Diketopiperazine Derivatives from the Marine-Derived Actinomycete Streptomyces sp. FXJ7.328


Abstract
:1. Introduction
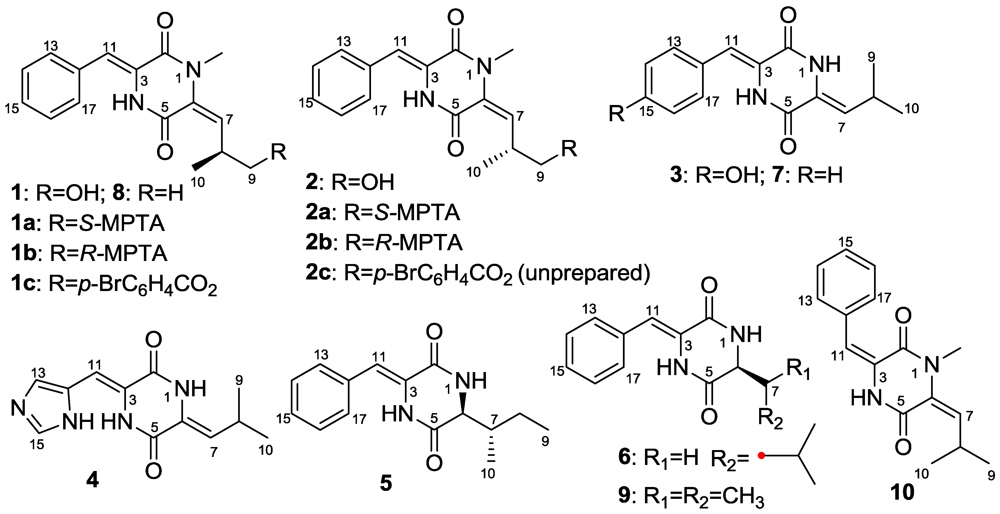
2. Results and Discussion
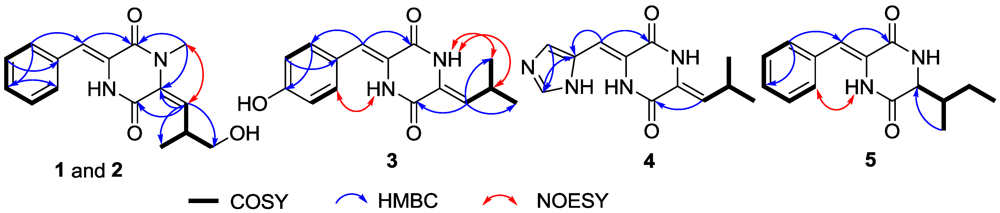
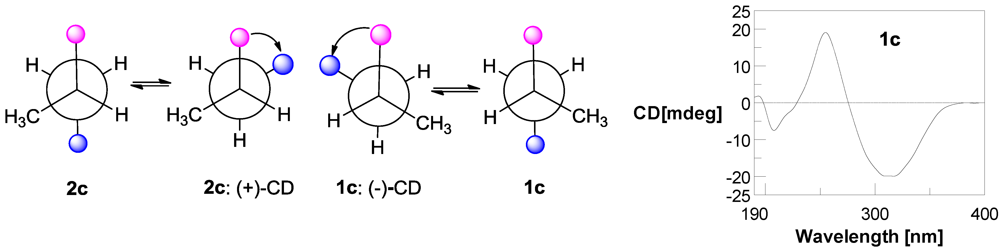
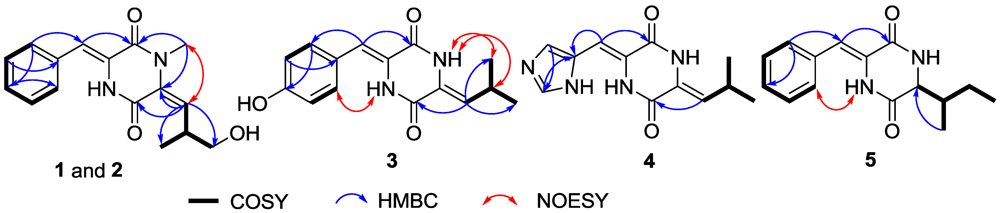
2.1. Structure Elucidation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 and 2 | 3 | 4 | 5 | ||||
|---|---|---|---|---|---|---|---|---|
| δC, type | δH, mult. (J in Hz) | δC, type | δH, mult. (J in Hz) | δC, type | δH, mult. (J in Hz) | δC, type | δH, mult. (J in Hz) | |
| 1 | 31.3, NCH3 | 3.17, s | 10.24, s | 8.47, s | ||||
| 2 | 158.5, qC | 158.0, qC | 157.8, qC | 161.0, qC | ||||
| 3 | 126.5, qC | 125.3, qC | 125.2, qC | 127.3, qC | ||||
| 4 | 9.82, s | 9.93, s | ||||||
| 5 | 158.9, qC | 157.4, qC | 156.8, qC | 166.8, qC | ||||
| 6 | 130.4, qC | 125.2, qC | 125.9, qC | 60.2, CH | 3.86, t, (3.3) | |||
| 7 | 129.7, CH | 5.56, d, (9.9) | 125.1, CH | 5.66, d, (10.4) | 125.5, CH | 5.68, d, (9.9) | 40.9, CH | 1.80, m |
| 8 | 34.9, CH | 3.59, m | 23.9, CH | 2.93, m | 24.4, CH | 2.95, m | 24.8, CH2 | 1.46, m; 1.18, m |
| 9 | 66.7, CH2 | 3.36 | 22.2, CH3 | 0.96, d, (6.5) | 22.8, CH3 | 0.97, d, (6.6) | 15.3, CH3 | 0.91, d, (7.1) |
| 10 | 17.9, CH3 | 1.01, d, (7.1) | 22.2, CH3 | 0.96, d, (6.5) | 22.8, CH3 | 0.97, d, (6.6) | 12.1, CH3 | 0.86, t, (7.7) |
| 11 | 116.1, CH | 6.75, s | 115.0, CH | 6.66, s | 105.1, CH | 6.60, s | 114.6, CH | 6.66, s |
| 12 | 133.9, qC | 123.9, CH | 137.0, qC | 133.9, qC | ||||
| 13/17 | 129.9, CH | 7.52, d, (7.7) | 130.9, CH | 7.36, d, (8.5) | 119.8, CH | 7.52, s | 129.3, CH | 7.45, d, (7.7) |
| 14/16 | 129.2, CH | 7.40, t, (7.7) | 115.6, CH | 6.79, d, (8.5) | 129.7, CH | 7.39, t, (7.7) | ||
| 15 | 128.7, CH | 7.31, t, (7.2) | 157.5, qC | 137.1, CH | 7.94, s | 128.5, CH | 7.29, t, (7.7) | |
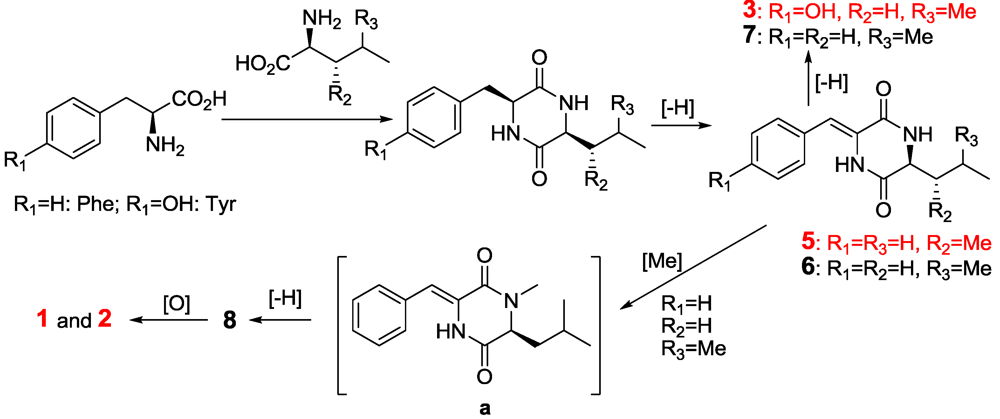
2.2. The Postulated Biosynthesis Pathway of Compounds 1–10
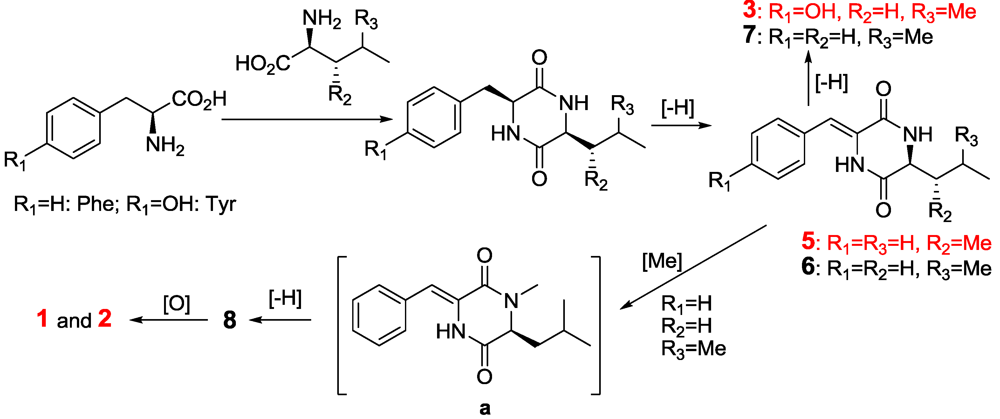
2.3. The Bioactivities of Compounds 1–10 from Streptomyces sp. FXJ7.328
3. Experimental Section
3.1. General Experimental Procedures
3.2. Actinomycete Material
3.3. Fermentation and Extraction
3.4. Purification and Identification
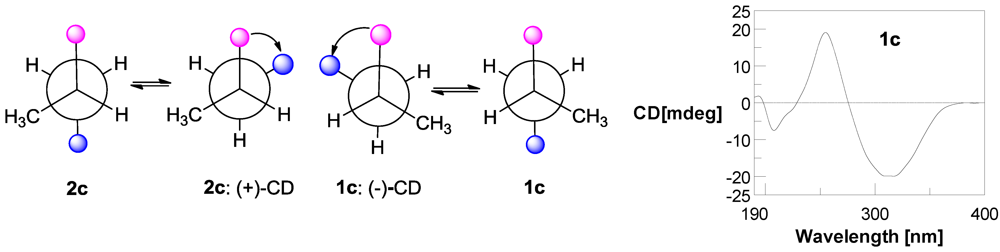
3.5. Preparation of p-Bromobenzoate (1c) of Compound 1 [14]
3.6. Preparation of S-MTPA and R-MTPA Esters 1a, 1b, 2a, and 2b of Compounds 1 and 2 [15,16]
3.7. Preparation of FDAA Derivatives of the Acid Hydrolysates of 5 and Four Authentic Isoleucine Samples (l-, l-allo-, d- and d-allo-) and Marfey’s Analysis [18] and C3 Marfey’s Analysis [19,20]
3.8. Bioassays
4. Conclusions
Acknowledgements
References
- Blunt, J.W.; Copp, B.R.; Munro, M.H.G.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2011, 28, 196–268. [Google Scholar] [CrossRef]
- Romero, F.; Espliego, F.; Baz, J.P.; Quesada, T.G.D.; Grávalos, D.; Calle, F.L.D.; Fernández-Puentes, J.L. Thiocoraline, a new depsipeptide with antitumor activity produced by a marine Micromonospora. I. Taxonomy, fermentation, isolation, and biological activities. J. Antibiot. 1997, 50, 734–737. [Google Scholar] [CrossRef]
- Renner, M.K.; Shen, Y.C.; Cheng, X.C.; Jensen, P.R.; Frankmoelle, W.; Kauffman, C.A.; Fenical, W.; Lobkovsky, E.; Clardy, J. Cyclomarins A–C, new antiinflammatory cyclic peptides produced by a marine bacterium (Streptomyces sp.). J. Am. Chem. Soc. 1999, 121, 11273–11276. [Google Scholar] [CrossRef]
- Fu, P.; Yang, C.L.; Wang, Y.; Liu, P.P.; Ma, Y.M.; Xu, L.; Su, M.B.; Hong, K.; Zhu, W.M. Streptocarbazoles A and B, two novel indolocarbazoles from the marine-derived actinomycete strain Streptomyces sp. F MA. Org. Lett. 2012, 14, 2422–2425. [Google Scholar]
- Fu, P.; Liu, P.P.; Li, X.; Wang, Y.; Wang, S.X.; Hong, K.; Zhu, W.M. Cyclic bipyridine glycosides from the marine-derived actinomycete Actinoalloteichus cyanogriseus WH1-2216-6. Org. Lett. 2011, 13, 5948–5951. [Google Scholar]
- Fu, P.; Liu, P.P.; Qu, H.J.; Wang, Y.; Chen, D.F.; Wang, H.; Li, J.; Zhu, W.M. α-Pyrones and diketopiperazine derivatives from the marine-derived actinomycete Nocardiopsis dassonvillei HR10-5. J. Nat. Prod. 2011, 74, 2219–2223. [Google Scholar] [CrossRef]
- Fu, P.; Wang, S.X.; Hong, K.; Li, X.; Liu, P.P.; Wang, Y.; Zhu, W.M. Cytotoxic bipyridines from the marine-derived actinomycete Actinoalloteichus cyanogriseus WH1-2216-6. J. Nat. Prod. 2011, 74, 1751–1756. [Google Scholar] [CrossRef]
- Kanzaki, H.; Yanagisawa, S.; Nitoda, T. Biosynthetic intermediates of the tetradehydro cyclic dipeptide albonoursin produced by Streptomyces albulus KO-23. J. Antibiot. 2000, 53, 1257–1264. [Google Scholar] [CrossRef]
- Wang, H.S.; Yeo, S.L.; Xu, J.; Xu, X.L.; He, H.; Ronca, F.; Ting, A.E.; Wang, Y.; Yu, V.C.; Sim, M.M. Isolation of streptonigrin and its novel derivative from Micromonospora as inducing agents of p53-dependent cell apoptosis. J. Nat. Prod. 2002, 65, 721–724. [Google Scholar] [CrossRef]
- Kanzaki, H.; Imura, D.; Nitoda, T.; Kawazu, K. Enzymatic dehydrogenation of cyclo l-Phe–l-Leu to a bioactive derivative, albonoursin. J. Mol. Catal. B Enzym. 1999, 6, 265–270. [Google Scholar] [CrossRef]
- Gurney, K.A.; Mantle, P.G. Biosynthesis of 1-N-methylalbonoursin by an endophytic Streptomyces sp. isolated from perennial ryegrass. J. Nat. Prod. 1993, 56, 1194–1198. [Google Scholar] [CrossRef]
- Davies, S.G.; Rodríguez-Solla, H.; Tamayo, J.A.; Cowley, A.R.; Concellón, C.; Garner, A.C.; Parkes, A.L.; Smith, A.D. Asymmetric conjugate reductions with samarium diiodide: Asymmetric synthesis of (2S,3R)- and (2S,3S)-[2-2H,3-2H]-leucine-(S)-phenylalanine dipeptides and (2S,3R)-[2-2H,3-2H]-phenylalanine methyl ester. Org. Biomol. Chem. 2005, 3, 1435–1447. [Google Scholar] [CrossRef]
- Tuntiwachwuttikul, P.; Taechowsian, T.; Wanbanjob, A.; Thadaniti, S.; Taylor, W.C. Lansai A–D, secondary metabolites from Streptomyces sp. SUC1. Tetrahedron 2008, 64, 7583–7586. [Google Scholar] [CrossRef]
- Gardoso, C.L.; Bolzani, V.D.S.; Silva, D.H.S.; Ishii, H.; Berova, N.; Nakanish, K. The Absolute configuration of 1-(3′,4′-dihydroxycinnamoyl)cyclopentane-2,3-diol from the amazonian tree Chimarrhis turbinata. J. Nat. Prod. 2006, 69, 1046–1050. [Google Scholar] [CrossRef]
- Tsuda, M.; Toriyabe, Y.; Endo, T.; Kobayashi, J. Application of modified Mosher’s method for primary alcohols with a methyl group at C2 position. Chem. Pharm. Bull. 2003, 51, 448–451. [Google Scholar] [CrossRef]
- Finamore, E.; Minale, L.; Riccio, R.; Rinaldo, G.; Zollo, F. Novel Marine polyhydroxylated steroids from the starfish Myxoderma platyacanthum. J. Org. Chem. 1991, 56, 1146–1153. [Google Scholar] [CrossRef]
- Takagi, M.; Motohashi, K.; Shin-Ya, K. Isolation of 2 new metabolites, JBIR-74 and JBIR-75, from the sponge-derived Aspergillus sp. fS14. J. Antibiot. 2010, 63, 393–395. [Google Scholar] [CrossRef]
- Marfey, P. Determination of d-amino acids. II. Use of a bifunctional reagent, 1,5-difluoro-2, 4-dinitrobenzene. Carlsberg Res. Commum. 1984, 49, 591–596. [Google Scholar] [CrossRef]
- Ratnayake, R.; Fremlin, L.J.; Lacey, E.; Gill, J.H.; Capon, R.J. Acremolides A–D, lipodepsipeptides from an Australian marine-derived fungus, Acremonium sp. J. Nat. Prod. 2008, 71, 403–408. [Google Scholar] [CrossRef]
- Fremlin, L.J.; Piggott, A.M.; Ernest, L.; Capon, R.J. Cottoquinazoline A and cotteslosins A and B, metabolites from an Australian marine-derived strain of Aspergillus versicolor. J. Nat. Prod. 2009, 72, 666–670. [Google Scholar] [CrossRef]
- Grassauer, A.; Weinmuellner, R.; Meier, C.; Pretsch, A.; Prieschl-Grassauer, E.; Unger, H. Iota-carrageenan is a potent inhibitor of rhinovirus infection. Virol. J. 2008, 5, 107. [Google Scholar] [CrossRef]
- Hung, H.C.; Tseng, C.P.; Yang, J.M.; Ju, Y.W.; Tseng, S.N.; Chen, Y.F.; Chao, Y.S.; Hsieh, H.P.; Shih, S.R.; Hsu, J.T. Aurintricarboxylic acid inhibits influenza virus neuraminidase. Antivir. Res. 2009, 81, 123–131. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistaica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R.; et al. New colorimetric cytotoxicity assay for anti-cancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Aquila, S.; Weng, Z.Y.; Zeng, Y.Q.; Sun, H.D.; Rios, J.L. Inhibition of NF-κB activation and iNOS induction by ent-kaurane diterpenoids in LPS-stimulated RAW264.7 murine macrophages. J. Nat. Prod. 2009, 72, 1269–1272. [Google Scholar] [CrossRef]
- Fu, P.; Kong, F.D.; Wang, Y.F.; Wang, Y.; Liu, P.P.; Zuo, G.Y.; Zhu, W.M. Antibiotic metabolites from the coral-associated actinomycete Streptomyces sp. OUCMDZ-1703. Chin. J. Chem. 2013, 31, 100–104. [Google Scholar]
- Li, W.R.; Peng, S.Z. Rational design and synthesis of unsaturated 2,5-dioxopiperazine derivatives as potential protein tyrosine kinase inhibitors. Tetrahedron Lett. 1988, 39, 7373–7376. [Google Scholar]
- Kanzaki, H.; Yanagisawas, S.; Kanoh, K.; Nitoda, T. A novel potent cell cycle inhibitor dehydrophenylahistin-enzymatic synthesis and inhibitory activity toward sea urchin embryo. J. Antibiot. 2002, 55, 1042–1047. [Google Scholar] [CrossRef]
- Shigeki, S. Preparation of 2,5-piperazinediones as blood platelet aggregation inhibitors. Jpn. Patent 01013074A, January 1989. [Google Scholar]
- Fukushima, K.; Yazawa, K.; Arai, T. Bological activities of albonoursin. J. Antibiot. 1973, 26, 175–176. [Google Scholar] [CrossRef]
- Taechowisan, T.; Wanbanjob, A.; Tuntiwachwuttikul, P.; Liu, J.K. Anti-inflammatory activity of lansais from endophytic Streptomyces sp. SUC1 in LPS-induced RAW 264.7 cells. Food Agric. Immunol. 2009, 20, 67–77. [Google Scholar] [CrossRef]
- Küster, E. Outline of a comparative study of criteria used in characterization of the actinomycetes. Int. Bull. Bacteriol. Nomencl. Taxon. 1959, 9, 97–104. [Google Scholar]
- Chun, J.; Goodfellow, M. A phylogenetic analysis of the genus Nocardia with 16s rRNA gene sequences. Int. J. Syst. Bacteriol. 1995, 2, 240–245. [Google Scholar] [CrossRef]
- Samples Availability: Available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, P.; Xi, L.; Liu, P.; Wang, Y.; Wang, W.; Huang, Y.; Zhu, W. Diketopiperazine Derivatives from the Marine-Derived Actinomycete Streptomyces sp. FXJ7.328. Mar. Drugs 2013, 11, 1035-1049. https://doi.org/10.3390/md11041035
Wang P, Xi L, Liu P, Wang Y, Wang W, Huang Y, Zhu W. Diketopiperazine Derivatives from the Marine-Derived Actinomycete Streptomyces sp. FXJ7.328. Marine Drugs. 2013; 11(4):1035-1049. https://doi.org/10.3390/md11041035
Chicago/Turabian StyleWang, Pei, Lijun Xi, Peipei Liu, Yi Wang, Wei Wang, Ying Huang, and Weiming Zhu. 2013. "Diketopiperazine Derivatives from the Marine-Derived Actinomycete Streptomyces sp. FXJ7.328" Marine Drugs 11, no. 4: 1035-1049. https://doi.org/10.3390/md11041035
APA StyleWang, P., Xi, L., Liu, P., Wang, Y., Wang, W., Huang, Y., & Zhu, W. (2013). Diketopiperazine Derivatives from the Marine-Derived Actinomycete Streptomyces sp. FXJ7.328. Marine Drugs, 11(4), 1035-1049. https://doi.org/10.3390/md11041035