Evaluation of the Antioxidant Activity of Cell Extracts from Microalgae


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Microorganisms

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Origin | Culture Medium | Antioxidant Power | |
|---|---|---|---|---|
| Intracellular | Extracellular | |||
| Unicellular | ||||
| Chlorococcus giganteus | ACOI 768 | BG11 | 2.95 ± 0.92 | nd |
| OHM | 1.01 ± 0.19 | nd | ||
| Cyanothece sp. | ATCC 51142 | BG11 | 9.78 ± 0.73 | nd |
| Gloeobacter violaceus | PCC 7421 | BG11 | 38.10 ± 6.03 a,b,c | 0.07 ± 0.01 b |
| Gloeothece sp. | ATCC 27152 | BG110 | 5.12 ± 1.09 | 0.03 ± 0.00 |
| Synechocystis sp. | PCC 6803 | BG11 | 2.97 ± 0.46 | nd |
| Synechocystis salina | ACOI 48 | BG11 | 7.98 ± 1.01 | nd |
| Filamentous | ||||
| Arthrospira platensis | ATCC 29408 | ASW-BG11 | 3.23 ± 0.59 | nd |
| Leptolyngbya sp. | PCC 73110 | BG11 | 2.68 ± 0.43 | nd |
| Leptolyngbya sp. | PCC 7410 | BG11 | 2.32 ± 0.31 | nd |
| M2-7 (Limnothrix sp.) * | Aquaculture biofilters | BG11 | 17.00 ± 8.47 a,b,c,d | 0.01 ± 0.00 a,c |
| Lyngbya majuscula | CCAP 1446/4 | BG11 | 3.46 ± 1.04 | nd |
| Filamentous heterocystous | ||||
| Anabaena variabilis | ATCC 29413 | BG110 | 2.78 ± 0.48 | nd |
| BG11 | 3.29 ± 0.89 | nd | ||
| J52 (Anabaena planctonica) * | Maranhão lagoon | BG11 | 35.90 ± 6.99 a,b,c | 0.01 ± 0.00 b |
| Nodularia harveyana | ACOI 729 | BG11 | 1.84 ± 0.57 | 0.01 ± 0.00 |
| Nostoc carneum | ACOI 650 | BG110 | 6.78 ± 1.33 | nd |
| BG11 | 5.00 ± 0.60 | nd | ||
| Nostoc muscorum | CCAP 1453/12 | BG110 | 2.02 ± 0.46 | nd |
| BG11 | 3.06 ± 0.32 | nd | ||
| Nostoc punctiforme | PCC 73102 | BG110 | 2.22 ± 0.36 | 0.07 ± 0.01 |
| BG11 | 9.42 ± 1.34 | nd | ||
| Scytonema obscurum | ACOI 573 | BG11 | 14.80 ± 1.76 c,d | nd |
| Class/Species | Origin | Culture Medium | Antioxidant Power | |
|---|---|---|---|---|
| Intracellular | Extracellular | |||
| Bacillariophyceae | ||||
| Phaeodactilum tricornutum | SERI-S/PHAEO-1 (TFX-1) | ASW | 8.8 ± 1.84 | nd |
| Chlorophyceae | ||||
| Chlorella vulgaris | CBSC | OHM | 10.2 ± 1.93 | nd |
| C. vulgaris | ACOI 879 | OHM | 7.48 ± 1.24 | nd |
| Haematococcus pluvialis | CCAP 34/7 | OHM | 49.80 ± 10.10 g | nd |
| Scenedesmus maximus | ACOI 318 | OHM | 6.42 ± 1.15 | nd |
| M2-1 (Scenedesmus obliquus) 1 | Aquaculture biofilters | OHM | 149.00 ± 46.60 e | 0.01 ± 0.00 b |
| M2-6 (S. obliquus) 1 | OHM | 25.10 ± 6.26 f | nd | |
| M3-9 (S. obliquus) 1 | OHM | 15.60 ± 2.83 f | nd | |
| M4-3 (S. obliquus) 1 | OHM | 2.66 ± 0.47 | 1.86 ± 0.32 | |
| M4-5 (S. obliquus) 1 | OHM | 63.10 ± 4.39 a,b | nd | |
| M2-5 (S. obliquus) 1 | OHM | 21.40 ± 3.91 c,d | nd | |
| M2-18 (S. obliquus) 1 | OHM | 10.5 ± 2.54 | nd | |
| Scenedesmus obliquus B | Estarreja wetlands | OHM | 4.35 ± 0.46 | nd |
| S. obliquus F | OHM | 6.04 ± 0.66 | nd | |
| S. obliquus G | OHM | 3.34 ± 0.50 | nd | |
| Desmodesmus pleiomorphus A | OHM | 1.54 ± 0.05 | nd | |
| D. pleiomorphus E | OHM | 0.76 ± 0.25 | nd | |
| D. pleiomorphus H | OHM | 2.92 ± 0.86 | nd | |
| D. pleiomorphus M | OHM | 1.81 ± 0.52 | nd | |
| Scenedesmus quadricauda | CBSC | OHM | 8.27 ± 1.91 | nd |
| Eustigmatophyceae | ||||
| Nannochloropsis sp. | SERI: NANNO-1 (GBSTICHO) | ASW 2 | 7.79 ± 2.11 | nd |
| Prymnesiophyceae | ||||
| Pavlova lutheri | IPIMAR: SMBA 60 | ASW 2 | 8.13 ± 1.83 | nd |
| Rhodophyceae | ||||
| Porphyridium aerugineum | ACOI 1332 | BG11 | 4.76 ± 1.31 | nd |
2.2. Antioxidant Capacity as ABTS Scavenging
2.3. Antioxidant Capacity as Deoxyribose Protection
| a | |
|---|---|
| Cyanobacterium Strains | Antioxidant Power |
| Gloeobacter violaceus | 113.68 ± 2.81 |
| M2-7 (Limnothrix sp.) | 9.32 ± 0.63 |
| J52 (Anabaena planctonica) | 51.45 ± 5.04 |
| Scytonema obscurum | 16.48 ± 0.65 |
| b | |
| Microalga Species | Antioxidant Power |
| Haematococcus pluvialis | 20.34 ± 0.79 |
| M2-1 (S. obliquus) | 477.91 ± 161.95 |
| M2-6 (S. obliquus) | 138.46 ± 30.86 |
| M2-5 (S. obliquus) | 122.92 ± 9.57 |
| M3-9 (S. obliquus) | 11.75 ± 2.60 |
| M4-5 (S. obliquus) | 121.85 ± 46.97 |
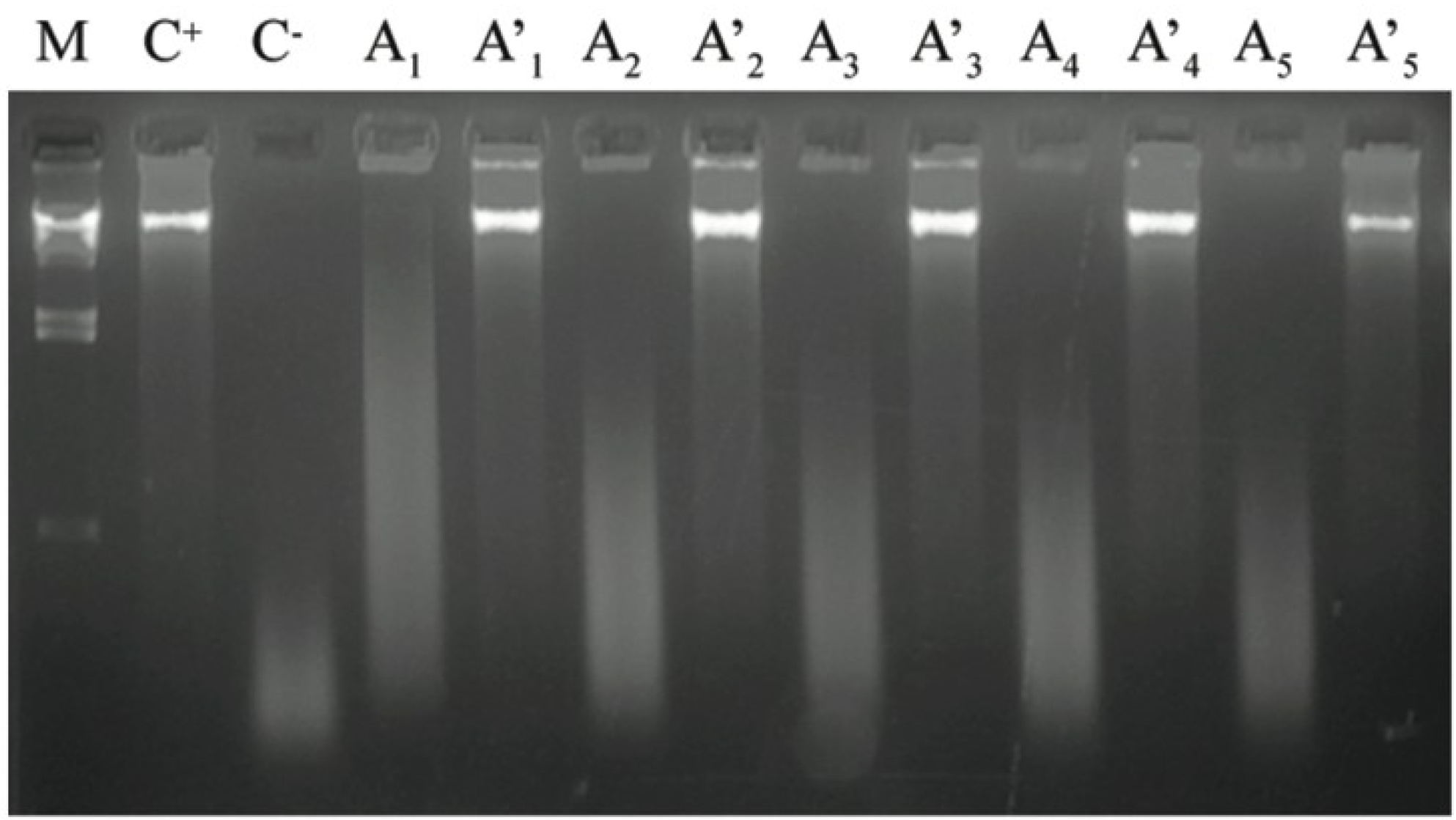
2.4. Antioxidant Capacity as DNA Protection
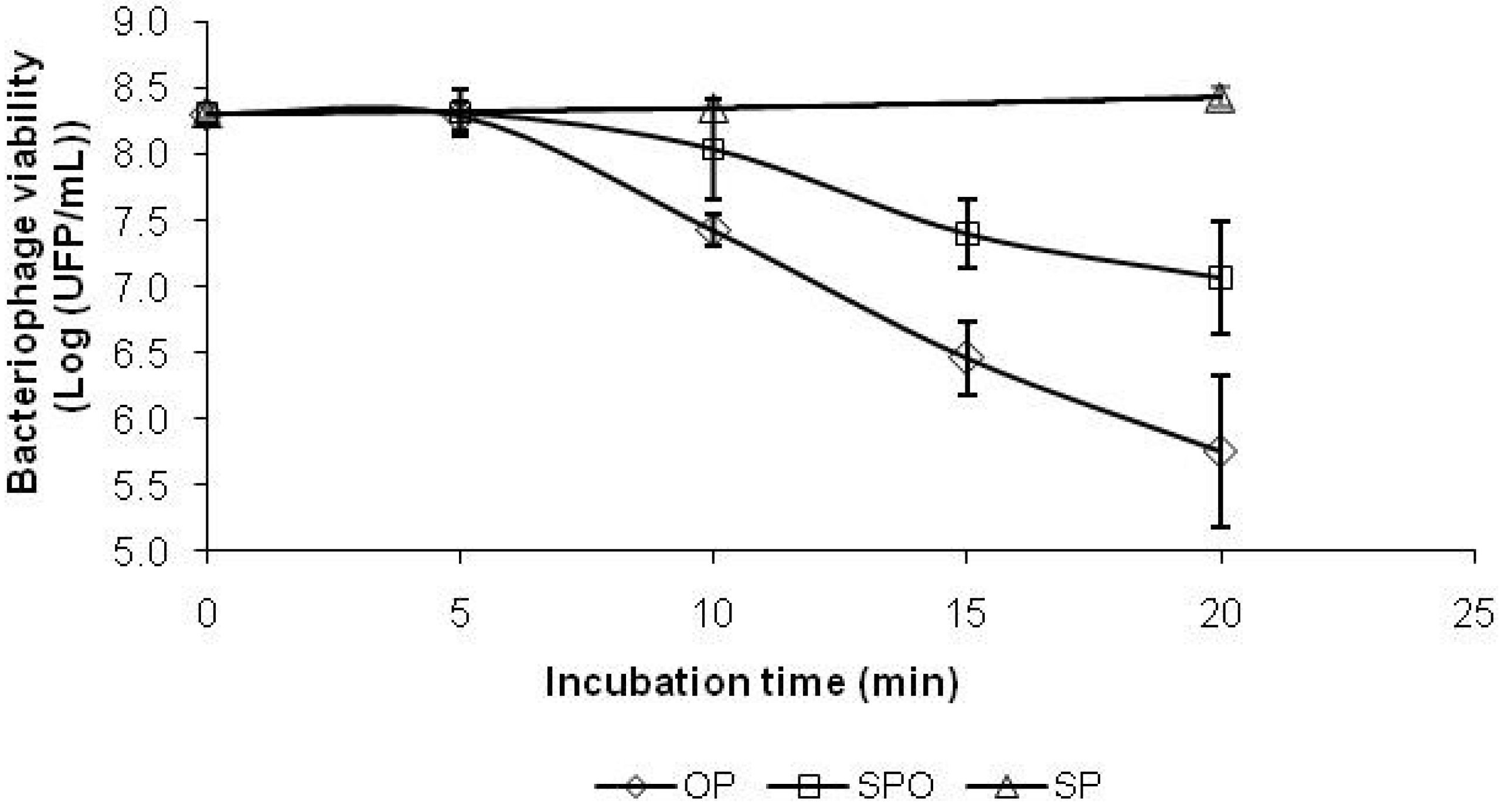
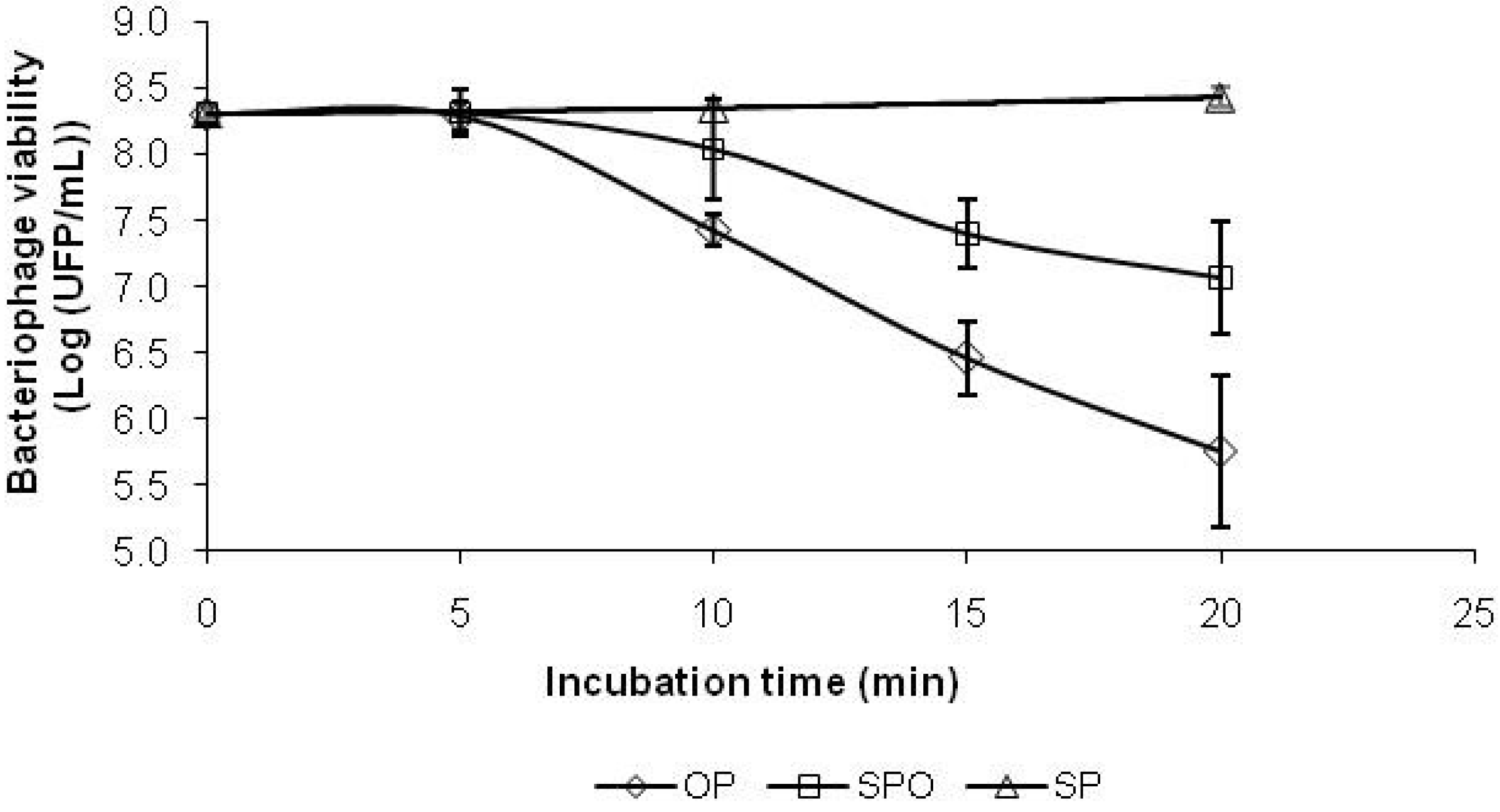
2.5. Antioxidant Capacity as Bacteriophage Protection
2.6. Antioxidant Compounds
| Compound | Elution Time (min) | Antioxidant Concentration (mgequivalent lutein/gmicroalga) |
|---|---|---|
| Neoxanthin | 5.9 | 0.56 ± 0.02 |
| Violaxanthin | 6.5 | 0.14 ± 0.01 |
| Lutein | 14.6 | 2.69 ± 0.09 |
| Zeaxanthin | 15.2 | nd |
| β-Apo-8′-carotenal | 20.1 | (internal standard) |
| β-Carotene | 34.5 | 0.40 ± 0.03 |
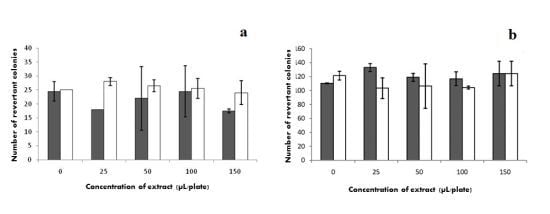
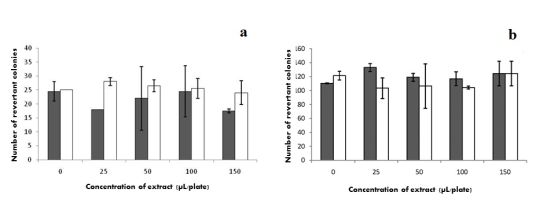
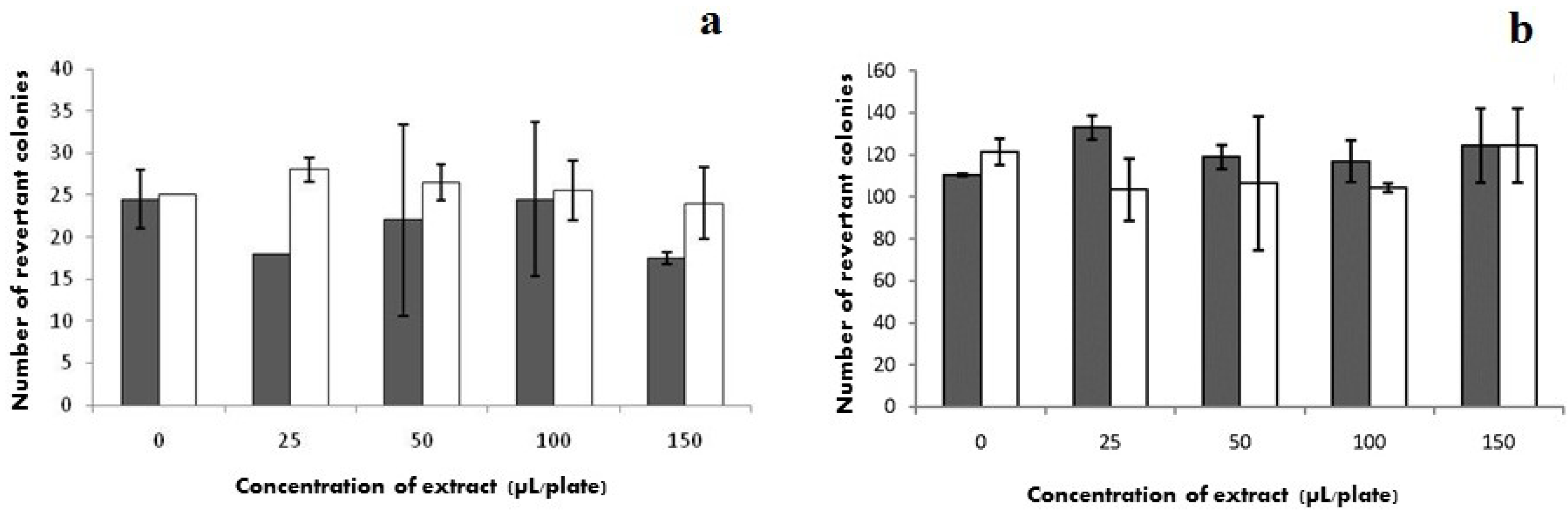
2.7. Mutagenicity
 ) without S9 and (□) with S9.
) without S9 and (□) with S9.
) without S9 and (□) with S9.
) without S9 and (□) with S9.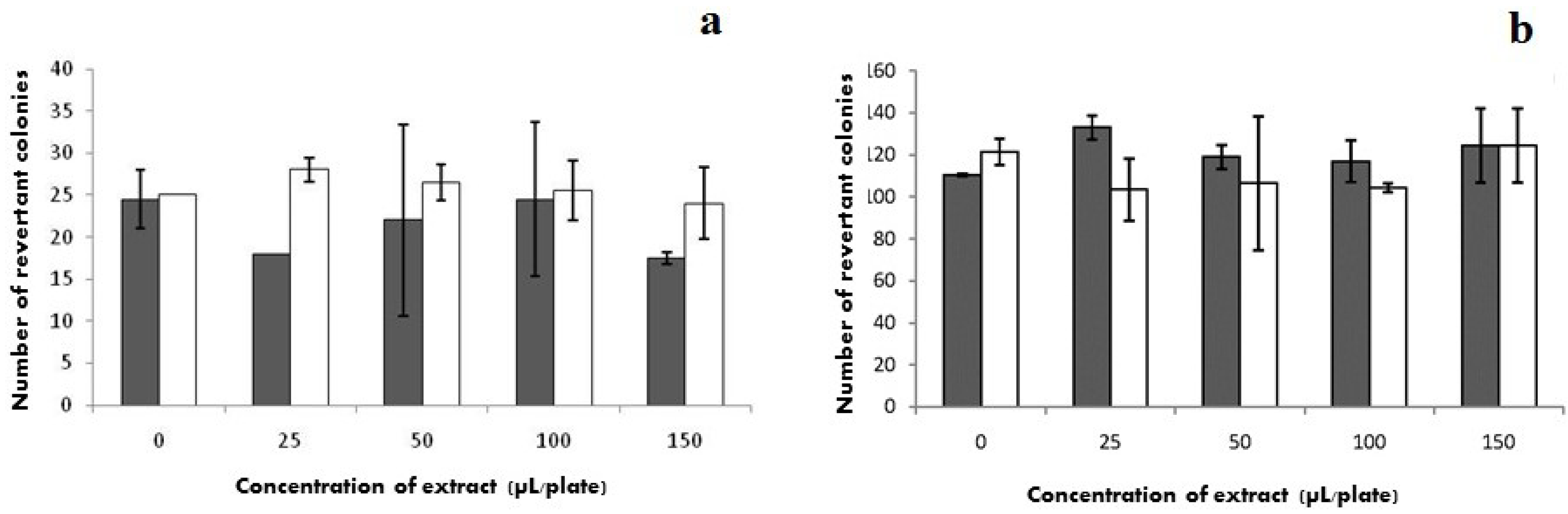
3. Experimental Section
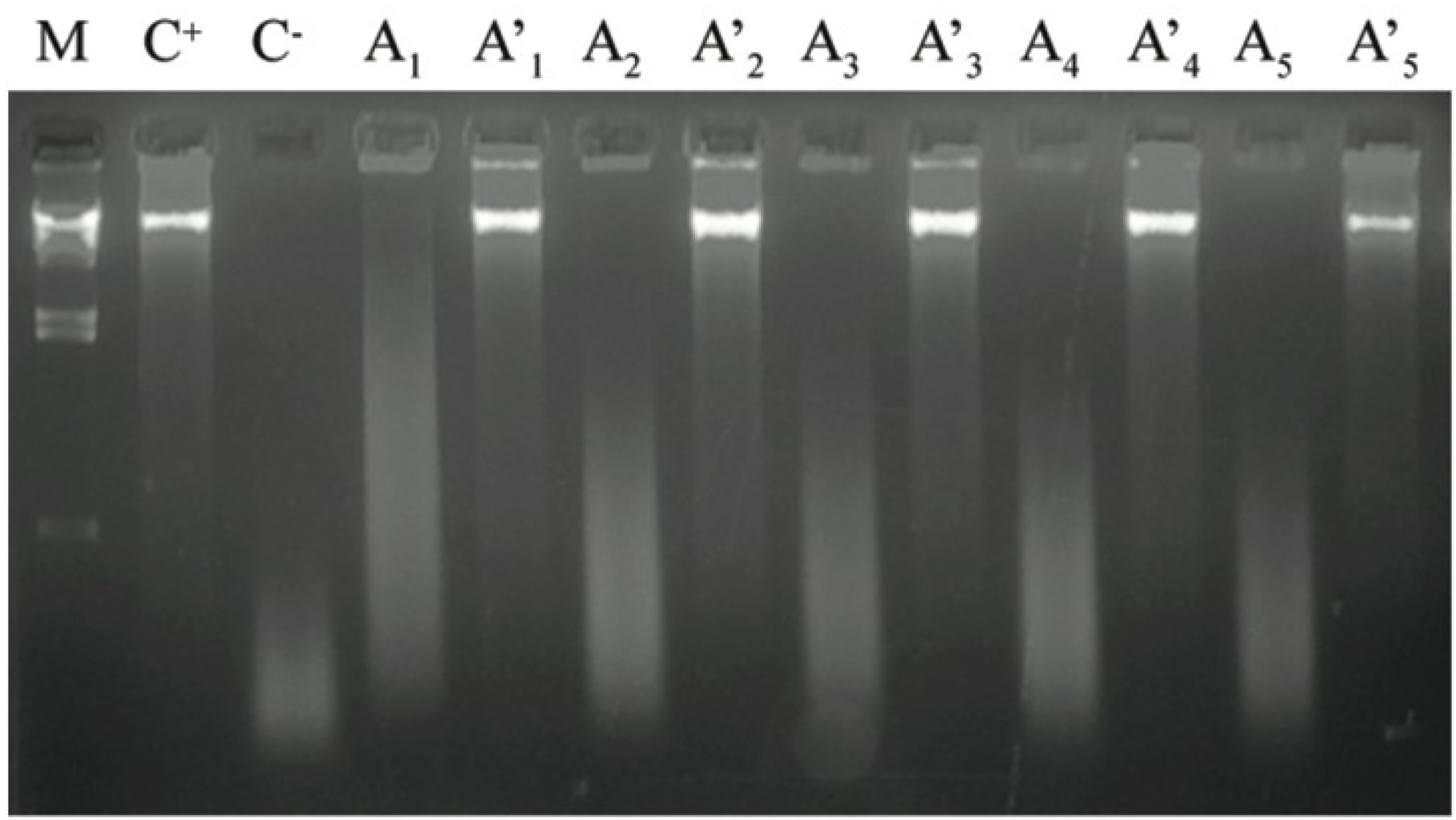
3.1. DNA Extraction and Agarose Gel Electrophoresis
3.2. DNA Amplification, Purification, Cloning and Sequencing
| Isolate | GenBank Accession Number | Closest Cultured Relative (% similarity, accession number) a |
|---|---|---|
| J52 | EU073188 | Anabaena planctonica strain 71 (99%, AJ293108) |
| M2-7 | EF634458 | Limnothrix sp. strain CENA110 (99%, EF088338) |
| M2-1 | EU073189 | Scenedesmus obliquus strain UTEX 393 (98%, DQ396875) |
| M2-6 | EU073191 | S. obliquus strain UTEX 393 (92%, DQ396875) |
| M3-9 | EU073193 | S. obliquus strain UTEX 393 (92%, DQ396875) |
| M4-3 | EU073194 | S. obliquus strain UTEX 393 (92%, DQ396875) |
| M4-5 | EU073195 | S. obliquus strain UTEX 393 (92%, DQ396875) |
| M2-5 | EU073190 | S. obliquus strain UTEX 393 (99%, DQ396875) |
| M2-18 | EU073192 | S. obliquus strain UTEX 393 (99%, DQ396875) |
3.3. Growth Conditions and Microorganisms
3.4. Extracellular and Intracellular Extraction
3.5. Chlorophyll a Content
3.6. ABTS Scavenging Assay
3.7. Deoxyribose Protection Assay
3.8. DNA Protection Assay
3.9. Bacteriophage Protection Assay
3.10. Antioxidant Identification
3.11. Mutagenicity Assessment
3.12. Statistical Analyses
4. Conclusions
Acknowledgments
References
- Abe, K.; Hattor, H.; Hiran, M. Accumulation and antioxidant activity of secondary carotenoids in the aerial microalga Coelastrella striolata var. multistriata. Food Chem. 2005, 100, 656–661. [Google Scholar]
- Guerin, M.; Huntley, M.E.; Olaizola, M. Haematococcus astaxanthin: Applications for human health and nutrition. Trends Biotechnol. 2003, 21, 210–216. [Google Scholar] [CrossRef]
- Wang, H.M.; Pan, J.L.; Chen, C.Y.; Chiu, C.C.; Yang, M.H.; Chang, H.W.; Chang, J.S. Identification of anti-lung cancer extract from Chlorella vulgaris C-C by antioxidant property using supercritical carbon dioxide extraction. Process Biochem. 2010, 45, 1865–1872. [Google Scholar] [CrossRef]
- Guedes, A.C.; Amaro, H.M.; Malcata, F.X. Microalgae as sources of high added-value compounds—A brief review of recent work. Biotechnol. Prog. 2011, 27, 597–613. [Google Scholar] [CrossRef]
- Tandeau-de-Marsac, N.; Houmard, J. Adaptation of cyanobacteria to environmental stimuli: New steps towards molecular mechanisms. FEMS Microbiol. Rev. 1993, 104, 119–190. [Google Scholar] [CrossRef]
- León, R.; Martín, M.; Vigara, J.; Vilchez, C.; Vega, J. Microalgae-mediated photoproduction of β-carotene in aqueous organic two-phase systems. Biomol. Eng. 2003, 20, 177–182. [Google Scholar] [CrossRef]
- Srivastava, A.; Bhargava, P.; Rai, L. Salinity and copper-induced oxidative damage and changes in the antioxidative defense systems of Anabaena doliolum. World J. Microbiol. Biotechnol. 2005, 21, 1291–1298. [Google Scholar] [CrossRef]
- Honga, Y.; Hua, H.Y.; Xiea, X.; Li, F.M. Responses of enzymatic antioxidants and non-enzymatic antioxidants in the cyanobacterium Microcystis aeruginosa to the allelochemical ethyl 2-methyl acetoacetate (EMA) isolated from reed (Phragmites communis). J. Plant Physiol. 2008, 165, 1264–1273. [Google Scholar] [CrossRef]
- Chin, H.C.; Tien, L.J.; Jou, L.F.; Pi, C.F.; Jye, Y.D. Determination of carotenoids in Dunaliella salina cultivated in Taiwan and antioxidant capacity of the algal carotenoid extract. Food Chem. 2008, 109, 439–446. [Google Scholar] [CrossRef]
- Halliwell, B.; Aeschbach, R.; Löliger, J.; Aruoma, I. The characterization of antioxidants. Food Chem. Toxic. 1995, 33, 601–617. [Google Scholar] [CrossRef]
- Perez, M.D.R.; Magariño, S.P.; Sanjosé, M.L.G.; Valls, V.; Cordoñer, P.; Muñiz, P. Inhibition of induced DNA oxidative damage by beers: Correlation with the content of polyphenols and melanoidins. J. Agric. Food Chem. 2005, 53, 3637–3642. [Google Scholar]
- Fernandes, J.C.; Eaton, P.; Nascimento, H.; Gião, M.S.; Ramos, O.S.; Belo, L.; Silva, A.S.; Pintado, M.E.; Malcata, F.X. Antioxidant activity of chitooligosaccharides upon two biological systems: Erythrocytes and bacteriophages. Carbohydr. Polym. 2010, 79, 1101–1106. [Google Scholar] [CrossRef]
- Gião, M.S.; Borges, A.B.; Guedes, C.J.; Hogg, T.A.; Pintado, M.E.; Malcata, F.X. Determination of antioxidant capacity using the biological system bacteriophage P22/bacterium Salmonella Typhimurium. J. Agric. Food Chem. 2009, 57, 22–25. [Google Scholar]
- Liu, D.; Shi, J.; Ibarra, A.C.; Kakuda, Y.; Xue, S.J. The scavenging capacity and synergistic effects of lycopene, vitamin E, vitamin C, and β-carotene mixtures on the DPPH free radical. Lebensm. Wiss. Technol. 2006, 41, 1344–1349. [Google Scholar]
- Ames, B.N.; Durston, W.E.; Yamasaki, D.E.; Frank, D.L. Carcinogens are mutagens: A simple test system containing liver homogenates for activation and bacteria for detection. Proc. Natl. Acad. USA 1973, 70, 2281–2285. [Google Scholar] [CrossRef]
- Maron, D.M.; Ames, B.N. Revised methods for the Salmonella mutagenicity test. Mutat. Res. 1983, 113, 173–215. [Google Scholar] [CrossRef]
- Tamagnini, P.; Troshina, O.; Oxelfelt, F.; Salema, R.; Lindblad, P. Hydrogenases in Nostoc sp. strain PCC 73102, a strain lacking a bidirectional enzyme. Appl. Environ. Microbiol. 1997, 63, 1801–1807. [Google Scholar]
- Burja, A.M.; Tamagnini, P.; Bustard, M.T.; Wright, P.C. Identification of the green alga, Chlorella vulgaris (SDC1), using cyanobacteria-derived 16S rDNA primers: Targeting the chloroplast. FEMS Microbiol. Lett. 2001, 202, 195–203. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, New York, NY, USA, 2001. [Google Scholar]
- Bishop, N.I.; Senger, H. Preparation and Properties of Synchronous Cultures of Scenedesmus. In Methods in Enzymology; Pietro, A.S., Ed.; Academic Press: New York, NY, USA, 1971; Volume 23A, pp. 53–66. [Google Scholar]
- Stanier, R.Y.; Kunisawa, R.; Mandel, M.; Cohen-Bazire, G. Purification and properties of unicellular blue-green algae (order Chlorococcales). Bacteriol. Rev. 1971, 35, 171–205. [Google Scholar]
- Borowitzka, M.A. Algal Growth Media and Sources of Algal Cultures. In Micro-Algal Biotechnology; Borowitzka, M.A., Borowitzka, J.L., Eds.; Cambridge University Press: Cambridge, UK, 1988; pp. 456–465. [Google Scholar]
- Allen, M.M. Simple conditions for growth of unicellular blue-green algae on plates. J. Phycol. 1968, 4, 1–4. [Google Scholar] [CrossRef]
- Guedes, A.C.; Amaro, H.M.; Pereira, R.D.; Malcata, F.X. Effects of temperature and pH on growth and antioxidant content of the microalga Scenedesmus obliquus. Biotechnol. Prog. 2011, 27, 1218–1224. [Google Scholar] [CrossRef]
- Meeks, J.C.; Castenholz, R.W. Growth and photosynthesis in an extreme thermophile, Synechococcus lividus (Cyanophyta). Arch. Microbiol. 1971, 78, 25–41. [Google Scholar]
- Re, R.; Pellegrini, N.; Proteggente, N.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Gião, M.S.; González-Sanjosé, M.L.; Pérez, M.D.R.; Pereira, C.I.; Pintado, M.E.; Malcata, F.X. Infusions of Portuguese medicinal plants: Dependence of final antioxidant capacity and phenolic content on extraction features. J. Sci. Food Agric. 2007, 87, 2638–2647. [Google Scholar] [CrossRef]
- Guimarães, C.M.; Martinez, S.S.; Pintado, A.I.; Gião, M.S.; Pintado, M.E.; Bento, L.S. Antioxidant activity—Including protective effect against DNA oxidative damage, of sugar molasses. J. Food Sci. 2007, 72, 39–43. [Google Scholar]
- Halliwell, B.; Gutteridge, J.M.C.; Aruoma, O.I. The deoxyribose method: A simple “test tube” assay for determination of rate constants for reactions of hydroxyl radicals. Anal. Biochem. 1987, 165, 215–219. [Google Scholar] [CrossRef]
- Muñiz, P.; Saéz, P.; Iradi, A.; Viña, J.; Oliva, R.; Sáez, G. Differences between cysteine and homocysteine in the induction of deoxyribose degradation and DNA damage. Free Radic. Biol. Med. 2001, 30, 354–362. [Google Scholar] [CrossRef]
- Pinto, M.M.M.; Ferreira, A.C.S.; Caris-Veyrat, C.; Pinho, P.G. Carotenoid, chlorophyll, and chlorophyll-derived compounds in grapes and Port wines. J. Agric. Food Chem. 2005, 53, 10034–10041. [Google Scholar] [CrossRef]
- SPSS Software, version 16.0.0, SPSS Inc.: Chicago, IL, USA, 2007.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Guedes, A.C.; Gião, M.S.; Seabra, R.; Ferreira, A.C.S.; Tamagnini, P.; Moradas-Ferreira, P.; Malcata, F.X. Evaluation of the Antioxidant Activity of Cell Extracts from Microalgae. Mar. Drugs 2013, 11, 1256-1270. https://doi.org/10.3390/md11041256
Guedes AC, Gião MS, Seabra R, Ferreira ACS, Tamagnini P, Moradas-Ferreira P, Malcata FX. Evaluation of the Antioxidant Activity of Cell Extracts from Microalgae. Marine Drugs. 2013; 11(4):1256-1270. https://doi.org/10.3390/md11041256
Chicago/Turabian StyleGuedes, A. Catarina, Maria S. Gião, Rui Seabra, A. C. Silva Ferreira, Paula Tamagnini, Pedro Moradas-Ferreira, and F. Xavier Malcata. 2013. "Evaluation of the Antioxidant Activity of Cell Extracts from Microalgae" Marine Drugs 11, no. 4: 1256-1270. https://doi.org/10.3390/md11041256
APA StyleGuedes, A. C., Gião, M. S., Seabra, R., Ferreira, A. C. S., Tamagnini, P., Moradas-Ferreira, P., & Malcata, F. X. (2013). Evaluation of the Antioxidant Activity of Cell Extracts from Microalgae. Marine Drugs, 11(4), 1256-1270. https://doi.org/10.3390/md11041256