and their congeners. Part 2. Synthesis of lyngbyatoxin A (teleocidin A-1), teleocidin A-2, pendolmycin, and (R,E)-and (S,E)-7-(3,7,11-trimethyl-1,6,10-dodecatrien-3-yl)-(−)-indolactams V. Tetrahedron 1991, 47, 8545–8558. [Google Scholar]
- Jiang, W.; Zhou, W.; Uchida, H.; Kikumori, M.; Irie, K.; Watanabe, R.; Suzuki, T.; Sakamoto, B.; Kamio, M.; Nagai, H. A new lyngbyatoxin from the Hawaiian cyanobacterium Moorea producens. Mar. Drugs 2014, 12, 2748–2759. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.E.; Patterson, G.M.; Entzeroth, M.; Morimoto, H.; Suganuma, M.; Hakii, H.; Fujiki, H.; Sugimura, T. Binding studies of [3H] lyngbyatoxin A and [3H] debromoaplysiatoxin to the phorbol ester receptor in a mouse epidermal particulate fraction. Carcinogenesis 1986, 7, 641–644. [Google Scholar] [PubMed]
- Ono, Y.; Fujii, T.; Igarashi, K.; Kuno, T.; Tanaka, C.; Kikkawa, U.; Nishizuka, Y. Phorbol ester binding to protein kinase C requires a cysteine-rich zinc-finger-like sequence. Proc. Natl. Acad. Sci. USA 1989, 86, 4868–4871. [Google Scholar] [CrossRef] [PubMed]
- Szállási, Z.; Denning, M.F.; Smith, C.B.; Dlugosz, A.A.; Yuspa, S.H.; Pettit, G.R.; Blumberg, P.M. Bryostatin 1 protects protein kinase C-delta from down-regulation in mouse keratinocytes in parallel with its inhibition of phorbol ester-induced differentiation. Mol. Pharmacol. 1994, 46, 840–850. [Google Scholar] [PubMed]
- Irie, K.; Nakahara, A.; Nakagawa, Y.; Ohigashi, H.; Shindo, M.; Fukuda, H.; Konishi, H.; Kikkawa, U.; Kashiwagi, K.; Saito, N. Establishment of a binding assay for protein kinase C isozymes using synthetic C1 peptides and development of new medicinal leads with protein kinase C isozyme and C1 domain selectivity. Pharmacol. Ther. 2002, 93, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, T.; Lindsay, D.J.; Kitamura, M.; Konishi, S.; Nishikawa, J.; Nishida, S.; Kamio, M.; Nagai, H. Evaluation of the bioactivities of water-soluble extracts from twelve deep-sea jellyfish species. Fish Sci. 2013, 79, 487–494. [Google Scholar] [CrossRef]
- Irie, K.; Oie, K.; Nakahara, A.; Yanai, Y.; Ohigashi, H.; Wender, P.A.; Fukuda, H.; Konishi, H.; Kikkawa, U. Molecular basis for protein kinase C isozyme-selective binding: The synthesis, folding, and phorbol ester binding of the cysteine-rich domains of all protein kinase C isozymes. J. Am. Chem. Soc. 1998, 120, 9159–9167. [Google Scholar] [CrossRef]
- Shindo, M.; Irie, K.; Nakahara, A.; Ohigashi, H.; Konishi, H.; Kikkawa, U.; Fukuda, H.; Wender, P.A. Toward the identification of selective modulators of protein kinase C (PKC) isozymes: Establishment of a binding assay for PKC isozymes using synthetic C1 peptide receptors and identification of the critical residues involved in the phorbolester binding. Bioorg. Med. Chem. 2001, 9, 2073–2081. [Google Scholar] [CrossRef] [PubMed]
- Irie, K.; Isaka, T.; Iwata, Y.; Yanai, Y.; Nakamura, Y.; Koizumi, F.; Ohigashi, H.; Wender, P.A.; Satomi, Y.; Nishino, H. Synthesis and biological activities of new conformationally restricted analogues of (−)-indolactam-V: Elucidation of the biologically active conformation of the tumor-promoting teleocidins. J. Am. Chem. Soc. 1996, 118, 10733–10743. [Google Scholar] [CrossRef]
- Sharkey, N.A.; Blumberg, P.M. Highly lipophilic phorbolesters as inhibitors of specific [3H]phorbol 12,13-dibutyrate binding. Cancer Res. 1985, 45, 19–24. [Google Scholar] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, W.; Tan, S.; Hanaki, Y.; Irie, K.; Uchida, H.; Watanabe, R.; Suzuki, T.; Sakamoto, B.; Kamio, M.; Nagai, H. Two New Lyngbyatoxin Derivatives from the Cyanobacterium, Moorea producens. Mar. Drugs 2014, 12, 5788-5800. https://doi.org/10.3390/md12125788
Jiang W, Tan S, Hanaki Y, Irie K, Uchida H, Watanabe R, Suzuki T, Sakamoto B, Kamio M, Nagai H. Two New Lyngbyatoxin Derivatives from the Cyanobacterium, Moorea producens. Marine Drugs. 2014; 12(12):5788-5800. https://doi.org/10.3390/md12125788
Chicago/Turabian StyleJiang, Weina, Satoshi Tan, Yusuke Hanaki, Kazuhiro Irie, Hajime Uchida, Ryuichi Watanabe, Toshiyuki Suzuki, Bryan Sakamoto, Michiya Kamio, and Hiroshi Nagai. 2014. "Two New Lyngbyatoxin Derivatives from the Cyanobacterium, Moorea producens" Marine Drugs 12, no. 12: 5788-5800. https://doi.org/10.3390/md12125788