2. Results and Discussion
The sliced bodies of the Taiwanese octocoral
B. excavatum were exhaustively extracted with acetone. The combined acetone extracts were concentrated to a brown gum, which was further partitioned between H
2O and EtOAc. The EtOAc-soluble portion was concentrated under reduced pressure and the residue was fractionated over silica gel 60 by column chromatography. The resulting fractions were further purified by RP-18 HPLC to yield the new compounds
1–
3 (
Figure 2).
Briacavatolide D (
1) was obtained as a white powder. Its HRMS and NMR spectral data established a molecular formula of C
24H
32O
11, implying the existence of eight double bond equivalents. The
1H and
13C NMR spectra (
Table 1) of
1 indicated the presence of two acetoxyls (
δH 1.97, 2.03;
δC 21.2, 21.1, 171.8, 170.6), a γ-lactone (
δH 6.22;
δC 172.2), and a trisubstituted olefin (
δH 5.71;
δC 149.3, 124.0). A tetrasubstituted epoxide containing a methyl substituent was indicateded by the signals of two quaternary oxygenated carbons (
δC 62.6, 71.1) and a methyl (
δC 9.3;
δH 1.67, 3H, s). A trisubstituted epoxide containing a methyl substituent was revealed from the signals of an oxymethine (
δC 61.4;
δH 3.05) and a quaternary oxygenated carbons (
δC 63.5) and a methyl (
δC 24.6;
δH 1.33, 3H, s). From the above data, metabolite
1 was found to be a pentacyclic compound. The structure and the
1H and
13C chemical shifts of
1 were assigned by the assistance of 2D NMR studies, including
1H–
1H COSY and HMBC experiments (
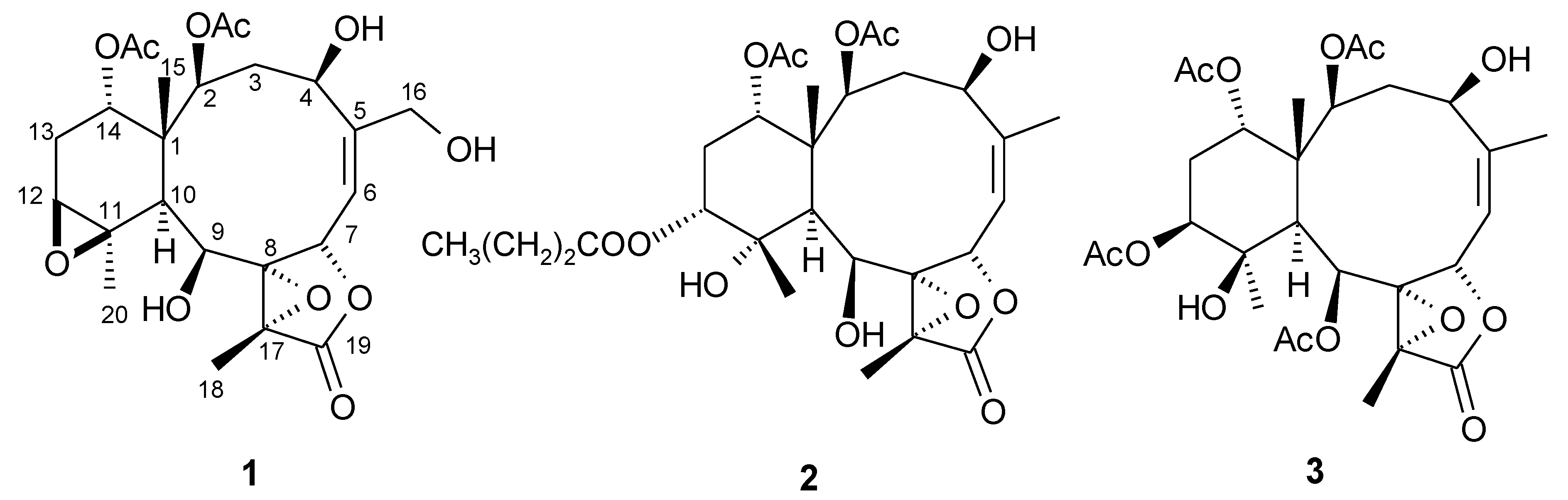
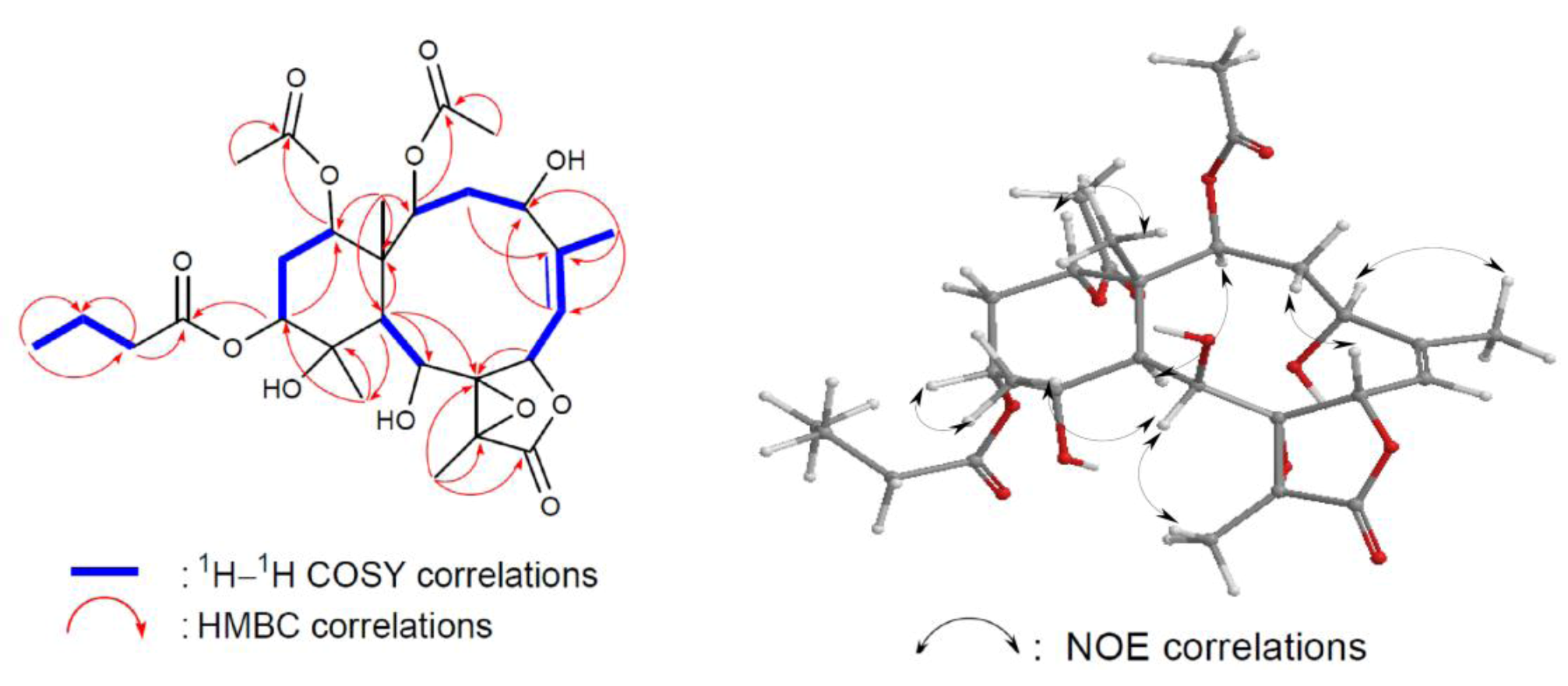
Figure 3). By analysis of
1H–
1H COSY correlations (
Figure 3), it was possible to establish four partial structures of consecutive proton systems extending from H-2 to H-4; H
3-16 to H-7 through H-6; H-6 to H-7; H-9 to H-10; and H-12 to H-14 through H-13. HMBC correlations (
Figure 3) further led to the connectivities of the gross structure. According to the above observations, metabolite
1 seemed to be very similar to 16-hydroxystecholide C acetate [
6], which was previously isolated from the soft coral
Solenopodium excavatum. By means of 1D and 2D NMR data it was found that the acetoxy groups at the C-4 and C-9 positions in 16-hydroxystecholide C acetate were replaced by hydroxy groups
1. On the basis of the above finding, and by the NOE correlations of
1 (
Figure 3), bricavatolide D (
1) was found to be the 4-
O-deacetyl-9-
O-deacetyl derivative of 16-hydroxystecholide C acetate.
Figure 3.
2D NMR correlations of compound 1.
Figure 3.
2D NMR correlations of compound 1.
Briacavatolide E (
2) was isolated as a white solid and had the molecular formula C
28H
40O
12, as determined by HRESIMS. The presence of hydroxyl, γ-lactone, and ester groups were evident from IR absorptions at 3447, 1773, and 1734 cm
−1, respectively.
13C NMR spectral data (
Table 1) revealed that
2 contains a trisubstituted double bond (
δC 145.2, s, C-5; 122.0, d, C-6) and four carbonyl resonances (
δC 171.0, 170.4, 170.2, and 172.5). Three esters were identified as two acetates and one
n-butyrate by the presence of resonances in the
1H NMR spectrum of
2 at
δH 1.97 (3H, s), 1.96 (3H, s), 0.96 (3H, t), 1.60 (1H, m) and 2.27 (H, m) (
Table 1).
The gross structure of
2 and all of the
1H and
13C chemical shifts associated with the molecule were determined by a series of 2D NMR experiments (
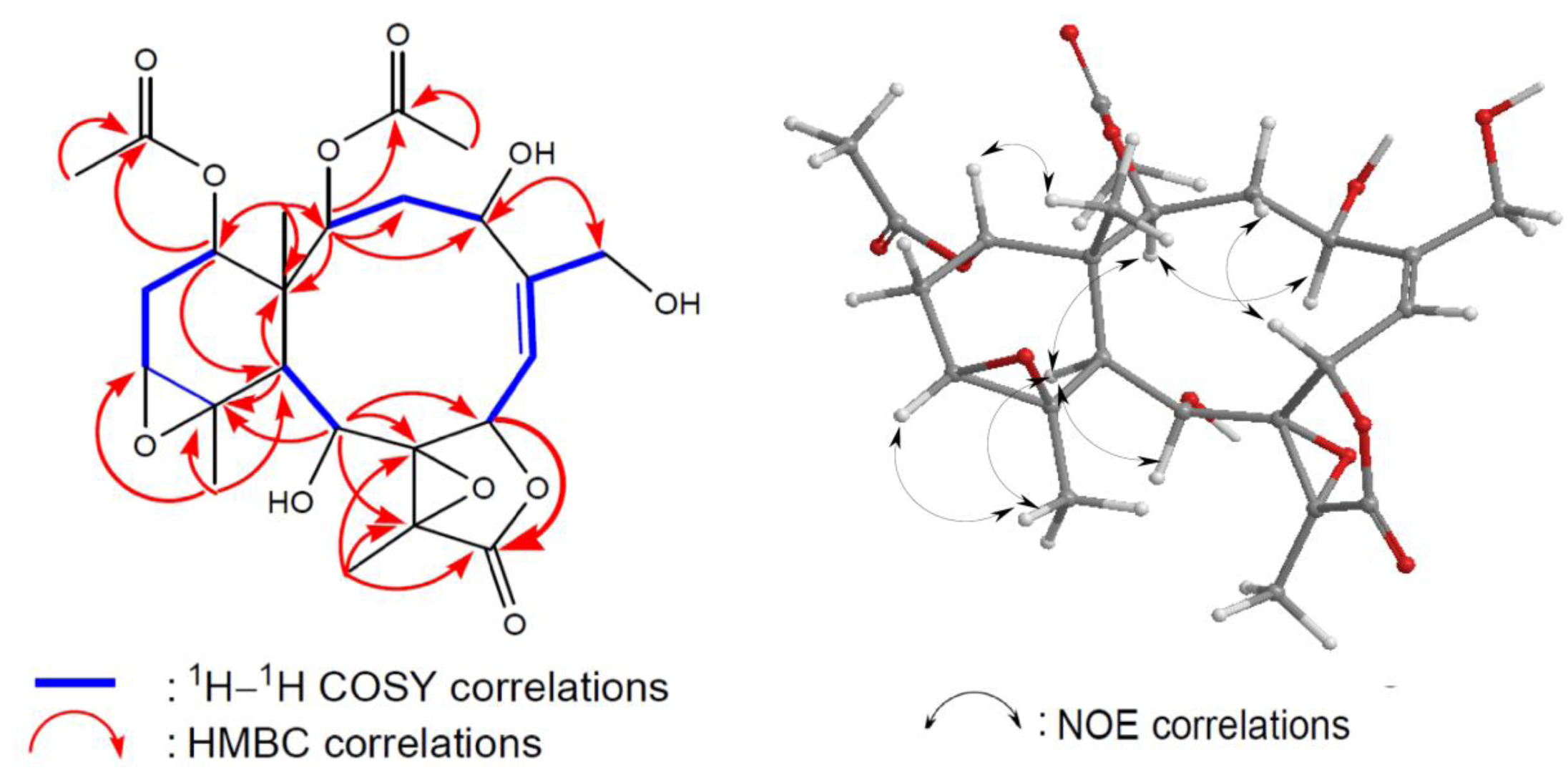
Figure 4). In the HMBC spectrum of
2, the
n-butyrate positioned at C-12 was confirmed from the long-range coupling between H-12 (
δH 4.82) with the carbonyl carbon (
δC 171.0) of the
n-butyryloxyl group. Furthermore, the HMBC correlations also revealed that two acetates were attached to C-2 and C-14. These data, together with the other
1H-
13C long-range correlations (
Table 1), unambiguously established the molecular framework of 2. The relative configurations of
2 were identical to those of briaexcavatin L except that of C-12. H
3-20 were found to exhibit NOE correlations (
Figure 4) with H-12 and H-9, revealing the β-orientation of H-12 [
7]. Splitting patterns of H-12 and H-14 as triplets (3.0 Hz) indicated that both are β equatorial.
Table 1.
NMR spectroscopic data of 1-3.
Table 1.
NMR spectroscopic data of 1-3.
| Position | 1 | 2 | 3 |
|---|
| δH (J in Hz) a | δC b | δH (J in Hz) a | δC b | δH (J in Hz) a | δC b |
|---|
| 1 | | 45.6 | | 47.5 | | 46.4 |
| 2 | 4.65 d (6.8) c | 74.5 | 5.00 d (8.8) | 74.9 | 4.86 d (7.6) | 74.1 |
| 3 | 1.95 m | 40.5 | 1.89 m | 39.7 | 2.03 m | 40.8 |
| 3.17 dd (15.6, 12.8) | | 3.38 dd (15.0, 12.8) | | 2.92 dd (15.6, 12.0) | |
| 4 | 4.34 dd (12.8, 4.8) | 68.0 | 4.09 dd (12.0, 5.2) | 71.2 | 4.21 dd (12.0, 5.6) | 71.0 |
| 5 | | 149.3 | | 145.2 | | 147.4 |
| 6 | 5.71 d (9.6) | 124.0 | 5.35 d (9.0) | 122.0 | 5.40 d (9.6) | 121.5 |
| 7 | 6.22 d (9.6) | 73.4 | 6.19 d (9.0) | 75.3 | 6.00 d (9.6) | 73.7 |
| 8 | | 71.1 | | 71.9 | | 70.3 |
| 9 | 4.43 br s | 71.6 | 4.73 d (3.6) | 64.9 | 5.88 d (2.4) | 67.5 |
| 10 | 2.21 d (3.6) | 42.5 | 2.14 s | 45.7 | 2.19 br s | 47.8 |
| 11 | | 63.5 | | 74.1 | | 75.5 |
| 12 | 3.05 d (5.2) | 61.4 | 4.82 t (3.0) | 73.9 | 4.91 dd (12.0, 5.2) | 73.0 |
| 13 | 2.10 m | 25.3 | 2.07 m 2.19 m | 25.8 | 1.92 m 1.99 m | 25.6 |
| 14 | 4.75 br s | 73.7 | 4.66 t (3.0) | 73.6 | 4.88 t (3.0) | 75.7 |
| 15 | 1.20 s | 16.0 | 1.34 s | 14.2 | 1.34 s | 15.8 |
| 16 | 4.31 br s | 67.1 | 2.05 d (0.8) | 25.4 | 2.12 d (1.6) | 25.2 |
| 17 | | 62.6 | | 64.9 | | 64.4 |
| 18 | 1.67 s | 9.3 | 1.65 s | 9.3 | 1.74 s | 10.3 |
| 19 | | 172.2 | | 172.5 | | 170.9 |
| 20 | 1.33 s | 24.6 | 1.42 s | 23.5 | 1.24 s | 27.8 |
| OAc | 2.03 s | 21.2 | 1.97 s | 21.5 | 2.00 s | 21.2 |
| 1.97 s | 21.1 | 1.96 s | 21.3 | 2.07 s | 21.5 |
| | 171.8 | | 170.4 | 2.22 s | 21.1 |
| | 170.6 | | 170.2 | 2.04 s | 21.3 |
| | | | | | 170.5 |
| | | | | | 168.1 |
| | | | | | 169.6 |
| | | | | | 170.4 |
| OCOPr | | | 0.96 t (7.2) | 13.7 | | |
| | | 1.60 m | 18.3 | | |
| | | 2.27 m | 36.3 | | |
| | | | 171.0 | | |
Figure 4.
2D NMR correlations of compound 2.
Figure 4.
2D NMR correlations of compound 2.
Briacavatolide F (
3) had a molecular formula of C
28H
38O
13 as deduced by HRESIMS. The IR spectrum of
3 indicated the presence of hydroxy (3501 cm
−1), γ-lactone (1778 cm
−1), and ester (1736 cm
−1) groups. From the
13C NMR data of
3 (
Table 1), a trisubstituted olefin (
δ 147.4, C
q, C-5; 121.5, CH, C-6) and five carbonyl resonances (
δ 170.5, 170.4, 169.6, 168.1, 170.9) were derived. Four esters were identified as acetates by the presence of four methyl resonances in the
1H NMR spectrum of
3 at
δ 2.22 (3H, s), 2.07 (3H, s), 2.04 (3H, s), and 2.00 (3H, s) (
Table 1). The planar structure of
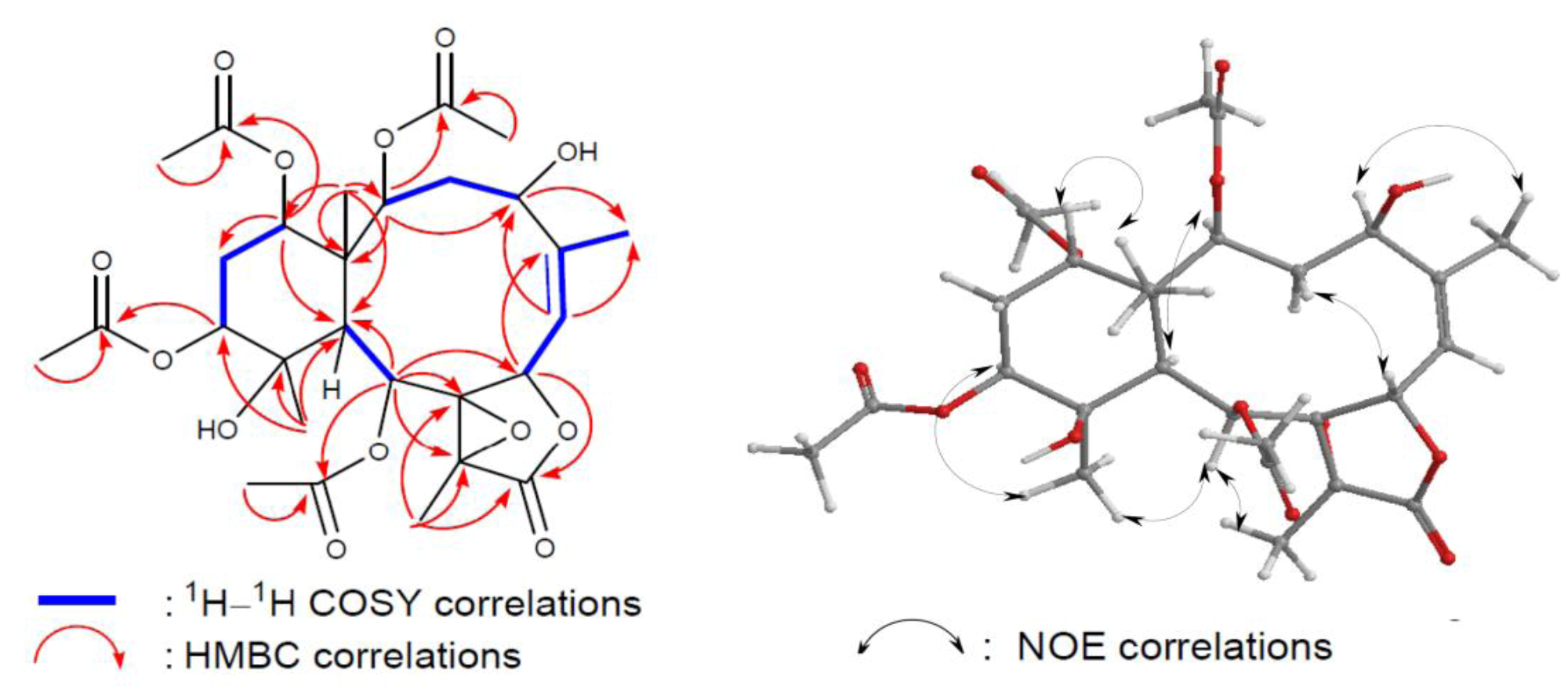
3 was determined by 2D NMR experiments (
Figure 5). The coupling information in the
1H–
1H COSY experiment of
3 enabled identification of the C-2/3/4, C-6/16 (by allylic coupling), C-6/7, C-9/10, and C-12/13/14 units. From these data, together with the results of an HMBC experiment of
3, the molecular framework of
3 could be further established. The HMBC data also revealed that the acetate groups are attached at C-2, C-9, C-12, and C-14; thus, the remaining hydroxy groups should be positioned at C-4 and C-11. The relative configurations of
3 elucidated from the NOE correlations (
Figure 5) were the same as those of briaexcavatin L except C-11. NOE correlations from H
3-20 to H-10/H-12 and from H-10 to H-12 revealed a β-axial orientation of hydroxy at C-11 for
3 [
7]. The dd splitting pattern of H-12 (12.0, 5.2 Hz) indicated that H-12 is α axial. The triplet splitting pattern of H-14 is (3.0 Hz) indicated that H-14 is an β equatorial position; this is confirmed by a cross signal with Me-15.
Figure 5.
2D NMR correlations of compound 3.
Figure 5.
2D NMR correlations of compound 3.
The cytotoxicity toward P-388 (mouse lymphocytic leukemia), HT-29 (human colon adenocarcinoma), A-549 (human lung epithelial carcinoma) tumor cells, and human embryonic lung (HEL) cells as well as anti-HCMV activity of briacavatolides D–F (
1–
3) are shown in
Table 2. Compound
3 showed moderate anti-HCMV activity.
Table 2.
Cytotoxicity and anti- human cytomegalovirus (HCMV) activity of 1–3.
Table 2.
Cytotoxicity and anti- human cytomegalovirus (HCMV) activity of 1–3.
| Compounds | ED50 (μM) | Anti-HCMV |
|---|
| A549 | HT-29 | P-388 | HEL |
|---|
| 1 | >100 | >100 | >100 | >100 | >100 |
| 2 | >100 | >100 | >100 | >100 | >100 |
| 3 | >100 | >100 | >100 | >100 | 22 |
3. Experimental Section
3.1. General Experimental Procedures
Optical rotations were determined with a JASCO P1020 digital polarimeter. UV and IR spectra were obtained on JASCO V-650 and JASCO FT/IR-4100 spectrophotometers, respectively. NMR spectra were recorded on a Varian MR 400 NMR spectrometer at 400 MHz for 1H and 100 MHz for 13C, respectively. 1H NMR chemical shifts are expressed in δ values referring to the solvent peak δH 7.27 for CDCl3, and coupling constants are expressed in Hz. 13C NMR chemical shifts are expressed in δ values referring to the solvent peak δC 77.0 for CDCl3. MS were recorded by a Bruker APEX II mass spectrometer. Calculated values for positively charged ions were calibrated. Silica gel 60 (Merck, Germany, 230–400 mesh) and LiChroprep RP-18 (Merck, 40–63 μm) were used for column chromatography. Precoated silica gel plates (Merck, Kieselgel 60 F254, 0.25 mm) and precoated RP-18 F254s plates (Merck) were used for thin-layer chromatography (TLC) analysis. High-performance liquid chromatography (HPLC) was carried out using a Hitachi L-7100 pump equipped with a Hitachi L-7400 UV detector at 220 nm together with a semi-preparative reversed-phase column (Merck, Hibar LiChrospher RP-18e, 5 μm, 250 × 25 mm).
3.2. Biological Material
The octocoral B. excavatum was collected by hand using scuba at Orchid Island off Taiwan, in July 2008 at a depth of 12 m and stored in a freezer until extraction. The voucher specimen (LY-05) was identified by Prof. Chang-Feng Dai, National Taiwan University and deposited at the Department of Marine Biotechnology and Resources, National Sun Yat-Sen University, Taiwan.
3.3. Extraction and Isolation
A specimen of octocoral B. excavatum (1.5 kg) was minced and extracted with acetone (2 L × 5) at room temperature. The combined acetone extracts was then partitioned between H2O and EtOAc. The resulting EtOAc extract (30.5 g) was subjected to gravity silica gel 60 column chromatography (Si 60 CC) using n-hexane and n-hexane/EtOAc of increasing polarity, to give 20 fractions. Fraction 12 (3.0 g), eluted with n-hexane/EtOAc (1:10), was further subjected to Si 60 CC (n-hexane/EtOAc, 10:1) to give 9 subfractions. A subfraction 12-5 (360 mg) was separated by a RP-18 flash column (MeOH/H2O, 50:50 to 100% MeOH) to give 6 fractions. In turn, a subfraction 12-5-2, eluted with MeOH/H2O (65:35), was further purified by RP-18 HPLC (MeOH/H2O, 60:40) to afford 3 (2.0 mg). Likewise, the subfraction 13-9 (62 mg), was separated by a RP-18 flash column (MeOH/H2O, 40:60 to 100% MeOH) to give 7 fractions. In turn, a subfraction 13-9-2, eluted with MeOH/H2O (55:45), was further purified by RP-18 HPLC (MeOH/H2O, 37:50) to afford 1 (1.5 mg). The fraction 14 (0.14 g), eluted with EtOAc/MeOH (70:1), was further subjected to a RP-18 flash column (MeOH/H2O, 40:60 to 100% MeOH) to give 7 fractions. The subfraction 14-1, eluted with MeOH/H2O (40:60), was purified by RP-18 HPLC (MeOH/H2O, 37:63) to afford 2 (1.5 mg).
Briacavatolide D (
1): White amorphous powder; [α]
D25 −33.6 (
c 0.2, CHCl
3); IR (neat) ν
max 3419, 2925, 1776, 1727, 1372, 1260, 1024 cm
−1;
1H NMR (CDCl
3, 400 MHz) and
13C NMR (CDCl
3, 100 MHz) data in
Table 2; HRESIMS
m/z 519.1839 [M + Na]
+ (calcd for C
24H
32O
11Na, 519.1842).
Briacavatolide E (
2): White amorphous powder; [α]
D25 +22.3 (
c 0.1, CHCl
3); IR (neat) ν
max 3447, 2964, 1773, 1734, 1371, 1262, 1022 cm
−1;
1H NMR (CDCl
3, 400 MHz) and
13C NMR (CDCl
3, 100 MHz) data in
Table 2; HRESIMS
m/z 591.2420 [M + Na]
+ (calcd for C
28H
40O
12Na, 591.2417).
Briacavatolide F (
3): White amorphous powder; [α]
D25 −27.6 (
c 0.1, CHCl
3); IR (neat) ν
max 3501, 2941, 1778, 1736, 1372, 1252, 1016 cm
−1;
1H NMR (CDCl
3, 400 MHz) and
13C NMR (CDCl
3, 100 MHz) data in
Table 2; HRESIMS
m/z 605.2214 [M + Na]
+ (calcd for C
28H
38O
13Na, 605.2210).
3.4. Cytotoxicity Assay
Cytotoxicity was determined on P-388 (mouse lymphocytic leukemia), HT-29 (human colon adenocarcinoma), and A-549 (human lung epithelial carcinoma) tumor cells using a modification of the MTT colorimetric method according to a previously described procedure [
8,
9,
10]. The provision of the P-388 cell line was supported by J. M. Pezzuto, formerly of the Department of Medicinal Chemistry and Pharmacognosy, University of Illinois at Chicago. HT-29 and A-549 cell lines were purchased from the American Type Culture Collection. To measure the cytotoxic activities of tested compounds, five concentrations with three replications were performed on each cell line. Mithramycin was used as a positive control.
3.5. Anti-HCMV Assay
To determine the effects of natural products upon HCMV cytopathic effect (CPE), confluent human embryonic lung (HEL) cells grown in 24-well plates were incubated for 1 h in the presence or absence of various concentrations of tested natural products with three replications. Ganciclovir was used as a positive control. Then, cells were infected with HCMV at an input of 1000 pfu (plaque forming units) per well of a 24-well dish. Antiviral activity was expressed as IC
50 (50% inhibitory concentration), or compound concentration required to reduce virus induced CPE by 50% after 7 days as compared with the untreated control. To monitor the cell growth upon treating with natural products, an MTT-colorimetric assay was employed [
8,
11].





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

