1. Introduction
Sulfated sterols are a well-known class of secondary metabolites, often found in sponges and echinoderms [
1,
2], that are emerging as a new potential class of lead compounds in the research for new drugs. A recent paper [
3] reports on the isolation from the sponge
Theonella swinhoei of solomonsterols A (
2) and B (
3), tri-sulfated sterols having the cholestan skeleton. They differ from each other in the length of the side chain, and are among the few examples of truncated steroid derivatives isolated from marine sources. Both compounds have shown an important pharmacological activity, in that they are agonists of the PXR nuclear receptor [
4]. PXR receptor is a transcription factor which is able to bind to a wide spectrum of structurally distinct endobiotic substrates and xenobiotic compounds, and is involved in innate immunity, xenobiotic metabolism, and detoxification [
5,
6]. PXR is proving to be an attractive target for small molecule drug discovery. In recent years, potential applications for exogenous PXR ligands have emerged in the treatment of important pathologies such as cancer [
7] and inflammatory diseases [
8].
As part of our research program focused on the search for new anti-inflammatory and anti-cancer lead compounds from marine sponges [
9,
10], we analyzed the chemical composition of the Caribbean sponge
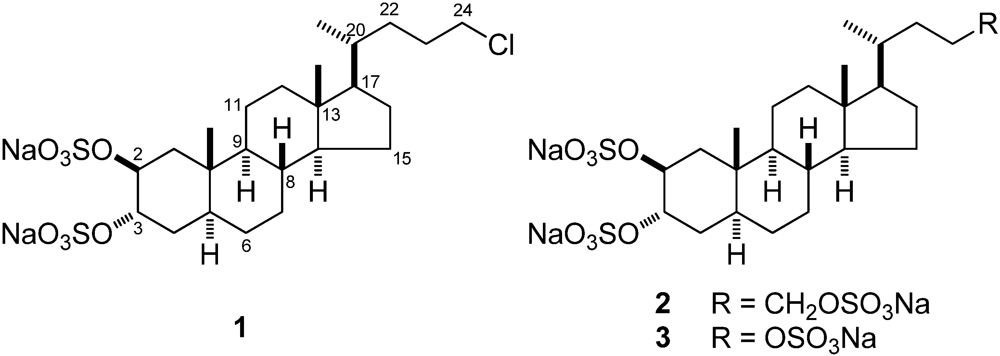
Chalinula molitba (de Laubenfels, 1949), a light purple sponge that habits the mangroves of Little San Salvador (Bahamas Islands). This study led to isolation and structural identification of chalinulasterol (
1) a new chlorinated sterol disulfate (
Figure 1). Compound
1 has a close structural relationship with
2, differing from the latter compound in having a chlorine atom instead of a sulfate function at position C-24 of the side chain. This relationship prompted us to investigate the possible role of
1 as modulator of the PXR receptors.
Figure 1.
Structures of chalinulasterol (1) and of solomonsterol A (2) and B (3).
Figure 1.
Structures of chalinulasterol (1) and of solomonsterol A (2) and B (3).
2. Results and Discussion
The positive-ion high-resolution ESI mass spectrum of 1 displayed a [M + Na]+ pseudomolecular ion peak at m/z 623.1447, in accordance with the formula C24H39ClS2O8Na3+ for this ion (calcd. 623.1462). The intensity of the (M + 2) isotopic peak in the MS spectrum (45%, calcd. 46.5%) and the peak at m/z 587.1675 ([M − HCl + Na]+) in the HRMS/MS spectrum confirmed the presence of a chlorine atom in the molecule. The MS/MS fragmentation pattern of 1 also revealed the presence of two sulfate groups from the peaks at m/z 503.1946 [M − NaHSO4 + Na]+, 262.8870 [2NaHSO4 + Na]+, and 244.8765 [Na2S2O7 + Na]+. The molecular formula was confirmed by the pseudomolecular ion peak at m/z 577.1669 observed in the negative-ion ESI mass spectrum, accounting for C24H39ClS2O8Na− (calcd. 577.1678).
Inspection of the
1H NMR spectrum of compound
1 showed two methyl singlets at δ 0.69 (H
3-18) and δ 1.01 (H
3-19) and one methyl doublet at δ 0.95 (H
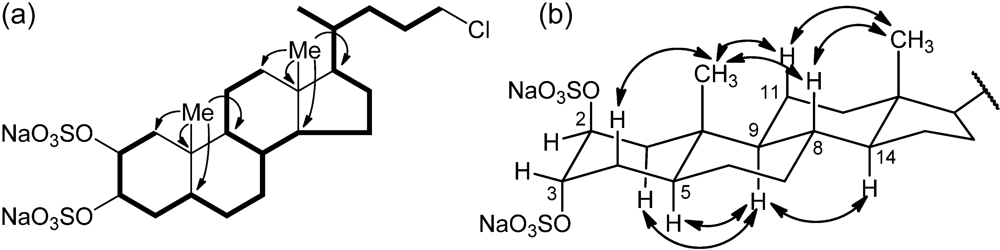
3-21) suggesting its steroidal nature. The steroidal backbone could be assembled through the interpretation of COSY, TOCSY, HSQC and HMBC 2D NMR experiments (
Figure 2 and
Table 1). Analysis of the COSY and TOCSY spectra allowed the sequential assignment of all the protons of the tetracyclic system. The sulfate groups were located at position C-2 and C-3 because of the low-field resonances of H-2 and H-3 (δ 4.72 and 4.69) and of the respective carbon atoms C-2 and C-3 (δ 76.5 and 76.3). The HMBC correlation peaks of the methyl protons H
3-19 with C-1, C-5, C-9, and C-10 and of H
3-18 with C-12, C-13, C-14, and C-17 located the A/B and C/D ring junctions and completed the planar structure determination of the steroid ring system.
Figure 2.
(a) Selected COSY and HMBC correlations of 1, represented as bold bonds and arrows, respectively. (b) Selected ROESY correlations detected for 1.
Figure 2.
(a) Selected COSY and HMBC correlations of 1, represented as bold bonds and arrows, respectively. (b) Selected ROESY correlations detected for 1.
Table 1.
1H (700 MHz) and 13C (175 MHz) NMR data of chalinulasterol (1)in CD3OD.
Table 1.
1H (700 MHz) and 13C (175 MHz) NMR data of chalinulasterol (1)in CD3OD.
| Pos. | | δH [mult. a,
J (Hz)] | δC [mult.] | Pos. | | δH [mult. a,
J (Hz)] | δC [mult.] |
|---|
| 1 | α/ax | 1.38 (dd, 14.6, 3.5) | 39.1 (CH2) | 13 | | - | 43.8 (C) |
| | β/eq | 2.12 (dd, 14.6, 2.1) | | 14 | | 1.05 (m) | 57.8 (CH) |
| 2 | | 4.72 (q, 2.7) | 76.5 (CH) | 15 | α | 1.59 (m) | 25.2 (CH2) |
| 3 | | 4.69 (q, 2.7) | 76.3 (CH) | | β | 1.08 (m) | |
| 4 | α/eq | 1.66 (dt, 14.6, 2.7) | 30.4 (CH2) | 16 | α | 1.86 (ddd, 14.6, 9.4, 3.9) | 29.3 (CH2) |
| | β/ax | 1.79 (ddd, 14.6, 12.6, 2.7) | | | β | 1.28 a | |
| 5 | | 1.62 (tt, 12.6, 2.7) | 40.3 (CH) | 17 | | 1.12 (q, 9.7) | 57.5 (CH) |
| 6 | α/eq | 1.25 (br. d, 14.5) | 29.2 (CH2) | 18 | | 0.69 (s) | 12.8 (CH3) |
| | β/ax | 1.29 (qd, 12.7, 3.5) | | 19 | | 1.01 (s) | 14.2 (CH3) |
| 7 | α/ax | 0.95 (qd, 12.6, 4.6) | 33.3 (CH2) | 20 | | 1.44 (m) | 36.6 (CH) |
| | β/eq | 1.68 (dq, 13.0. 3.3) | | 21 | | 0.95 (d, 6.5) | 19.2 (CH3) |
| 8 | | 1.41 (qd, 11.2, 3.5) | 36.5 (CH) | 22 | a | 1.56 (dddd, 13.4, 10.6, 5.6, 2.9) | 34.4 (CH2) |
| 9 | | 0.72 (ddd, 13.2, 10.5, 3.8) | 56.7 (CH) | | b | 1.15 (dddd, 13.4, 10.6, 8.8, 4.3) | |
| 10 | | - | 36.4 (C) | 23 | a | 1.81 (ddtd, 13.9, 11.2, 7.1, 4.5) | 30.6 (CH2) |
| 11 | α/eq | 1.53 (dq, 14.1, 3.5) | 22.1 (CH2) | | b | 1.65 (ddq, 13.9, 10.9, 5.7) | |
| | β/ax | 1.33 (qd, 13.2, 3.7) | | 24 | a | 3.53 (dt, 10.7, 6.5) | 46.4 (CH2) |
| 12 | α/ax | 1.14 (td, 12.6, 3.8) | 41.4 (CH2) | | b | 3.51 (ddd, 10.7, 7.1, 6.4) | |
| | β/eq | 1.99 (dt, 12.2, 3.5) | | | | | |
Information on the side chain was also provided by analysis of 2D NMR data. The COSY correlation between H-17 and the multiplet at δ 1.44 identified H-20; the latter was coupled with the methyl H3-21 and the protons at δ 1.15 and 1.56 (H-22a and H-22b), in turn coupled with the methylene protons at δ 1.65 and 1.81 (H-23a and H-23b). The coupling of H-23a and H-23b and the two protons at δ 3.51 and 3.53 (H-24a, and H-24b) could be also evidenced from the COSY spectrum. The linkage to this latter methylene group of the chlorine atom required from the molecular formula was shown by its 1H (δ 3.51 and 3.53) and 13C (δ 46.4) chemical shifts.
Analysis of the ROESY spectrum, supported by coupling constant analysis, defined the A/B, B/C and C/D
trans ring junctions of a 5-α-cholane skeleton. The axial orientation of H-5, H-8, H-9, and H-14 was apparent from their respective coupling constants (
Table 1), that of the angular methyl groups from the ROESY correlations of both H
3-18 and H
3-19 with H-8 and the axial H-11β. On this skeleton, the small coupling constants showed by H-2 and H-3 illustrated their equatorial orientation, and therefore the diaxial (
i.e., 2β,3α) orientation of the two sulfate groups. According to this information, the structure of chalinulasterol (
1) was established as sodium 24-chloro-5α-cholane-2β,3α-diyl 2,3-disulfate.
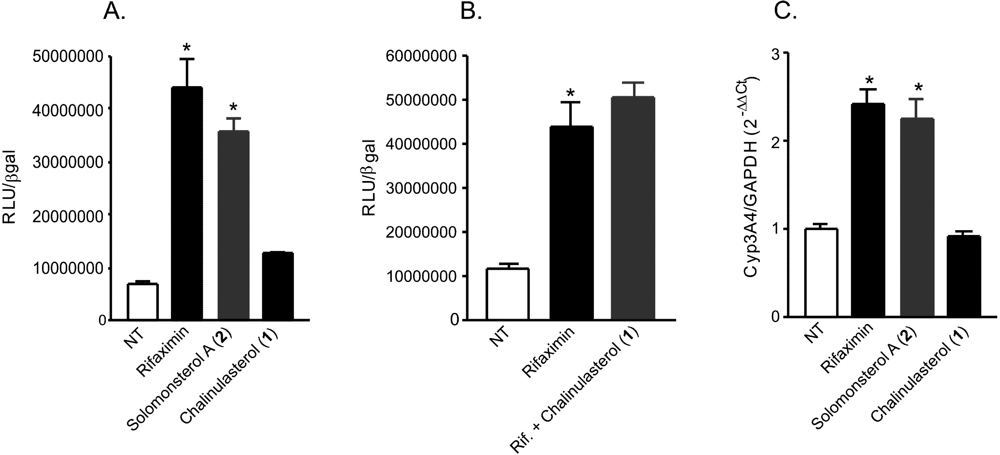
Figure 3.
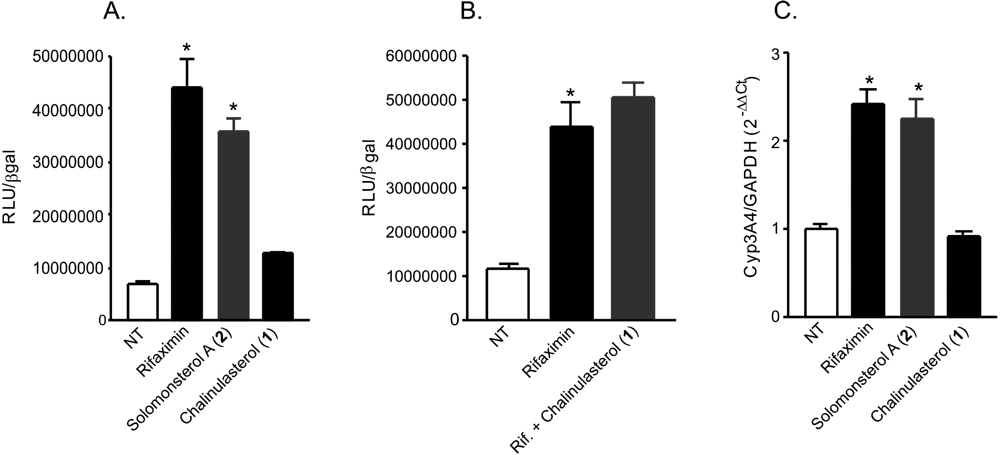
(A,B) Luciferase reporter assay. HepG2 cells, a hepatocarcinoma cell line, were transiently transfected with pSG5-PXR, pSG5-RXR, pCMV-βgalactosidase and p(CYP3A4)-TK-Luc vectors and then stimulated with (A) 10 µM rifaximin, solomonsterol A (2) or chalinulasterol (1) for 18 h, or (B) 10 µM rifaximin alone or in combination with 50 µM of compounds 1. Relative Luciferase Units were normalized with β-galactosidase Units (RLU/βgal). NT, not treated. *P<0.05 versus NT cells. Data are mean ± SE of three experiments. (C) Real-Time PCR of Cyp3A4. HepG2 cells were stimulated with 10 µM rifaximin, 2 or 1 for 18 h. Total RNA was extracted and the relative mRNA levels of PXR target gene Cyp3A4, was measured. Values were normalized relative to GAPDH mRNA and are expressed relative to those of untreated mice, which are arbitrarily set to 1. Analysis was carried out in triplicate and the experiment was repeated twice. NT, not treated. *P<0.05 versus NT cells.
Figure 3.
(A,B) Luciferase reporter assay. HepG2 cells, a hepatocarcinoma cell line, were transiently transfected with pSG5-PXR, pSG5-RXR, pCMV-βgalactosidase and p(CYP3A4)-TK-Luc vectors and then stimulated with (A) 10 µM rifaximin, solomonsterol A (2) or chalinulasterol (1) for 18 h, or (B) 10 µM rifaximin alone or in combination with 50 µM of compounds 1. Relative Luciferase Units were normalized with β-galactosidase Units (RLU/βgal). NT, not treated. *P<0.05 versus NT cells. Data are mean ± SE of three experiments. (C) Real-Time PCR of Cyp3A4. HepG2 cells were stimulated with 10 µM rifaximin, 2 or 1 for 18 h. Total RNA was extracted and the relative mRNA levels of PXR target gene Cyp3A4, was measured. Values were normalized relative to GAPDH mRNA and are expressed relative to those of untreated mice, which are arbitrarily set to 1. Analysis was carried out in triplicate and the experiment was repeated twice. NT, not treated. *P<0.05 versus NT cells.
Considering the structural similarity with the PXR agonist solomonsterol A (
2), we have investigated a possible role of chalinulasterol (
1) in regulating the pregnane-X receptor activity and carried out a transactivation assay on HepG2 cells, a human hepatocarcinoma cell line as described in the Experimental Part. As shown in
Figure 3A, despite the structural similarity with
2, compound
1 failed in transactivating PXR. We have also investigated the possibility that
1 could act as potential PXR antagonist. As shown in
Figure 3B,
1 failed to reverse the induction of luciferase caused by rifaximin, indicating that it was not a PXR antagonist. Similar results have been obtained by analyzing the effect exerted by
1 in terms of regulation of PXR mediated induction of Cyp3A4 gene. Indeed,
1 also failed to induce Cyp3A4. Although negative, these results have an important implication in terms of structure-activity relationship, because they suggest that the sulfate group present at position C-24 of compound
2 is essential in the ligand-receptor binding. This can be rationalized by the binding model of
2 to the PXR receptor proposed in [
3], in which a clear interaction of the 24-sulfate with the positively charged Lys210 is observed, and further supports this model.
Although halogen-containing secondary metabolites are well-known and abundant in nature, particularly in marine organisms, only a few natural chlorinated steroids have been reported so far [
12,
13], and there is only one example in the literature of a sulfated and chlorinated steroid [
14].
3. Experimental Section
3.1. General Experimental Procedures
High-resolution ESI-MS spectra were performed on a Thermo LTQ Orbitrap XL mass spectrometer. The spectra were recorded by infusion into the ESI source using MeOH as the solvent. Optical rotations were measured at 589 nm on a Jasco P-2000 polarimeter using a 10-cm microcell. CD spectra were recorded on a Jasco J-710 spectrophotometer using a 1-cm cell. NMR spectra were determined on Varian Unity Inova spectrometers at 700 and 500 MHz; chemical shifts were referenced to the residual solvent signal (CD3OD: δH 3.31, δC 49.00). For an accurate measurement of the coupling constants, the one-dimensional 1H NMR spectra were transformed at 64K points (digital resolution: 0.09 Hz). Through-space 1H connectivities were evidenced using a ROESY experiment with a mixing time of 450 ms. The HSQC spectra were optimized for 1JCH = 142 Hz, and the HMBC experiments for 2,3JCH = 8.3 Hz. High performance liquid chromatography (HPLC) separations were achieved on a Varian Prostar 210 apparatus equipped with a Varian 350 refractive index detector.
3.2. Collection, Extraction, and Isolation
Specimens of Chalinula molitba were collected in the mangroves of Little San Salvador (Bahamas Islands) during the 2010 Pawlik expedition. The samples were frozen immediately after collection and stored at −20 °C until extraction. The sponge (424 g of dry weight after extraction) was homogenized and extracted with MeOH (5 × 4 L) and then with CHCl3 (2 × 4 L). The MeOH extracts were partitioned between H2O and n-BuOH; the BuOH layer was combined with the CHCl3 extract and concentrated in vacuo.
The organic extract (10, 70 g) was chromatographed on a column packed with RP-18 silica gel. A fraction eluted with MeOH/H2O (8:2, 150 mg) was subjected to HPLC separation on a preparative RP-18 column [MeOH/H2O (6:4), Ascentis® C18–25 cm × 10 mm, 5 µm-SUPELCO®], thus affording a fraction (1.4 mg) mainly composed of 1.
Final purification was achieved by HPLC on an analytical RP-18 column (Ascentis® C18—25 cm × 4.6 mm, 5 µm-SUPELCO®), using MeOH/H2O (6:4) as eluent, which gave 1 mg of pure compound 1.
3.3. Chalinulasterol (1)
Colorless amorphous solid, [α]
D25 +11.4 (
c 0.1, MeOH); HRESIMS (positive ion mode, MeOH)
m/
z 623.1447 ([M + Na]
+, calcd. for C
24H
39ClS
2O
8Na
3+ 623.1462); MS isotope pattern: M (100%), M + 1 (27.5%, calcd. 27.8%), M + 2 (45.5%, calcd. 46.4%), M + 3 (11.5%, calcd. 12.2%,), M + 4 (4.9%, calcd. 5.4%);
1H and
13C NMR:
Table 1.
3.4. Cell Culture
HepG2 cells were maintained at 37 °C in E-MEM containing 10% fetal bovine serum (FBS), 1% L-glutamine and 1% penicillin/streptomycin. HepG2 cells were stimulated 18 h with 10 μM rifaximin, 1 and compound 2–10 and relative mRNA levels of CYP3A4 were analyzed by Real-Time PCR.
3.5. Transactivation Experiments
HepG2 cells were transfeted using Fugene HD transfection reagent (Roche). The plasmids used for luciferase assay were pSG5-PXR, pSG5-RXR, pCMV-βgalactosidase and the reporter vector p(CYP3A4)-TK-Luc. 48 h post-transfection cells were stimulated 18 h with 10 µM rifaximin, Solomonsterol A, compound 1 or with the combination of 10 µM rifaximin plus 50 µM compound 1. Cells were lysed in 100 µL diluted reporter lysis buffer (Promega). 20 µL of cellular lysates were read using the Luciferase Substrate (Promega). Luminescence was measured using the Glomax 10/10 luminometer (Promega). Luciferase activities were normalized for transfection efficiencies by dividing the relative light units by β-galactosidase activity expressed from cotransfected pCMV-βgal.
3.6. Real-Time PCR
Total RNA was extracted using the TRIzol reagent (Invitrogen), purified of the genomic DNA by DNAase I treatment (Invitrogen) and random reverse-transcribed with Superscript II (Invitrogen). 50 ng template was amplified using the following reagents: 0.2 µM of each primer and 12.5 µL of 2× SYBR Green qPCR master mix (Invitrogen). All reactions were performed in triplicate and the thermal cycling conditions were: 2 min at 95 °C, followed by 40 cycles of 95 °C for 20 s, 55 °C for 20 s and 72 °C for 30 s in iCycler iQ instrument (Bio-Rad). The relative mRNA expression was calculated and expressed as 2−(ΔΔCt). Primers used for qRT-PCR were: hGAPDH: gaaggtgaa ggtcggagt and catgggtggaatcatattggaa; hCYP3A4: CAAGACCCCTTTGTGG AAAA and CGAGGCGACTTTCTTTCATC.
3.7. Statistical Analysis
All values are expressed as means ± standard error (SE) of n observations/group. Comparisons of 2 groups were made with a one-wayANOVA with post hoc Tukey’s test. Differences were considered statistically significant at values of P < 0.05.

 ,
, 



{kind=link}
{kind=link}
{kind=link}