Two-Front War on Cancer—Targeting TAM Receptors in Solid Tumour Therapy

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. TAM Family in Carcinogenesis
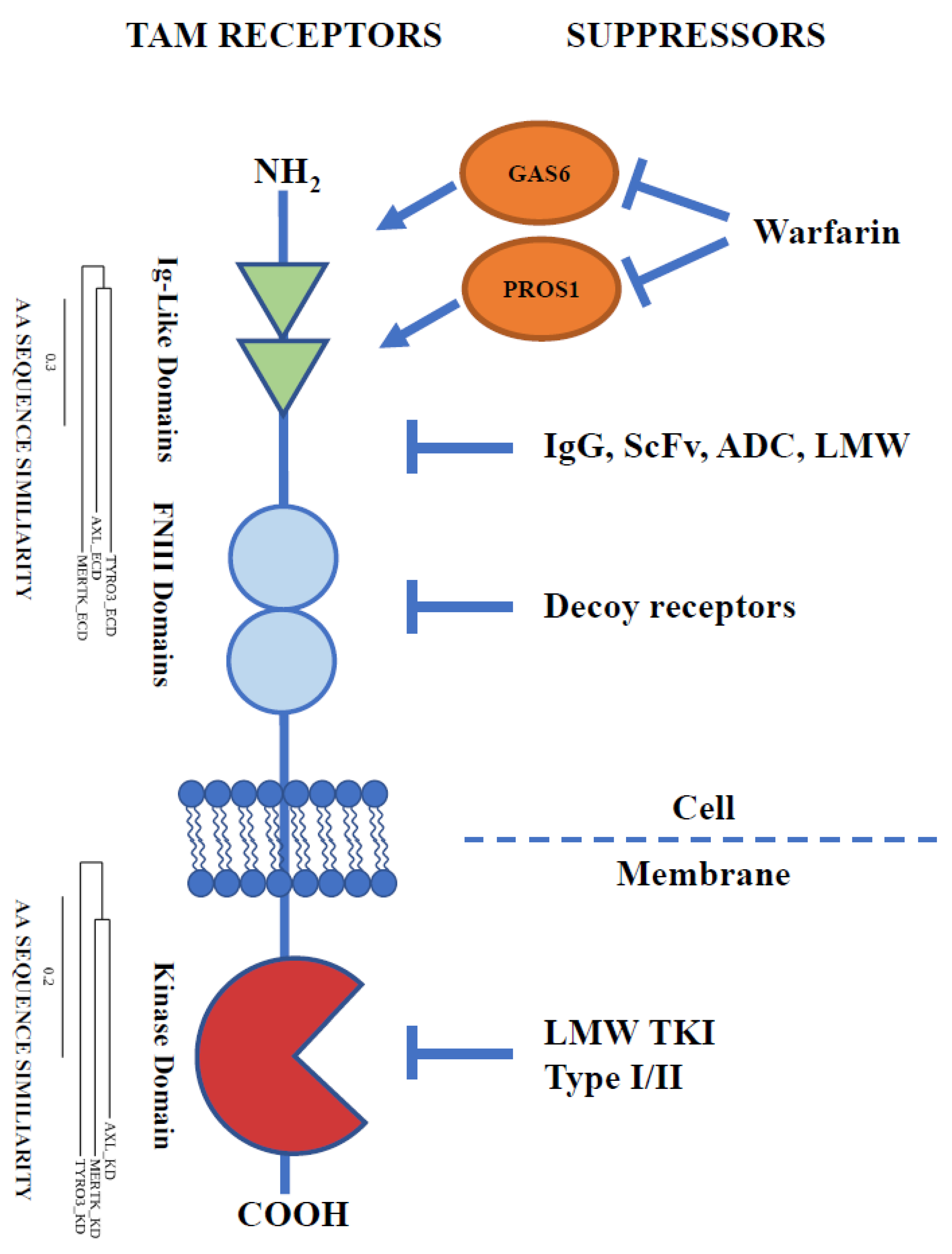
3. Extracellular Domain: An Approach from the Outside
3.1. Ligand Binding/Dimerisation Inhibition
3.2. Receptor Cleavage and Decoy Receptors
3.3. Low-Molecular-Weight Compounds Targeting ECD of TAMs
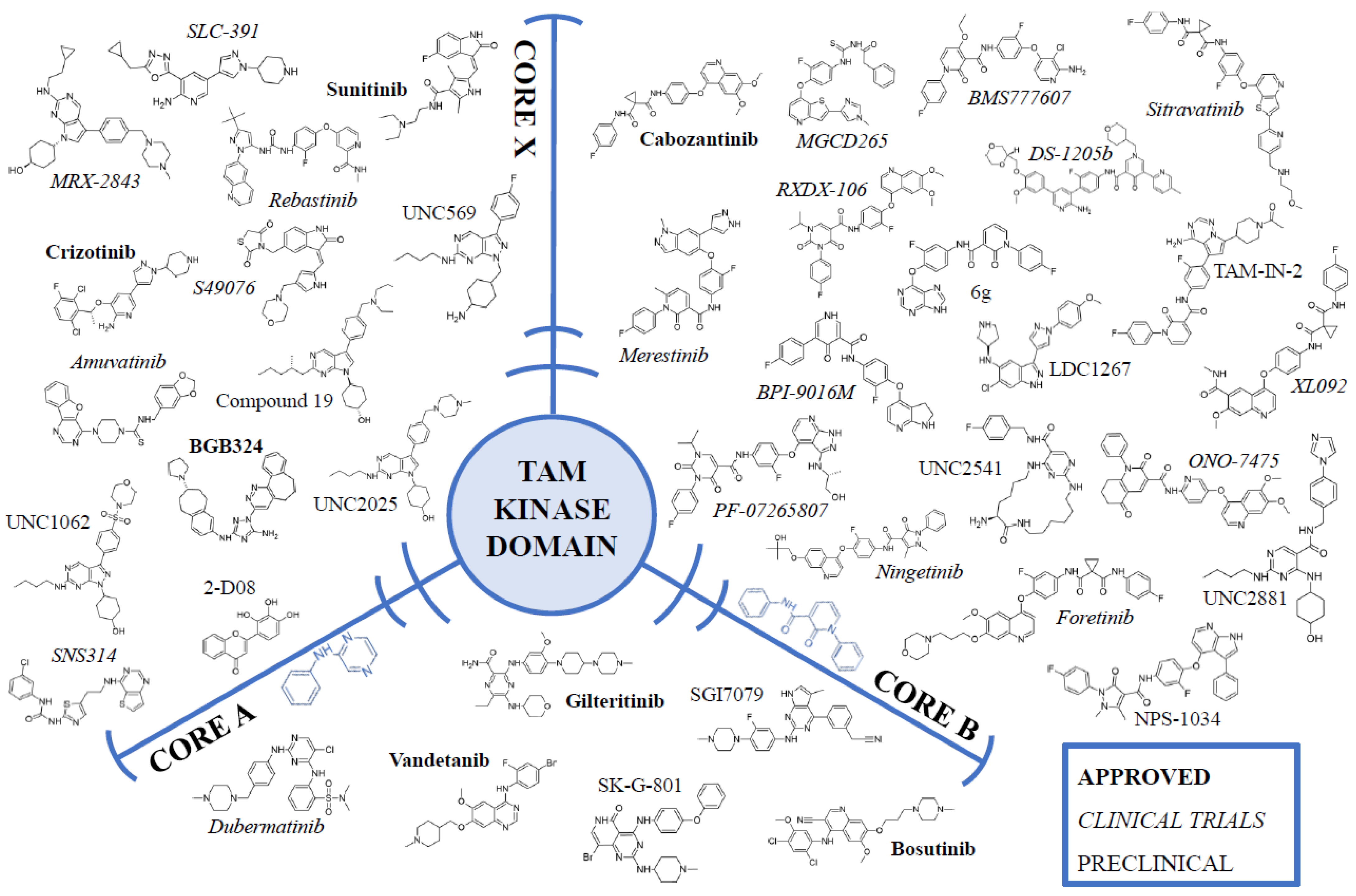
4. Kinase Domain: An Approach from the Inside
Tyrosine Kinase Inhibitors
5. Battles for the Future
5.1. Selectivity of TAM Family Inhibitors
5.2. Combination Therapy
5.3. Biomarkers
5.4. Potential Concerns
6. Future Directions
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Scott, R.S.; McMahon, E.J.; Pop, S.M.; Reap, E.A.; Caricchio, R.; Cohen, P.L.; Earp, H.S.; Matsushima, G.K. Phagocytosis and Clearance of Apoptotic Cells Is Mediated by MER. Nature 2001, 411, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Schulz, N.T.; Paulhiac, C.I.; Lee, L.; Zhou, R. Isolation and Expression Analysis of Tyro3, a Murine Growth Factor Receptor Tyrosine Kinase Preferentially Expressed in Adult Brain. Mol. Brain Res. 1995, 28, 273–280. [Google Scholar] [CrossRef]
- Weinger, J.G.; Brosnan, C.F.; Loudig, O.; Goldberg, M.F.; Macian, F.; Arnett, H.A.; Prieto, A.L.; Tsiperson, V.; Shafit-Zagardo, B. Loss of the Receptor Tyrosine Kinase Axl Leads to Enhanced Inflammation in the CNS and Delayed Removal of Myelin Debris during Experimental Autoimmune Encephalomyelitis. J. Neuroinflamm. 2011, 8, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavet, M.E.; Smolock, E.M.; Ozturk, O.H.; World, C.; Pang, J.; Konishi, A.; Berk, B.C. Gas6–Axl Receptor Signaling Is Regulated by Glucose in Vascular Smooth Muscle Cells. ATVB 2008, 28, 886–891. [Google Scholar] [CrossRef] [Green Version]
- O’Bryan, J.P.; Frye, R.A.; Cogswell, P.C.; Neubauer, A.; Kitch, B.; Prokop, C.; Iii, R.E.; Beau, M.M.L.; Earp, H.S.; LIUl, E.T. Axl, a Transforming Gene Isolated from Primary Human Myeloid Leukemia Cells, Encodes a Novel Receptor Tyrosine Kinase. Mol. Cell. Biol. 1991, 11, 5016–5031. [Google Scholar] [PubMed] [Green Version]
- Graham, D.; Bowman, G.; Dawson, T.; Stanford, W.; Earp, H.; Snodgrass, H. Cloning and Developmental Expression Analysis of the Murine C-Mer Tyrosine Kinase. Oncogene 1995, 10, 2349–2359. [Google Scholar]
- Ohashi, K.; Nagata, K.; Toshima, J.; Nakano, T.; Arita, H.; Tsuda, H.; Suzuki, K.; Mizuno, K. Stimulation of Sky Receptor Tyrosine Kinase by the Product of Growth Arrest-Specific Gene 6. J. Biol. Chem. 1995, 270, 22681–22684. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, T.; Knyazev, P.G.; Clout, N.J.; Cheburkin, Y.; Göhring, W.; Ullrich, A.; Timpl, R.; Hohenester, E. Structural Basis for Gas6–Axl Signalling. EMBO J. 2006, 25, 80–87. [Google Scholar] [CrossRef] [Green Version]
- Tsou, W.-I.; Nguyen, K.-Q.N.; Calarese, D.A.; Garforth, S.J.; Antes, A.L.; Smirnov, S.V.; Almo, S.C.; Birge, R.B.; Kotenko, S.V. Receptor Tyrosine Kinases, TYRO3, AXL, and MER, Demonstrate Distinct Patterns and Complex Regulation of Ligand-Induced Activation. J. Biol. Chem. 2014, 289, 25750–25763. [Google Scholar] [CrossRef] [Green Version]
- Stitt, T.N.; Conn, G.; Gore, M.; Lai, C.; Bruno, J.; Radziejewski, C.; Mattsson, K.; Fisher, J.; Gies, D.R.; Jones, P.F.; et al. The Anticoagulation Factor Protein S and Its Relative, Gas6, Are Ligands for the Tyro 3/Axl Family of Receptor Tyrosine Kinases. Cell 1995, 80, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Varnum, B.; Young, C.; Elliott, G.; Garcia, A.; Bartley, T.; Fridell, Y.; Hunt, R.; Trail, G.; Clogston, C.; Toso, R. Axl Receptor Tyrosine Kinase Stimulated by the Vitamin K-Dependent Protein Encoded by Growth-Arrest-Specific Gene 6. Nature 1995, 373, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Helenius, A. Vaccinia Virus Uses Macropinocytosis and Apoptotic Mimicry to Enter Host Cells. Science 2008, 320, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, Q.; Darrow, A.L.; Wang, Y.; Derian, C.K.; Yang, J.; de Garavilla, L.; Andrade-Gordon, P.; Damiano, B.P. Mer Receptor Tyrosine Kinase Signaling Participates in Platelet Function. ATVB 2004, 24, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Geng, K.; Kumar, S.; Kimani, S.G.; Kholodovych, V.; Kasikara, C.; Mizuno, K.; Sandiford, O.; Rameshwar, P.; Kotenko, S.V.; Birge, R.B. Requirement of Gamma-Carboxyglutamic Acid Modification and Phosphatidylserine Binding for the Activation of Tyro3, Axl, and Mertk Receptors by Growth Arrest-Specific 6. Front. Immunol. 2017, 8, 1521. [Google Scholar] [CrossRef] [PubMed]
- Aehnlich, P.; Powell, R.M.; Peeters, M.J.W.; Rahbech, A.; thor Straten, P. TAM Receptor Inhibition–Implications for Cancer and the Immune System. Cancers 2021, 13, 1195. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Chen, J.; Hammonds, G.; Phillips, H.; Armanini, M.; Wood, P.; Bunge, R.; Godowski, P.; Sliwkowski, M.; Mather, J. Identification of Gas6 as a Growth Factor for Human Schwann Cells. J. Neurosci. 1996, 16, 2012–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linger, R.M.A.; Cohen, R.A.; Cummings, C.T.; Sather, S.; Migdall-Wilson, J.; Middleton, D.H.G.; Lu, X.; Barón, A.E.; Franklin, W.A.; Merrick, D.T.; et al. Mer or Axl Receptor Tyrosine Kinase Inhibition Promotes Apoptosis, Blocks Growth and Enhances Chemosensitivity of Human Non-Small Cell Lung Cancer. Oncogene 2013, 32, 3420–3431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demarchi, F.; Verardo, R.; Varnum, B.; Brancolini, C.; Schneider, C. Gas6 Anti-Apoptotic Signaling Requires NF-ΚB Activation. J. Biol. Chem. 2001, 276, 31738–31744. [Google Scholar] [CrossRef] [Green Version]
- Paccez, J.D.; Vasques, G.J.; Correa, R.G.; Vasconcellos, J.F.; Duncan, K.; Gu, X.; Bhasin, M.; Libermann, T.A.; Zerbini, L.F. The Receptor Tyrosine Kinase Axl Is an Essential Regulator of Prostate Cancer Proliferation and Tumor Growth and Represents a New Therapeutic Target. Oncogene 2013, 32, 689–698. [Google Scholar] [CrossRef] [Green Version]
- Goruppi, S.; Ruaro, E.; Varnum, B.; Schneider, C. Requirement of Phosphatidylinositol 3-Kinase-Dependent Pathway and Src for Gas6-Axl Mitogenic and Survival Activities in NIH 3T3 Fibroblasts. Mol. Cell. Biol. 1997, 17, 4442–4453. [Google Scholar] [CrossRef] [Green Version]
- Shinh, Y.-S.; Lai, C.-Y.; Kao, Y.-R.; Shiah, S.-G.; Chu, Y.-W.; Lee, H.-S.; Wu, C.-W. Expression of Axl in Lung Adenocarcinoma and Correlation with Tumor Progression. Neoplasia 2005, 7, 1058–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knubel, K.H.; Pernu, B.M.; Sufit, A.; Nelson, S.; Pierce, A.M.; Keating, A.K. MerTK Inhibition Is a Novel Therapeutic Approach for Glioblastoma Multiforme. Oncotarget 2014, 5, 1338–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlegel, J.; Sambade, M.J.; Sather, S.; Moschos, S.J.; Tan, A.-C.; Winges, A.; DeRyckere, D.; Carson, C.C.; Trembath, D.G.; Tentler, J.J.; et al. MERTK Receptor Tyrosine Kinase Is a Therapeutic Target in Melanoma. J. Clin. Investig. 2013, 123, 2257–2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davra, V.; Kumar, S.; Geng, K.; Calianese, D.; Mehta, D.; Gadiyar, V.; Kasikara, C.; Lahey, K.C.; Chang, Y.; Wichroski, M.; et al. Axl and Mertk Receptors Cooperate to Promote Breast Cancer Progression by Combined Oncogenic Signaling and Evasion of Host Antitumor Immunity. Cancer Res. 2021, 81, 698–712. [Google Scholar] [CrossRef]
- Jansen, F.H.; van Rijswijk, A.; Teubel, W.; van Weerden, W.M.; Reneman, S.; van den Bemd, G.-J.; Roobol, M.J.; Bangma, C.H.; Staal, F.J.T.; Jenster, G. Profiling of Antibody Production against Xenograft-Released Proteins by Protein Microarrays Discovers Prostate Cancer Markers. J. Proteome Res. 2012, 11, 728–735. [Google Scholar] [CrossRef]
- Ben-Batalla, I.; Schultze, A.; Wroblewski, M.; Erdmann, R.; Heuser, M.; Waizenegger, J.S.; Riecken, K.; Binder, M.; Schewe, D.; Sawall, S.; et al. Axl, a Prognostic and Therapeutic Target in Acute Myeloid Leukemia Mediates Paracrine Crosstalk of Leukemia Cells with Bone Marrow Stroma. Blood 2013, 122, 2443–2452. [Google Scholar] [CrossRef]
- Schmitz, R.; Valls, A.F.; Yerbes, R.; von Richter, S.; Kahlert, C.; Loges, S.; Weitz, J.; Schneider, M.; de Almodovar, C.R.; Ulrich, A.; et al. TAM Receptors Tyro3 and Mer as Novel Targets in Colorectal Cancer. Oncotarget 2016, 7, 56355–56370. [Google Scholar] [CrossRef] [Green Version]
- Goyette, M.-A.; Duhamel, S.; Aubert, L.; Pelletier, A.; Savage, P.; Thibault, M.-P.; Johnson, R.M.; Carmeliet, P.; Basik, M.; Gaboury, L.; et al. The Receptor Tyrosine Kinase AXL Is Required at Multiple Steps of the Metastatic Cascade during HER2-Positive Breast Cancer Progression. Cell Rep. 2018, 23, 1476–1490. [Google Scholar] [CrossRef]
- Onken, J.; Torka, R.; Korsing, S.; Radke, J.; Krementeskaia, I.; Nieminen, M.; Bai, X.; Ullrich, A.; Heppner, F.; Vajkoczy, P. Inhibiting Receptor Tyrosine Kinase AXL with Small Molecule Inhibitor BMS-777607 Reduces Glioblastoma Growth, Migration, and Invasion in Vitro and in Vivo. Oncotarget 2016, 7, 9876–9889. [Google Scholar] [CrossRef] [Green Version]
- Uribe, D.J.; Mandell, E.K.; Watson, A.; Martinez, J.D.; Leighton, J.A.; Ghosh, S.; Rothlin, C.V. The Receptor Tyrosine Kinase AXL Promotes Migration and Invasion in Colorectal Cancer. PLoS ONE 2017, 12, e0179979. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Zhang, Z.; Miao, L.; Yang, Z.; Yang, J.; Wang, Y.; Qian, D.; Cai, H.; Wang, Y. Anexelekto (AXL) Increases Resistance to EGFR-TKI and Activation of AKT and ERK1/2 in Non-Small Cell Lung Cancer Cells. Oncol. Res. 2016, 24, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Giles, K.M.; Kalinowski, F.C.; Candy, P.A.; Epis, M.R.; Zhang, P.M.; Redfern, A.D.; Stuart, L.M.; Goodall, G.J.; Leedman, P.J. Axl Mediates Acquired Resistance of Head and Neck Cancer Cells to the Epidermal Growth Factor Receptor Inhibitor Erlotinib. Mol. Cancer Ther. 2013, 12, 2541–2558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, K.F.; Du, W.; Sorrelle, N.B.; Wnuk-Lipinska, K.; Topalovski, M.; Toombs, J.E.; Cruz, V.H.; Yabuuchi, S.; Rajeshkumar, N.V.; Maitra, A.; et al. Small-Molecule Inhibition of Axl Targets Tumor Immune Suppression and Enhances Chemotherapy in Pancreatic Cancer. Cancer Res. 2018, 78, 246–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, D.; Parker, R.E.; Wang, X.; Frye, S.V.; Earp, H.S.; DeRyckere, D.; Graham, D.K. MERTK Promotes Resistance to Irreversible EGFR Tyrosine Kinase Inhibitors in Non–Small Cell Lung Cancers Expressing Wild-Type EGFR Family Members. Clin. Cancer Res. 2018, 24, 6523–6535. [Google Scholar] [CrossRef] [Green Version]
- Gjerdrum, C.; Tiron, C.; Høiby, T.; Stefansson, I.; Haugen, H.; Sandal, T.; Collett, K.; Li, S.; McCormack, E.; Gjertsen, B.T.; et al. Axl Is an Essential Epithelial-to-Mesenchymal Transition-Induced Regulator of Breast Cancer Metastasis and Patient Survival. Proc. Natl. Acad. Sci. USA 2010, 107, 1124–1129. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL Kinase Causes Resistance to EGFR-Targeted Therapy in Lung Cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef]
- Thomson, S.; Petti, F.; Sujka-Kwok, I.; Mercado, P.; Bean, J.; Monaghan, M.; Seymour, S.L.; Argast, G.M.; Epstein, D.M.; Haley, J.D. A Systems View of Epithelial–Mesenchymal Transition Signaling States. Clin. Exp. Metastasis 2011, 28, 137–155. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, M.; Horikoshi, Y.; Nakaso, K.; Kurashiki, T.; Kitagawa, Y.; Hanaki, T.; Sakamoto, T.; Honjo, S.; Umekita, Y.; Fujiwara, Y.; et al. Oncogenic Role of TYRO3 Receptor Tyrosine Kinase in the Progression of Pancreatic Cancer. Cancer Lett. 2020, 470, 149–160. [Google Scholar] [CrossRef]
- Lu, Q.; Lemke, G. Homeostatic Regulation of the Immune System by Receptor Tyrosine Kinases of the Tyro 3 Family. Science 2001, 293, 306–311. [Google Scholar] [CrossRef]
- Carrera Silva, E.A.; Chan, P.Y.; Joannas, L.; Errasti, A.E.; Gagliani, N.; Bosurgi, L.; Jabbour, M.; Perry, A.; Smith-Chakmakova, F.; Mucida, D.; et al. T Cell-Derived Protein S Engages TAM Receptor Signaling in Dendritic Cells to Control the Magnitude of the Immune Response. Immunity 2013, 39, 160–170. [Google Scholar] [CrossRef] [Green Version]
- Lemke, G.; Rothlin, C.V. Immunobiology of the TAM Receptors. Nat. Rev. Immunol. 2008, 8, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.A.; Lemke, G. TAM Receptors Are Pleiotropic Inhibitors of the Innate Immune Response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Q.; Gore, M.; Zhang, Q.; Camenisch, T.; Boast, S.; Casagranda, F.; Lai, C.; Skinner, M.K.; Klein, R.; Matsushima, G.K.; et al. Tyro-3 Family Receptors Are Essential Regulators of Mammalian Spermatogenesis. Nature 1999, 398, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.L.; Caricchio, R.; Abraham, V.; Camenisch, T.D.; Jennette, J.C.; Roubey, R.A.S.; Earp, H.S.; Matsushima, G.; Reap, E.A. Delayed Apoptotic Cell Clearance and Lupus-like Autoimmunity in Mice Lacking the c-Mer Membrane Tyrosine Kinase. J. Exp. Med. 2002, 196, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Tirado-Gonzalez, I.; Descot, A.; Soetopo, D.; Nevmerzhitskaya, A.; Schäffer, A.; Kur, I.-M.; Czlonka, E.; Wachtel, C.; Tsoukala, I.; Müller, L.; et al. AXL Inhibition in Macrophages Stimulates Host-versus-Leukemia Immunity and Eradicates Naïve and Treatment-Resistant Leukemia. Cancer Discov. 2021, 11, 2924–2943. [Google Scholar] [CrossRef]
- Zhou, Y.; Fei, M.; Zhang, G.; Liang, W.-C.; Lin, W.; Wu, Y.; Piskol, R.; Ridgway, J.; McNamara, E.; Huang, H.; et al. Blockade of the Phagocytic Receptor MerTK on Tumor-Associated Macrophages Enhances P2X7R-Dependent STING Activation by Tumor-Derived CGAMP. Immunity 2020, 52, 357–373.e9. [Google Scholar] [CrossRef]
- Paolino, M.; Choidas, A.; Wallner, S.; Pranjic, B.; Uribesalgo, I.; Loeser, S.; Jamieson, A.M.; Langdon, W.Y.; Ikeda, F.; Fededa, J.P.; et al. The E3 Ligase Cbl-b and TAM Receptors Regulate Cancer Metastasis via Natural Killer Cells. Nature 2014, 507, 508–512. [Google Scholar] [CrossRef]
- Giroud, P.; Renaudineau, S.; Gudefin, L.; Calcei, A.; Menguy, T.; Rozan, C.; Mizrahi, J.; Caux, C.; Duong, V.; Valladeau-Guilemond, J. Expression of TAM-R in Human Immune Cells and Unique Regulatory Function of MerTK in IL-10 Production by Tolerogenic DC. Front. Immunol. 2020, 11, 564133. [Google Scholar] [CrossRef]
- Cook, R.S.; Jacobsen, K.M.; Wofford, A.M.; DeRyckere, D.; Stanford, J.; Prieto, A.L.; Redente, E.; Sandahl, M.; Hunter, D.M.; Strunk, K.E.; et al. MerTK Inhibition in Tumor Leukocytes Decreases Tumor Growth and Metastasis. J. Clin. Investig. 2013, 123, 3231–3242. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, T.A.; Rafat, M.; Castellini, L.; Shehade, H.; Kariolis, M.S.; Hui, A.B.-Y.; Stehr, H.; von Eyben, R.; Jiang, D.; Ellies, L.G.; et al. Reprogramming the Immunological Microenvironment through Radiation and Targeting Axl. Nat. Commun. 2016, 7, 13898. [Google Scholar] [CrossRef] [Green Version]
- Peeters, M.J.W.; Dulkeviciute, D.; Draghi, A.; Ritter, C.; Rahbech, A.; Skadborg, S.K.; Seremet, T.; Carnaz Simões, A.M.; Martinenaite, E.; Halldórsdóttir, H.R.; et al. MERTK Acts as a Costimulatory Receptor on Human CD8+ T Cells. Cancer Immunol. Res. 2019, 7, 1472–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantl, W.J.; Johnson, D.E.; Williams, L.T. Signalling by Receptor Tyrosine Kinases. Annu. Rev. Biochem. 1993, 62, 453–481. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, A.; Schlessinger, J. Signal Transduction by Receptors with Tyrosine Kinase Activity. Cell 1990, 61, 203–212. [Google Scholar] [CrossRef]
- Lew, E.D.; Oh, J.; Burrola, P.G.; Lax, I.; Zagórska, A.; Través, P.G.; Schlessinger, J.; Lemke, G. Differential TAM Receptor–Ligand–Phospholipid Interactions Delimit Differential TAM Bioactivities. eLife 2014, 3, e03385. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Ling, L.; Templeton, D.; Kung, H.-J. Identification of the Major Autophosphorylation Sites of Nyk/Mer, an NCAM-Related Receptor Tyrosine Kinase. J. Biol. Chem. 1996, 271, 18355–18362. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Wei, Y.; Wei, X. AXL Receptor Tyrosine Kinase as a Promising Anti-Cancer Approach: Functions, Molecular Mechanisms and Clinical Applications. Mol. Cancer 2019, 18, 153. [Google Scholar] [CrossRef] [Green Version]
- Braunger, J.; Schleithoff, L.; Schulz, A.S.; Kessler, H.; Lammers, R.; Ullrich, A.; Bartram, C.R.; Janssen, J.W. Intracellular Signaling of the Ufo/Axl Receptor Tyrosine Kinase Is Mediated Mainly by a Multi-Substrate Docking-Site. Oncogene 1997, 14, 2619–2631. [Google Scholar] [CrossRef] [Green Version]
- Auyez, A.; Sayan, A.E.; Kriajevska, M.; Tulchinsky, E. AXL Receptor in Cancer Metastasis and Drug Resistance: When Normal Functions Go Askew. Cancers 2021, 13, 4864. [Google Scholar] [CrossRef]
- Bellosta, P.; Costa, M.; Lin, D.A.; Basilico, C. The Receptor Tyrosine Kinase ARK Mediates Cell Aggregation by Homophilic Binding. Mol. Cell. Biol. 1995, 15, 614–625. [Google Scholar] [CrossRef] [Green Version]
- Burchert, A.; Attar, E.C.; McCloskey, P.; Fridell, Y.-W.C.; Liu, E.T. Determinants for Transformation Induced by the Axl Receptor Tyrosine Kinase. Oncogene 1998, 16, 3177–3187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korshunov, V.A. Axl-Dependent Signalling: A Clinical Update. Clin. Sci. 2012, 122, 361–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierce, A.; Bliesner, B.; Xu, M.; Nielsen-Preiss, S.; Lemke, G.; Tobet, S.; Wierman, M.E. Axl and Tyro3 Modulate Female Reproduction by Influencing Gonadotropin-Releasing Hormone Neuron Survival and Migration. Mol. Endocrinol. 2008, 22, 2481–2495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seitz, H.M.; Camenisch, T.D.; Lemke, G.; Earp, H.S.; Matsushima, G.K. Macrophages and Dendritic Cells Use Different Axl/Mertk/Tyro3 Receptors in Clearance of Apoptotic Cells. J. Immunol. 2007, 178, 5635–5642. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.E.; Krodel, M.; Pazos, M.; Lai, C.; Prieto, A.L. Cross-Phosphorylation, Signaling and Proliferative Functions of the Tyro3 and Axl Receptors in Rat2 Cells. PLoS ONE 2012, 7, e36800. [Google Scholar] [CrossRef] [Green Version]
- Vouri, M.; Croucher, D.R.; Kennedy, S.P.; An, Q.; Pilkington, G.J.; Hafizi, S. Axl-EGFR Receptor Tyrosine Kinase Hetero-Interaction Provides EGFR with Access to pro-Invasive Signalling in Cancer Cells. Oncogenesis 2016, 5, e266. [Google Scholar] [CrossRef] [Green Version]
- McShane, L.; Tabas, I.; Lemke, G.; Kurowska-Stolarska, M.; Maffia, P. TAM Receptors in Cardiovascular Disease. Cardiovasc. Res. 2019, 115, 1286–1295. [Google Scholar] [CrossRef] [Green Version]
- Mudduluru, G.; Ceppi, P.; Kumarswamy, R.; Scagliotti, G.V.; Papotti, M.; Allgayer, H. Regulation of Axl Receptor Tyrosine Kinase Expression by MiR-34a and MiR-199a/b in Solid Cancer. Oncogene 2011, 30, 2888–2899. [Google Scholar] [CrossRef]
- Kurowska-Stolarska, M.; Alivernini, S.; Melchor, E.G.; Elmesmari, A.; Tolusso, B.; Tange, C.; Petricca, L.; Gilchrist, D.S.; Di Sante, G.; Keijzer, C.; et al. MicroRNA-34a Dependent Regulation of AXL Controls the Activation of Dendritic Cells in Inflammatory Arthritis. Nat. Commun. 2017, 8, 15877. [Google Scholar] [CrossRef]
- Thorp, E.; Vaisar, T.; Subramanian, M.; Mautner, L.; Blobel, C.; Tabas, I. Shedding of the Mer Tyrosine Kinase Receptor Is Mediated by ADAM17 Protein through a Pathway Involving Reactive Oxygen Species, Protein Kinase Cδ, and P38 Mitogen-Activated Protein Kinase (MAPK). J. Biol. Chem. 2011, 286, 33335–33344. [Google Scholar] [CrossRef] [Green Version]
- Merilahti, J.A.M.; Ojala, V.K.; Knittle, A.M.; Pulliainen, A.T.; Elenius, K. Genome-Wide Screen of Gamma-Secretase–Mediated Intramembrane Cleavage of Receptor Tyrosine Kinases. MBoC 2017, 28, 3123–3131. [Google Scholar] [CrossRef] [PubMed]
- Orme, J.J.; Du, Y.; Vanarsa, K.; Mayeux, J.; Li, L.; Mutwally, A.; Arriens, C.; Min, S.; Hutcheson, J.; Davis, L.S.; et al. Heightened Cleavage of Axl Receptor Tyrosine Kinase by ADAM Metalloproteases May Contribute to Disease Pathogenesis in SLE. Clin. Immunol. 2016, 169, 58–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sather, S.; Kenyon, K.D.; Lefkowitz, J.B.; Liang, X.; Varnum, B.C.; Henson, P.M.; Graham, D.K. A Soluble Form of the Mer Receptor Tyrosine Kinase Inhibits Macrophage Clearance of Apoptotic Cells and Platelet Aggregation. Blood 2007, 109, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Weinger, J.G.; Omari, K.M.; Marsden, K.; Raine, C.S.; Shafit-Zagardo, B. Up-Regulation of Soluble Axl and Mer Receptor Tyrosine Kinases Negatively Correlates with Gas6 in Established Multiple Sclerosis Lesions. Am. J. Pathol. 2009, 175, 283–293. [Google Scholar] [CrossRef] [Green Version]
- Gal, A.; Li, Y.; Thompson, D.A.; Weir, J.; Orth, U.; Jacobson, S.G.; Apfelstedt-Sylla, E.; Vollrath, D. Mutations in MERTK, the Human Orthologue of the RCS Rat Retinal Dystrophy Gene, Cause Retinitis Pigmentosa. Nat. Genet. 2000, 26, 270–271. [Google Scholar] [CrossRef]
- D’Cruz, P.M. Mutation of the Receptor Tyrosine Kinase Gene Mertk in the Retinal Dystrophic RCS Rat. Hum. Mol. Genet. 2000, 9, 645–651. [Google Scholar] [CrossRef] [Green Version]
- Gay, C.M.; Balaji, K.; Byers, L.A. Giving AXL the Axe: Targeting AXL in Human Malignancy. Br. J. Cancer 2017, 116, 415–423. [Google Scholar] [CrossRef]
- Niederst, M.J.; Engelman, J.A. Bypass Mechanisms of Resistance to Receptor Tyrosine Kinase Inhibition in Lung Cancer. Sci. Signal. 2013, 6, re6. [Google Scholar] [CrossRef] [Green Version]
- Manfioletti, G.; Brancolini, C.; Avanzi, G.; Schneider, C. The Protein Encoded by a Growth Arrest-Specific Gene (Gas6) Is a New Member of the Vitamin K-Dependent Proteins Related to Protein S, a Negative Coregulator in the Blood Coagulation Cascade. Mol. Cell. Biol. 1993, 13, 10. [Google Scholar]
- Nagata, K.; Ohashi, K.; Nakano, T.; Arita, H.; Zong, C.; Hanafusa, H.; Mizuno, K. Identification of the Product of Growth Arrest-Specific Gene 6 as a Common Ligand for Axl, Sky, and Mer Receptor Tyrosine Kinases. J. Biol. Chem. 1996, 271, 30022–30027. [Google Scholar] [CrossRef] [Green Version]
- Mark, M.R.; Chen, J.; Hammonds, R.G.; Sadick, M.; Godowsk, P.J. Characterization of Gas6, a Member of the Superfamily of G Domain-Containing Proteins, as a Ligand for Rse and Axl. J. Biol. Chem. 1996, 271, 9785–9789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zagórska, A.; Través, P.G.; Lew, E.D.; Dransfield, I.; Lemke, G. Diversification of TAM Receptor Tyrosine Kinase Function. Nat. Immunol. 2014, 15, 920–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadahiro, H.; Kang, K.-D.; Gibson, J.T.; Minata, M.; Yu, H.; Shi, J.; Chhipa, R.; Chen, Z.; Lu, S.; Simoni, Y.; et al. Activation of the Receptor Tyrosine Kinase AXL Regulates the Immune Microenvironment in Glioblastoma. Cancer Res. 2018, 78, 3002–3013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uehara, S.; Fukuzawa, Y.; Matuyama, T.; Gotoh, K. Role of Tyro3, Axl, and Mer Receptors and Their Ligands (Gas6, and Protein S) in Patients with Hepatocellular Carcinoma. JCT 2017, 8, 112–130. [Google Scholar] [CrossRef] [Green Version]
- Davra, V.; Kimani, S.; Calianese, D.; Birge, R. Ligand Activation of TAM Family Receptors-Implications for Tumor Biology and Therapeutic Response. Cancers 2016, 8, 107. [Google Scholar] [CrossRef] [PubMed]
- Uehara, H.; Shacter, E. Auto-Oxidation and Oligomerization of Protein S on the Apoptotic Cell Surface Is Required for Mer Tyrosine Kinase-Mediated Phagocytosis of Apoptotic Cells. J. Immunol. 2008, 180, 2522–2530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemke, G. Biology of the TAM Receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009076. [Google Scholar] [CrossRef]
- Vouri, M.; An, Q.; Birt, M.; Pilkington, G.J.; Hafizi, S. Small Molecule Inhibition of Axl Receptor Tyrosine Kinase Potently Suppresses Multiple Malignant Properties of Glioma Cells. Oncotarget 2015, 6, 16183–16197. [Google Scholar] [CrossRef] [Green Version]
- Di Stasi, R.; De Rosa, L.; D’Andrea, L.D. Therapeutic Aspects of the Axl/Gas6 Molecular System. Drug Discov. Today 2020, 25, 2130–2148. [Google Scholar] [CrossRef]
- Wu, X.; Ma, W.; Zhou, Q.; Yan, H.; Lim, Z.-F.; Huang, M.; Deng, C.; Yu, X.; Su, H.; Komo, S.; et al. AXL–GAS6 Expression Can Predict for Adverse Prognosis in Non-Small Cell Lung Cancer with Brain Metastases. J. Cancer Res. Clin. Oncol. 2017, 143, 1947–1957. [Google Scholar] [CrossRef] [Green Version]
- Mao, S.; Wu, Y.; Wang, R.; Guo, Y.; Bi, D.; Ma, W.; Zhang, W.; Zhang, J.; Yan, Y.; Yao, X. Overexpression of GAS6 Promotes Cell Proliferation and Invasion in Bladder Cancer by Activation of the PI3K/AKT Pathway. OTT 2020, 13, 4813–4824. [Google Scholar] [CrossRef] [PubMed]
- Buehler, M.; Tse, B.; Leboucq, A.; Jacob, F.; Caduff, R.; Fink, D.; Goldstein, D.R.; Heinzelmann-Schwarz, V. Meta-Analysis of Microarray Data Identifies GAS6 Expression as an Independent Predictor of Poor Survival in Ovarian Cancer. BioMed Res. Int. 2013, 2013, 238284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitman, S.P.; Kohlschmidt, J.; Maharry, K.; Volinia, S.; Mrózek, K.; Nicolet, D.; Schwind, S.; Becker, H.; Metzeler, K.H.; Mendler, J.H.; et al. GAS6 Expression Identifies High-Risk Adult AML Patients: Potential Implications for Therapy. Leukemia 2014, 28, 1252–1258. [Google Scholar] [CrossRef] [Green Version]
- Cardone, C.; Liguori, G.; Troiani, T.; Nappi, A.; Amoroso, N.; Iaffaioli, V.R.; Romano, C.; Botti, G.; Vitagliano, D.; Martini, G.; et al. Expression of Axl Receptor and Its Ligand Gas6 in Colorectal Cancer (Crc). Ann. Oncol. 2014, 25, iv73. [Google Scholar] [CrossRef]
- Loges, S.; Schmidt, T.; Tjwa, M.; van Geyte, K.; Lievens, D.; Lutgens, E.; Vanhoutte, D.; Borgel, D.; Plaisance, S.; Hoylaerts, M.; et al. Malignant Cells Fuel Tumor Growth by Educating Infiltrating Leukocytes to Produce the Mitogen Gas6. Blood 2010, 115, 2264–2273. [Google Scholar] [CrossRef] [Green Version]
- Al Kafri, N.; Hafizi, S. Tumour-Secreted Protein S (ProS1) Activates a Tyro3-Erk Signalling Axis and Protects Cancer Cells from Apoptosis. Cancers 2019, 11, 1843. [Google Scholar] [CrossRef] [Green Version]
- Ubil, E.; Caskey, L.; Holtzhausen, A.; Hunter, D.; Story, C.; Earp, H.S. Tumor-Secreted Pros1 Inhibits Macrophage M1 Polarization to Reduce Antitumor Immune Response. J. Clin. Investig. 2018, 128, 2356–2369. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.J.; Mydel, P.; Benedyk-Machaczka, M.; Kamińska, M.; Kalucka, U.; Blø, M.; Furriol, J.; Gausdal, G.; Lorens, J.; Osman, T.; et al. AXL Targeting by a Specific Small Molecule or Monoclonal Antibody Inhibits Renal Cell Carcinoma Progression in an Orthotopic Mice Model. Physiol. Rep. 2021, 9, e15140. [Google Scholar] [CrossRef]
- Leconet, W.; Larbouret, C.; Chardès, T.; Thomas, G.; Neiveyans, M.; Busson, M.; Jarlier, M.; Radosevic-Robin, N.; Pugnière, M.; Bernex, F.; et al. Preclinical Validation of AXL Receptor as a Target for Antibody-Based Pancreatic Cancer Immunotherapy. Oncogene 2014, 33, 5405–5414. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Liu, R.; Ma, B.; Li, X.; Yen, H.; Zhou, Y.; Krasnoperov, V.; Xia, Z.; Zhang, X.; Bove, A.M.; et al. Axl Receptor Tyrosine Kinase Is a Potential Therapeutic Target in Renal Cell Carcinoma. Br. J. Cancer 2015, 113, 616–625. [Google Scholar] [CrossRef] [Green Version]
- Leconet, W.; Chentouf, M.; du Manoir, S.; Chevalier, C.; Sirvent, A.; Aït-Arsa, I.; Busson, M.; Jarlier, M.; Radosevic-Robin, N.; Theillet, C.; et al. Therapeutic Activity of Anti-AXL Antibody against Triple-Negative Breast Cancer Patient-Derived Xenografts and Metastasis. Clin. Cancer Res. 2017, 23, 2806–2816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Y.; Luo, L.; Qiao, C.; Li, X.; Wang, J.; Liu, H.; Zhou, T.; Shen, B.; Lv, M.; Feng, J. A Novel Human Anti-AXL Monoclonal Antibody Attenuates Tumour Cell Migration. Scand. J. Immunol. 2019, 90, e12777. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Li, Y.; Stawicki, S.; Couto, S.; Eastham-Anderson, J.; Kallop, D.; Weimer, R.; Wu, Y.; Pei, L. An Anti-Axl Monoclonal Antibody Attenuates Xenograft Tumor Growth and Enhances the Effect of Multiple Anticancer Therapies. Oncogene 2010, 29, 5254–5264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarado, D.; Vitale, L.; Murphy, M.; O’Neill, T.; Natoli, E.; Lillquist, J.; Crew, L.; Wasiuk, A.; Weidlick, J.; Sisson, C.; et al. 550 An Axl-Targeting Monoclonal Antibody That Inhibits Axl Activity and Potently Stimulates the Innate Immune Response. J. ImmunoTherapy of Cancer 2020, 8, A334.1. [Google Scholar]
- Chien, C.-W.; Hou, P.-C.; Wu, H.-C.; Chang, Y.-L.; Lin, S.-C.; Lin, S.-C.; Lin, B.-W.; Lee, J.-C.; Chang, Y.-J.; Sun, H.S.; et al. Targeting TYRO3 Inhibits Epithelial–Mesenchymal Transition and Increases Drug Sensitivity in Colon Cancer. Oncogene 2016, 35, 5872–5881. [Google Scholar] [CrossRef]
- Takeda, S.; Andreu-Agullo, C.; Sridhar, S.; Halberg, N.; Lorenz, I.C.; Tavazoie, S.; Kurth, I.; Tavazoie, M. Abstract LB-277: Characterization of the Anti-Cancer and Immunologic Activity of RGX-019, a Novel Pre-Clinical Stage Humanized Monoclonal Antibody Targeting the MERTK Receptor. Cancer Res. 2019, 79, LB-277. [Google Scholar] [CrossRef]
- Ballantine, L.; Midgley, A.; Harris, D.; Richards, E.; Burgess, S.; Beresford, M.W. Increased Soluble Phagocytic Receptors SMer, STyro3 and SAxl and Reduced Phagocytosis in Juvenile-Onset Systemic Lupus Erythematosus. Pediatr. Rheumatol. 2015, 13, 10. [Google Scholar] [CrossRef] [Green Version]
- Kariolis, M.S.; Miao, Y.R.; Diep, A.; Nash, S.E.; Olcina, M.M.; Jiang, D.; Jones, D.S.; Kapur, S.; Mathews, I.I.; Koong, A.C.; et al. Inhibition of the GAS6/AXL Pathway Augments the Efficacy of Chemotherapies. J. Clin. Investig. 2016, 127, 183–198. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Hu, B.; Qiao, C.; Luo, L.; Li, X.; Wang, J.; Liu, H.; Zhou, T.; Shen, B.; Lv, M.; et al. Engineered AXL-ECD-Fc Variants That Abolish the AXL/Gas6 Interaction Suppress Tumor Cell Migration. Oncol. Lett. 2019, 17, 5784–5792. [Google Scholar] [CrossRef] [Green Version]
- Kariolis, M.S.; Miao, Y.R.; Jones, D.S.; Kapur, S.; Mathews, I.I.; Giaccia, A.J.; Cochran, J.R. An Engineered Axl “decoy Receptor” Effectively Silences the Gas6-Axl Signaling Axis. Nat. Chem. Biol. 2014, 10, 977–983. [Google Scholar] [CrossRef]
- Kimani, S.G.; Kumar, S.; Bansal, N.; Singh, K.; Kholodovych, V.; Comollo, T.; Peng, Y.; Kotenko, S.V.; Sarafianos, S.G.; Bertino, J.R.; et al. Small Molecule Inhibitors Block Gas6-Inducible TAM Activation and Tumorigenicity. Sci. Rep. 2017, 7, 43908. [Google Scholar] [CrossRef] [PubMed]
- Kirane, A.; Ludwig, K.F.; Sorrelle, N.; Haaland, G.; Sandal, T.; Ranaweera, R.; Toombs, J.E.; Wang, M.; Dineen, S.P.; Micklem, D.; et al. Warfarin Blocks Gas6-Mediated Axl Activation Required for Pancreatic Cancer Epithelial Plasticity and Metastasis. Cancer Res. 2015, 75, 3699–3705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tie, J.-K.; Stafford, D.W. Functional Study of the Vitamin K Cycle Enzymes in Live Cells. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2017; Volume 584, pp. 349–394. ISBN 978-0-12-812213-6. [Google Scholar]
- Linger, R.M.A.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM Receptor Tyrosine Kinases: Biologic Functions, Signaling, and Potential Therapeutic Targeting in Human Cancer. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2008; Volume 100, pp. 35–83. ISBN 978-0-12-374358-9. [Google Scholar]
- Liu, M.-H.; Chen, S.-B.; Yu, J.; Liu, C.-J.; Zhang, X.-J. Promiscuity and Selectivity of Small-Molecule Inhibitors across TAM Receptor Tyrosine Kinases in Pediatric Leukemia. J. Mol. Graph. Model. 2017, 75, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Baladi, T.; Abet, V.; Piguel, S. State-of-the-Art of Small Molecule Inhibitors of the TAM Family: The Point of View of the Chemist. Eur. J. Med. Chem. 2015, 105, 220–237. [Google Scholar] [CrossRef]
- Zhang, Y.-X.; Knyazev, P.G.; Cheburkin, Y.V.; Sharma, K.; Knyazev, Y.P.; Őrfi, L.; Szabadkai, I.; Daub, H.; Kéri, G.; Ullrich, A. AXL Is a Potential Target for Therapeutic Intervention in Breast Cancer Progression. Cancer Res. 2008, 68, 1905–1915. [Google Scholar] [CrossRef] [Green Version]
- Remsing Rix, L.L.; Rix, U.; Colinge, J.; Hantschel, O.; Bennett, K.L.; Stranzl, T.; Müller, A.; Baumgartner, C.; Valent, P.; Augustin, M.; et al. Global Target Profile of the Kinase Inhibitor Bosutinib in Primary Chronic Myeloid Leukemia Cells. Leukemia 2009, 23, 477–485. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive Analysis of Kinase Inhibitor Selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef]
- Mori, M.; Kaneko, N.; Ueno, Y.; Yamada, M.; Tanaka, R.; Saito, R.; Shimada, I.; Mori, K.; Kuromitsu, S. Gilteritinib, a FLT3/AXL Inhibitor, Shows Antileukemic Activity in Mouse Models of FLT3 Mutated Acute Myeloid Leukemia. Investig. New Drugs 2017, 35, 556–565. [Google Scholar] [CrossRef] [Green Version]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a Novel MET and VEGFR2 Inhibitor, Simultaneously Suppresses Metastasis, Angiogenesis, and Tumor Growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [Green Version]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J.; et al. R428, a Selective Small Molecule Inhibitor of Axl Kinase, Blocks Tumor Spread and Prolongs Survival in Models of Metastatic Breast Cancer. Cancer Res. 2010, 70, 1544–1554. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.J.; Tran-Dubé, M.; Shen, H.; Nambu, M.; Kung, P.-P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.; et al. Structure Based Drug Design of Crizotinib (PF-02341066), a Potent and Selective Dual Inhibitor of Mesenchymal–Epithelial Transition Factor (c-MET) Kinase and Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [Google Scholar] [CrossRef] [PubMed]
- Karaman, M.W.; Herrgard, S.; Treiber, D.K.; Gallant, P.; Atteridge, C.E.; Campbell, B.T.; Chan, K.W.; Ciceri, P.; Davis, M.I.; Edeen, P.T.; et al. A Quantitative Analysis of Kinase Inhibitor Selectivity. Nat. Biotechnol. 2008, 26, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Mollard, A.; Warner, S.L.; Call, L.T.; Wade, M.L.; Bearss, J.J.; Verma, A.; Sharma, S.; Vankayalapati, H.; Bearss, D.J. Design, Synthesis, and Biological Evaluation of a Series of Novel AXL Kinase Inhibitors. ACS Med. Chem. Lett. 2011, 2, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, G.M.; An, Y.; Cai, Z.-W.; Chen, X.-T.; Clark, C.; Cornelius, L.A.M.; Dai, J.; Gullo-Brown, J.; Gupta, A.; Henley, B.; et al. Discovery of N-(4-(2-Amino-3-Chloropyridin-4-Yloxy)-3-Fluorophenyl)-4-Ethoxy-1-(4-Fluorophenyl)-2-Oxo-1,2-Dihydropyridine-3-Carboxamide (BMS-777607), a Selective and Orally Efficacious Inhibitor of the Met Kinase Superfamily. J. Med. Chem. 2009, 52, 1251–1254. [Google Scholar] [CrossRef]
- Cui, X.; Zheng, X.; Jiang, J.; Tan, F.; Ding, L.; Hu, P. Simultaneous Determination of a Novel C-Met/AXL Dual-Target Small-Molecule Inhibitor BPI-9016M and Its Metabolites in Human Plasma by Liquid Chromatography-Tandem Mass Spectrometry: Application in a Pharmacokinetic Study in Chinese Advanced Solid Tumor Patients. J. Chromatogr. B 2017, 1068–1069, 33–40. [Google Scholar] [CrossRef]
- Jimbo, T.; Hatanaka, M.; Komatsu, T.; Taira, T.; Kumazawa, K.; Maeda, N.; Suzuki, T.; Ota, M.; Haginoya, N.; Isoyama, T.; et al. DS-1205b, a Novel Selective Inhibitor of AXL Kinase, Blocks Resistance to EGFR-Tyrosine Kinase Inhibitors in a Non-Small Cell Lung Cancer Xenograft Model. Oncotarget 2019, 10, 5152–5167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eder, J.P.; Shapiro, G.I.; Appleman, L.J.; Zhu, A.X.; Miles, D.; Keer, H.; Cancilla, B.; Chu, F.; Hitchcock-Bryan, S.; Sherman, L.; et al. A Phase I Study of Foretinib, a Multi-Targeted Inhibitor of c-Met and Vascular Endothelial Growth Factor Receptor 2. Clin. Cancer Res. 2010, 16, 3507–3516. [Google Scholar] [CrossRef] [Green Version]
- Yan, S.B.; Peek, V.L.; Ajamie, R.; Buchanan, S.G.; Graff, J.R.; Heidler, S.A.; Hui, Y.-H.; Huss, K.L.; Konicek, B.W.; Manro, J.R.; et al. LY2801653 Is an Orally Bioavailable Multi-Kinase Inhibitor with Potent Activity against MET, MST1R, and Other Oncoproteins, and Displays Anti-Tumor Activities in Mouse Xenograft Models. Investig. New Drugs 2013, 31, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Xi, N.; Zhang, Y.; Wang, Z.; Wu, Y.; Wang, T. Abstract 1755: CT053PTSA, a Novel c-MET and VEGFR2 Inhibitor, Potently Suppresses Angiogenesis and Tumor Growth. Cancer Res. 2014, 74, 1755. [Google Scholar] [CrossRef]
- Ruvolo, P.P.; Ma, H.; Ruvolo, V.R.; Zhang, X.; Mu, H.; Schober, W.; Hernandez, I.; Gallardo, M.; Khoury, J.D.; Cortes, J.; et al. Anexelekto/MER Tyrosine Kinase Inhibitor ONO-7475 Arrests Growth and Kills FMS-like Tyrosine Kinase 3-Internal Tandem Duplication Mutant Acute Myeloid Leukemia Cells by Diverse Mechanisms. Haematologica 2017, 102, 2048–2057. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, Y.; Lew, E.D.; Seelige, R.; Tindall, E.A.; Walsh, C.; Fagan, P.C.; Lee, J.Y.; Nevarez, R.; Oh, J.; Tucker, K.D.; et al. Immuno-Oncological Efficacy of RXDX-106, a Novel TAM (TYRO3, AXL, MER) Family Small-Molecule Kinase Inhibitor. Cancer Res. 2019, 79, 1996–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patwardhan, P.P.; Ivy, K.S.; Musi, E.; de Stanchina, E.; Schwartz, G.K. Significant Blockade of Multiple Receptor Tyrosine Kinases by MGCD516 (Sitravatinib), a Novel Small Molecule Inhibitor, Shows Potent Anti-Tumor Activity in Preclinical Models of Sarcoma. Oncotarget 2016, 7, 4093–4109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, J.; Chong, C.; Goon, L.; Balayan, J.; Wu, S.; Johnson, E.; Lorenzana, G.; Bannen, L.; Nguyen, L.; Scheffold, C.; et al. XL092, a Multi-Targeted Inhibitor of MET, VEGFR2, AXL and MER with an Optimized Pharmacokinetic Profile. Eur. J. Cancer 2020, 138, S16. [Google Scholar] [CrossRef]
- Minson, K.A.; Smith, C.C.; DeRyckere, D.; Libbrecht, C.; Lee-Sherick, A.B.; Huey, M.G.; Lasater, E.A.; Kirkpatrick, G.D.; Stashko, M.A.; Zhang, W.; et al. The MERTK/FLT3 Inhibitor MRX-2843 Overcomes Resistance-Conferring FLT3 Mutations in Acute Myeloid Leukemia. JCI Insight 2016, 1, e85630. [Google Scholar] [CrossRef] [PubMed]
- Burbridge, M.F.; Bossard, C.J.; Saunier, C.; Fejes, I.; Bruno, A.; Léonce, S.; Ferry, G.; Da Violante, G.; Bouzom, F.; Cattan, V.; et al. S49076 Is a Novel Kinase Inhibitor of MET, AXL, and FGFR with Strong Preclinical Activity Alone and in Association with Bevacizumab. Mol. Cancer Ther. 2013, 12, 1749–1762. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Siemann, D.W. Therapeutic Targeting of the Gas6/Axl Signaling Pathway in Cancer. Int. J. Mol. Sci. 2021, 22, 9953. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, W.; Liu, J.; He, J.; Cao, R.; Chen, X.; Peng, X.; Xu, H.; Zhao, Q.; Zhong, J.; et al. Therapeutic Activity of DCC-2036, a Novel Tyrosine Kinase Inhibitor, against Triple-negative Breast Cancer Patient-derived Xenografts by Targeting AXL/MET. Int. J. Cancer 2019, 144, 651–664. [Google Scholar] [CrossRef] [Green Version]
- Lai, S.; Li, R.; Raha, P.; Hu, Y.; Yan, J.; Zhang, H.; Marotta, A.; Zhang, Z. Abstract B148: Activity of the TAM Kinase-Targeting Compound, SLC-391, Is Mediated by the Engagement of the Immune System in CT-26 Syngeneic Mouse Model. Mol. Cancer Ther. 2018, 17, B148. [Google Scholar] [CrossRef]
- Rios-Doria, J.; Favata, M.; Lasky, K.; Feldman, P.; Lo, Y.; Yang, G.; Stevens, C.; Wen, X.; Sehra, S.; Katiyar, K.; et al. A Potent and Selective Dual Inhibitor of AXL and MERTK Possesses Both Immunomodulatory and Tumor-Targeted Activity. Front. Oncol. 2020, 10, 598477. [Google Scholar] [CrossRef]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An Epithelial–Mesenchymal Transition Gene Signature Predicts Resistance to EGFR and PI3K Inhibitors and Identifies Axl as a Therapeutic Target for Overcoming EGFR Inhibitor Resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.D.; Park, E.J.; Choi, E.K.; Song, S.Y.; Hoe, K.-L.; Kim, D.-U. G-749 Promotes Receptor Tyrosine Kinase TYRO3 Degradation and Induces Apoptosis in Both Colon Cancer Cell Lines and Xenograft Mouse Models. Front. Pharmacol. 2021, 12, 730241. [Google Scholar] [CrossRef] [PubMed]
- Suárez, R.M.; Chevot, F.; Cavagnino, A.; Saettel, N.; Radvanyi, F.; Piguel, S.; Bernard-Pierrot, I.; Stoven, V.; Legraverend, M. Inhibitors of the TAM Subfamily of Tyrosine Kinases: Synthesis and Biological Evaluation. Eur. J. Med. Chem. 2013, 61, 2–25. [Google Scholar] [CrossRef] [PubMed]
- Rho, J.K.; Choi, Y.J.; Kim, S.Y.; Kim, T.W.; Choi, E.K.; Yoon, S.-J.; Park, B.M.; Park, E.; Bae, J.H.; Choi, C.-M.; et al. MET and AXL Inhibitor NPS-1034 Exerts Efficacy against Lung Cancer Cells Resistant to EGFR Kinase Inhibitors Because of MET or AXL Activation. Cancer Res. 2014, 74, 253–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIver, A.L.; Zhang, W.; Liu, Q.; Jiang, X.; Stashko, M.A.; Nichols, J.; Miley, M.J.; Norris-Drouin, J.; Machius, M.; DeRyckere, D.; et al. Discovery of Macrocyclic Pyrimidines as MerTK-Specific Inhibitors. Chem. Med. Chem. 2017, 12, 207–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; McIver, A.L.; Stashko, M.A.; DeRyckere, D.; Branchford, B.R.; Hunter, D.; Kireev, D.; Miley, M.J.; Norris-Drouin, J.; Stewart, W.M.; et al. Discovery of Mer Specific Tyrosine Kinase Inhibitors for the Treatment and Prevention of Thrombosis. J. Med. Chem. 2013, 56, 9693–9700. [Google Scholar] [CrossRef] [Green Version]
- Fujino, N.; Kubo, H.; Maciewicz, R.A. Phenotypic Screening Identifies Axl Kinase as a Negative Regulator of an Alveolar Epithelial Cell Phenotype. Lab. Investig. 2017, 97, 1047–1062. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, W.; Stashko, M.A.; DeRyckere, D.; Cummings, C.T.; Hunter, D.; Yang, C.; Jayakody, C.N.; Cheng, N.; Simpson, C.; et al. UNC1062, a New and Potent Mer Inhibitor. Eur. J. Med. Chem. 2013, 65, 83–93. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; DeRyckere, D.; Hunter, D.; Liu, J.; Stashko, M.A.; Minson, K.A.; Cummings, C.T.; Lee, M.; Glaros, T.G.; Newton, D.L.; et al. UNC2025, a Potent and Orally Bioavailable MER/FLT3 Dual Inhibitor. J. Med. Chem. 2014, 57, 7031–7041. [Google Scholar] [CrossRef] [Green Version]
- Powell, N.A.; Hoffman, J.K.; Ciske, F.L.; Kohrt, J.T.; Baxi, S.M.; Peng, Y.-W.; Zhong, M.; Catana, C.; Ohren, J.; Perrin, L.A.; et al. Optimization of Highly Selective 2,4-Diaminopyrimidine-5-Carboxamide Inhibitors of Sky Kinase. Bioorganic Med. Chem. Lett. 2013, 23, 1051–1055. [Google Scholar] [CrossRef]
- Vijayan, R.S.K.; He, P.; Modi, V.; Duong-Ly, K.C.; Ma, H.; Peterson, J.R.; Dunbrack, R.L.; Levy, R.M. Conformational Analysis of the DFG-Out Kinase Motif and Biochemical Profiling of Structurally Validated Type II Inhibitors. J. Med. Chem. 2015, 58, 466–479. [Google Scholar] [CrossRef] [Green Version]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-Targeted Cancer Therapies: Progress, Challenges and Future Directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Kannaiyan, R.; Mahadevan, D. A Comprehensive Review of Protein Kinase Inhibitors for Cancer Therapy. Expert Rev. Anticancer Ther. 2018, 18, 1249–1270. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Classification of Small Molecule Protein Kinase Inhibitors Based upon the Structures of Their Drug-Enzyme Complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef]
- Myers, K.V.; Amend, S.R.; Pienta, K.J. Targeting Tyro3, Axl and MerTK (TAM Receptors): Implications for Macrophages in the Tumor Microenvironment. Mol. Cancer 2019, 18, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imran, M.; Asdaq, S.M.B.; Khan, S.A.; Unnikrishnan Meenakshi, D.; Alamri, A.S.; Alsanie, W.F.; Alhomrani, M.; Mohzari, Y.; Alrashed, A.; AlMotairi, M.; et al. Innovations and Patent Trends in the Development of USFDA Approved Protein Kinase Inhibitors in the Last Two Decades. Pharmaceuticals 2021, 14, 710. [Google Scholar] [CrossRef] [PubMed]
- Feneyrolles, C.; Spenlinhauer, A.; Guiet, L.; Fauvel, B.; Daydé-Cazals, B.; Warnault, P.; Chevé, G.; Yasri, A. Axl Kinase as a Key Target for Oncology: Focus on Small Molecule Inhibitors. Mol. Cancer Ther. 2014, 13, 2141–2148. [Google Scholar] [CrossRef] [Green Version]
- Myers, S.H.; Brunton, V.G.; Unciti-Broceta, A. AXL Inhibitors in Cancer: A Medicinal Chemistry Perspective: Miniperspective. J. Med. Chem. 2016, 59, 3593–3608. [Google Scholar] [CrossRef] [Green Version]
- McDaniel, N.K.; Cummings, C.T.; Iida, M.; Hülse, J.; Pearson, H.E.; Vasileiadi, E.; Parker, R.E.; Orbuch, R.A.; Ondracek, O.J.; Welke, N.B.; et al. MERTK Mediates Intrinsic and Adaptive Resistance to AXL-Targeting Agents. Mol. Cancer Ther. 2018, 17, 2297–2308. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of Human Triple-Negative Breast Cancer Subtypes and Preclinical Models for Selection of Targeted Therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, E.; Martini, G.; Cardone, C.; Troiani, T.; Liguori, G.; Vitagliano, D.; Napolitano, S.; Morgillo, F.; Rinaldi, B.; Melillo, R.M.; et al. AXL Is an Oncotarget in Human Colorectal Cancer. Oncotarget 2015, 6, 23281–23296. [Google Scholar] [CrossRef] [Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Christoph, S.; DeRyckere, D.; Schlegel, J.; Frazer, J.K.; Batchelor, L.A.; Trakhimets, A.Y.; Sather, S.; Hunter, D.M.; Cummings, C.T.; Liu, J.; et al. UNC569, a Novel Small-Molecule Mer Inhibitor with Efficacy against Acute Lymphoblastic Leukemia In Vitro and In Vivo. Mol. Cancer Ther. 2013, 12, 2367–2377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandao, L.N.; Winges, A.; Christoph, S.; Sather, S.; Migdall-Wilson, J.; Schlegel, J.; McGranahan, A.; Gao, D.; Liang, X.; DeRyckere, D.; et al. Inhibition of MerTK Increases Chemosensitivity and Decreases Oncogenic Potential in T-Cell Acute Lymphoblastic Leukemia. Blood Cancer J. 2013, 3, e101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, D.K.; Salzberg, D.B.; Kurtzberg, J.; Sather, S.; Matsushima, G.K.; Keating, A.K.; Liang, X.; Lovell, M.A.; Williams, S.A.; Dawson, T.L.; et al. Ectopic Expression of the Proto-Oncogene Mer in Pediatric T-Cell Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2006, 12, 2662–2669. [Google Scholar] [CrossRef] [Green Version]
- Summers, R.J.; Ryan, J.; Minson, K.A.; Frye, S.V.; Wang, X.; Earp, H.S., III.; DeRyckere, D.; Graham, D.K. Mertk Is a Therapeutic Target in Early T-Precursor Acute Lymphoblastic Leukemia. Blood 2017, 130, 2536. [Google Scholar]
- Tormoen, G.W.; Blair, T.C.; Bambina, S.; Kramer, G.; Baird, J.; Rahmani, R.; Holland, J.M.; McCarty, O.J.T.; Baine, M.J.; Verma, V.; et al. Targeting MerTK Enhances Adaptive Immune Responses After Radiation Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2020, 108, 93–103. [Google Scholar] [CrossRef]
- Lin, J.-Z.; Wang, Z.-J.; De, W.; Zheng, M.; Xu, W.-Z.; Wu, H.-F.; Armstrong, A.; Zhu, J.-G. Targeting AXL Overcomes Resistance to Docetaxel Therapy in Advanced Prostate Cancer. Oncotarget 2017, 8, 41064–41077. [Google Scholar] [CrossRef]
- Taniguchi, H.; Yamada, T.; Wang, R.; Tanimura, K.; Adachi, Y.; Nishiyama, A.; Tanimoto, A.; Takeuchi, S.; Araujo, L.H.; Boroni, M.; et al. AXL Confers Intrinsic Resistance to Osimertinib and Advances the Emergence of Tolerant Cells. Nat. Commun. 2019, 10, 259. [Google Scholar] [CrossRef]
- Xie, S.; Li, Y.; Li, X.; Wang, L.; Yang, N.; Wang, Y.; Wei, H. Mer receptor tyrosine kinase is frequently overexpressed in human non-small cell lung cancer, confirming resistance to erlotinib. Oncotarget 2015, 6, 9206–9219. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Bach, D.-H.; Fan, Y.-H.; Luu, T.-T.-T.; Hong, J.-Y.; Park, H.J.; Lee, S.K. AXL Degradation in Combination with EGFR-TKI Can Delay and Overcome Acquired Resistance in Human Non-Small Cell Lung Cancer Cells. Cell Death Dis. 2019, 10, 361. [Google Scholar] [CrossRef] [Green Version]
- Elkabets, M.; Pazarentzos, E.; Juric, D.; Sheng, Q.; Pelossof, R.A.; Brook, S.; Benzaken, A.O.; Rodon, J.; Morse, N.; Yan, J.J.; et al. AXL Mediates Resistance to PI3Kα Inhibition by Activating the EGFR/PKC/MTOR Axis in Head and Neck and Esophageal Squamous Cell Carcinomas. Cancer Cell 2015, 27, 533–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terry, S.; Abdou, A.; Engelsen, A.S.T.; Buart, S.; Dessen, P.; Corgnac, S.; Collares, D.; Meurice, G.; Gausdal, G.; Baud, V.; et al. AXL Targeting Overcomes Human Lung Cancer Cell Resistance to NK- and CTL-Mediated Cytotoxicity. Cancer Immunol. Res. 2019, 7, 1789–1802. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; Dalban, C.; Rioux-Leclercq, N.; Adam, J.; Meylan, M.; Buart, S.; Bougoüin, A.; Lespagnol, A.; Dugay, F.; Moreno, I.C.; et al. Association of AXL and PD-L1 Expression with Clinical Outcomes in Patients with Advanced Renal Cell Carcinoma Treated with PD-1 Blockade. Clin. Cancer Res. 2021, 27, 6749–6760. [Google Scholar] [CrossRef] [PubMed]
- Krebs, M.; Brunsvig, P.; Helland, Å.; Viñolas, N.; Aix, S.; Carcereny, E.; Gomez, M.D.; Perez, J.M.T.; Arriola, E.; Campelo, R.G.; et al. P1.01-72 A Phase II Study of Selective AXL Inhibitor Bemcentinib and Pembrolizumab in Patients with NSCLC Refractory to Anti-PD(L)1. J. Thorac. Oncol. 2019, 14, S388. [Google Scholar] [CrossRef]
- Oliva, M.; Chepeha, D.; Araujo, D.V.; Diaz-Mejia, J.J.; Olson, P.; Prawira, A.; Spreafico, A.; Bratman, S.V.; Shek, T.; de Almeida, J.; et al. Antitumor Immune Effects of Preoperative Sitravatinib and Nivolumab in Oral Cavity Cancer: SNOW Window-of-Opportunity Study. J. Immunother. Cancer 2021, 9, e003476. [Google Scholar] [CrossRef] [PubMed]
- Spicer, J.; Helland, Å.; Carcereny, E.; Arriola, E.; Gomez, M.D.; Trigo Perez, J.M.; Thompson, J.; Strauss, J.; Ortega Granados, A.L.; Felip, E.; et al. 362 A PhII Study of Bemcentinib, a First-in-Class Selective AXL Kinase Inhibitor, in Combination with Pembrolizumab in Pts with Previously-Treated Advanced NSCLC: Updated Clinical & Translational Analysis. Christie Sch. Oncol. 2020, A221. [Google Scholar]
- Colavito, S.A. AXL as a Target in Breast Cancer Therapy. J. Oncol. 2020, 2020, 5291952. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Xu, X.S.; Yang, J.X.; Guo, J.H.; Chao, T.F.; Tong, Y. The Prognostic Role of Gas6/Axl Axis in Solid Malignancies: A Meta-Analysis and Literature Review. OTT 2018, 11, 509–519. [Google Scholar] [CrossRef] [Green Version]
- Shiozawa, Y.; Pedersen, E.A.; Taichman, R.S. GAS6/Mer Axis Regulates the Homing and Survival of the E2A/PBX1-Positive B-Cell Precursor Acute Lymphoblastic Leukemia in the Bone Marrow Niche. Exp. Hematol. 2010, 38, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Schoumacher, M.; Burbridge, M. Key Roles of AXL and MER Receptor Tyrosine Kinases in Resistance to Multiple Anticancer Therapies. Curr. Oncol. Rep. 2017, 19, 19. [Google Scholar] [CrossRef] [Green Version]
- Caraux, A.; Lu, Q.; Fernandez, N.; Riou, S.; Di Santo, J.P.; Raulet, D.H.; Lemke, G.; Roth, C. Natural Killer Cell Differentiation Driven by Tyro3 Receptor Tyrosine Kinases. Nat. Immunol. 2006, 7, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Nandrot, E.F.; Dufour, E.M. Mertk in Daily Retinal Phagocytosis: A History in the Making. In Retinal Degenerative Diseases; Advances in Experimental Medicine and Biology; Anderson, R.E., Hollyfield, J.G., LaVail, M.M., Eds.; Springer: New York, NY, USA, 2010; Volume 664, pp. 133–140. ISBN 978-1-4419-1398-2. [Google Scholar]
- Inoue, S.; Yamane, Y.; Tsukamoto, S.; Azuma, H.; Nagao, S.; Murai, N.; Nishibata, K.; Fukushima, S.; Ichikawa, K.; Nakagawa, T.; et al. Discovery of a Potent and Selective Axl Inhibitor in Preclinical Model. Bioorganic Med. Chem. 2021, 39, 116137. [Google Scholar] [CrossRef] [PubMed]
- Vollrath, D.; Yasumura, D.; Benchorin, G.; Matthes, M.T.; Feng, W.; Nguyen, N.M.; Sedano, C.D.; Calton, M.A.; LaVail, M.M. Tyro3 Modulates Mertk-Associated Retinal Degeneration. PLoS Genet. 2015, 11, e1005723. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Inhibitor | Status 1 | Core | Type | Inhibitory Parameters | References |
|---|---|---|---|---|---|
| Bosutinib; SKI-606; PF5208763; Bosulif | APPROVED | A | I | AXL IC50 = 174 nM MERTK IC50 = 110 nM TYRO3 IC50 = 61 nM | [117,118,119] |
| Gilteritinib; ASP2215; Xospata | APPROVED | A | I | AXL IC50 = 0.73 nM MERTK IC50 = 5 nM | [120] |
| Vandetanib; ZD6474; Caprelsa | APPROVED | A | I | AXL IC50 = 250 nM MERTK IC50 = 1400 nM TYRO3 IC50 = 93 nM | [119] |
| Cabozantinib; XL 184; BMS-907351; Cabometyx | APPROVED | B | II | AXL IC50 = 7 nM | [121] |
| BGB324; R428; Bemcentinib | APPROVED | X | I | AXL IC50 = 14 nM | [122] |
| Crizotinib PF-02341066; Xalkori | APPROVED | X | I | AXL IC50 = 294 nM | [123] |
| Sunitinib; SU 11248; Sutent | APPROVED | X | I | AXL IC50 = 9 nM | [124] |
| TP-0903; Dubermatinib | CLINICAL TRIALS NCT04518345 | A | I | AXL IC50 = 27 nM | [125] |
| BMS777607; ASLAN002 | CLINICAL TRIALS NCT01721148 NCT00605618 | B | II | AXL IC50 = 1.1 nM MERTK IC50 = 14 nM TYRO3 IC50 = 4.3 nM | [126] |
| BPI-9016M | CLINICAL TRIALS NCT02929290 NCT02478866 | B | II | AXL IC50 = 9 nM | [127] |
| DS-1205b/c | CLINICAL TRIALS NCT03599518 NCT03255083 (TERMINATED) | B | II | AXL IC50 = 1.3 nM MERTK IC50 = 63 nM | [128] |
| Foretinib; XL880; GSK1363089 | CLINICAL TRIALS NCT00920192 NCT01147484 NCT01138384 NCT00742131 NCT00725764 NCT00726323 NCT00725712 NCT00743067 NCT01068587 | B | II | AXL IC50 = 11 nM | [129] |
| Merestinib; LY2801653 | CLINICAL TRIALS NCT03125239 NCT03027284 NCT02779738 NCT02745769 | B | II | AXL IC50 = 2 nM MERTK IC50 = 10 nM | [125,130] |
| MGCD265; Glesatinib | CLINICAL TRIALS NCT02954991 | B | II | n/a | |
| Ningetinib; CT053PTSA | CLINICAL TRIALS NCT04577703 NCT03758287 | B | II | AXL IC50 < 1.0 nM | [131] |
| ONO-7475 | CLINICAL TRIALS NCT03176277 | B | II | AXL IC50 = 0.7 nM MERTK IC50 = 1 nM TYRO3 IC50 = 1.9 nM | [132] |
| PF-07265807; ARRY-067; PF-5807 | CLINICAL TRIALS NCT04458259 | B | II | n/a | |
| RXDX-106; CEP-40783 | CLINICAL TRIALS NCT03454243 (TERMINATED) | B | II | AXL IC50 = 0.31 nM MERTK IC50 = 1.89 nM TYRO3 IC50 = 3.5 nM | [133] |
| Sitravatinib; MGCD516 | CLINICAL TRIALS NCT04123704 NCT03575598 NCT04472650 NCT04772612 NCT04921358 NCT04727996 NCT04800614 NCT04904302 NCT04925986 NCT05176925 NCT05104801 NCT05255276 NCT04887194 NCT04734262 NCT02954991 NCT03606174 NCT04887870 | B | II | AXL IC50 = 1.5 nM MERTK IC50 = 2 nM | [134] |
| XL092 | CLINICAL TRIALS NCT03845166 NCT05176483 | B | II | AXL IC50 = 3.4 nM MERTK IC50 = 7.2 nM | [135] |
| Amuvatinib; MP470 | CLINICAL TRIALS NCT01357395 NCT00894894 NCT00881166 | X | I | AXL IC50 = 10 nM | [57] |
| MRX-2843; UNC2371 | CLINICAL TRIALS NCT03510104 NCT04762199 NCT04872478 | X | I | AXL IC50 = 15 nM MERTK IC50 = 1.3 nM TYRO3 IC50 = 17 nM | [136] |
| S49076 | CLINICAL TRIALS ISRCTN11619481 | X | I | AXL IC50 = 7 nM MERTK IC50 = 2 nM | [137] |
| SNS314 | CLINICAL TRIALS NCT00519662 | X | I | AXL IC50 = 84 nM | [138] |
| Rebastinib; DCC-2036 | CLINICAL TRIALS NCT00827138 | X | II | AXL IC50 = 42 nM | [139] |
| SLC-391 | CLINICAL TRIALS NCT05278845 NCT03990454 | X | n/a | AXL IC50 = 9.6 nM MERTK IC50 = 42.3 nM TYRO3 IC50 = 44 nM | [140] |
| INCB081776 | CLINICAL TRIALS NCT03522142 | n/a | n/a | AXL IC50 = 0.61 nM MERTK IC50 = 3.17 nM TYRO3 IC50 = 101 nM | [141] |
| Q702 | CLINICAL TRIALS NCT04648254 | n/a | n/a | n/a | |
| SGI7079 | PRECLINICAL | A | I | AXL IC50 = 58 nM | [142] |
| SK-G-801; G-801 | PRECLINICAL | A | I | AXL IC50 = 20 nM | [143] |
| 6g; purine analogue of BMS777607 | PRECLINICAL | B | II | AXL IC50 = 39 nM MERTK IC50 = 42 nM TYRO3 IC50 = 65 nM | [144] |
| LDC1267 | PRECLINICAL | B | II | AXL IC50 = 29 nM MERTK IC50 = 5 nM TYRO3 IC50 = 8 nM | [47] |
| NPS-1034 | PRECLINICAL | B | II | AXL IC50 = 10.3 nM | [145] |
| TAM-IN-2 | PRECLINICAL | B | II | n/a | |
| UNC2541 | PRECLINICAL | B | II | MERTK IC50 = 4.4 nM | [146] |
| UNC2881 | PRECLINICAL | B | II | AXL IC50 = 360 nM MERTK IC50 = 4.3 nM TYRO3 IC50 = 250 nM | [147] |
| 2-D08 | PRECLINICAL | X | I | AXL IC50 = 0.49 nM | [148] |
| UNC1062 | PRECLINICAL | X | I | AXL IC50 = 85 nM MERTK IC50 = 1.1 nM TYRO3 IC50 = 60 nM | [23,149] |
| UNC2025 | PRECLINICAL | X | I | AXL IC50 = 14 nM MERTK IC50 = 0.7 nM TYRO3 IC50 = 18 nM | [150] |
| Compound 19 | PRECLINICAL | X | n/a | TYRO3 IC50 = 10 nM | [151] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikolajczyk, A.; Mitula, F.; Popiel, D.; Kaminska, B.; Wieczorek, M.; Pieczykolan, J. Two-Front War on Cancer—Targeting TAM Receptors in Solid Tumour Therapy. Cancers 2022, 14, 2488. https://doi.org/10.3390/cancers14102488
Mikolajczyk A, Mitula F, Popiel D, Kaminska B, Wieczorek M, Pieczykolan J. Two-Front War on Cancer—Targeting TAM Receptors in Solid Tumour Therapy. Cancers. 2022; 14(10):2488. https://doi.org/10.3390/cancers14102488
Chicago/Turabian StyleMikolajczyk, Agata, Filip Mitula, Delfina Popiel, Bozena Kaminska, Maciej Wieczorek, and Jerzy Pieczykolan. 2022. "Two-Front War on Cancer—Targeting TAM Receptors in Solid Tumour Therapy" Cancers 14, no. 10: 2488. https://doi.org/10.3390/cancers14102488
APA StyleMikolajczyk, A., Mitula, F., Popiel, D., Kaminska, B., Wieczorek, M., & Pieczykolan, J. (2022). Two-Front War on Cancer—Targeting TAM Receptors in Solid Tumour Therapy. Cancers, 14(10), 2488. https://doi.org/10.3390/cancers14102488