Abstract
Hepatic encephalopathy (HE) is a severe metabolic syndrome linked with acute/chronic hepatic disorders. HE is also a pernicious neuropsychiatric complication associated with cognitive decline, coma, and death. Limited therapies are available to treat HE, which is formidable to oversee in the clinic. Thus, determining a novel therapeutic approach is essential. The pathogenesis of HE has not been well established. According to various scientific reports, neuropathological symptoms arise due to excessive accumulation of ammonia, which is transported to the brain via the blood–brain barrier (BBB), triggering oxidative stress and inflammation, and disturbing neuronal-glial functions. The treatment of HE involves eliminating hyperammonemia by enhancing the ammonia scavenging mechanism in systemic blood circulation. Melatonin is the sole endogenous hormone linked with HE. Melatonin as a neurohormone is a potent antioxidant that is primarily synthesized and released by the brain’s pineal gland. Several HE and liver cirrhosis clinical studies have demonstrated impaired synthesis, secretion of melatonin, and circadian patterns. Melatonin can cross the BBB and is involved in various neuroprotective actions on the HE brain. Hence, we aim to elucidate how HE impairs brain functions, and elucidate the precise molecular mechanism of melatonin that reverses the HE effects on the central nervous system.
1. Introduction
The liver, which is a metabolic organ, is involved in detoxification, nutritional metabolism, maintenance of blood volume, and hormone regulation [1]. Hepatic disease and liver failure are the leading cause of death worldwide [2], and are involved in the development and pathogenesis of neurological illnesses [3]. Globally, 40% of liver cirrhosis cases transition to hepatic encephalopathy (HE) (also known as portosystemic encephalopathy (PSE)). HE is a severe metabolic disorder caused by end-stage liver disease [4] and associated with reversible neurological dysfunction ranging from personality changes to coma and death [5]. HE is categorized into two classes: 1. Covert HE/Minimal Hepatic encephalopathy (MHE), which is associated with neuropsychiatric symptoms, including alteration in mood, personality, memory, sleep, and motor coordination; and 2. Overt HE, which occurs when covert HE becomes chronic, causing a decline in the patient’s survival [6]. HE can also be classified into three types according to the causes: Type I, acute liver failure (ALF)-induced HE; Type II, Bypass shunts-induced HE; and Type III, Chronic liver disease-induced HE. Notably, HE does not have a single clinical symptom. HE may either be accompanied with reversible metabolic encephalopathy, atrophy, or edema in the brain [7].
The pathophysiology of HE is multifactorial and has not been clearly explained. Various in vivo and in vitro liver failure studies demonstrated that large amounts of ammonia crosses the blood–brain barrier (BBB), causing neuropathological disruptions, such as personality changes, altered cognition, locomotor ability, and consciousness [8]. Ammonia is the central metabolite and the principal neurotoxin related to HE [9]. Ammonia is synthesized in enterocytes from glutamine and metabolized by the liver [9]. According to previous studies, normal healthy individuals have 45 μM ammonia in arterial circulation [10]. The highest ammonia concentration is found in end-stage liver disease with irreversible brain damage (340 μM) [11]. In liver diseases, liver detoxification unexpectedly declines, causing hyperammonemia [9]. The circulatory ammonia enters into the brain and deposits in the brain and cerebrospinal fluid (CSF) [12]. The accumulated neurotoxin increases oxidative stress (OS) [13], generating proinflammatory cytokines [14], altering the synthesis and transmission of the neurotransmitters [15], impairing glucose and energy metabolism [16], and inducing astrocyte swelling [17] and brain edema [18] (Figure 1).
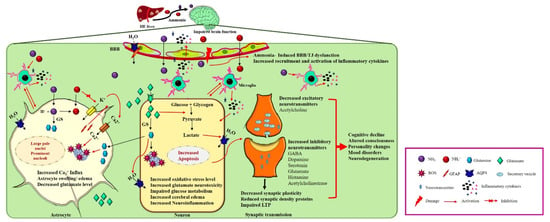
Figure 1.
Neuropathogenesis of HE on brain dysfunction. HE liver releases excess nitrogenous toxin (NH3, NH4+) that enters cerebral circulation. Ammonia can cross the BBB and trigger the other pathological response such as activation of aquaporin water channels and damage of BBB’s tight junctions. Astrocytes detoxify ammonia to form glutamine from glutamate by glutamine synthase (GS). Excess glutamine production increases oxidative stress, aquaporin channels’ activation, increases Ca2+ influx and GFAP production, and decreases glutamate uptake leading to accumulation of glutamate into the extracellular fluid. Activation of water channels, increased Ca2+ influx, and increased glutamine secretion cause astrocyte swelling. On the other hand, accumulated extracellular glutamate enters into neurons, causing glutamate neurotoxicity. Intracellular glutamate impairs glucose metabolism, activates microglial inflammatory cytokines, increases oxidative stress, and inhibits mitochondrial functions, leading to decrease in excitatory neurotransmitters synthesis and release into the synapses. In synaptic transmission increases synthesis and release of inhibitory neurotransmitters impairing the LTP, synaptic plasticity, and reducing synaptic density proteins, leading to cognitive decline and other neuropsychiatric illnesses.
A therapeutic approach for HE is currently emerging as an important issue. Thus, therapies that inhibit oxidative stress induced by hyperammonemia are markedly needed to inhibit neuronal damage caused by oxidative stress and enhance the prognosis of HE. Various HE clinical and experimental reports revealed several pharmacologic therapies, such as antibiotics [19] and nutritional supplements [20] for HE. However, other studies revealed the adverse effects of antibiotics in HE [21]. Currently, overt HE can only be treated, while covert/MHE does not have an appropriate therapeutic approach in modern medicine. Therefore, modern medicine focuses on the HE therapeutic agent that recovers the hepatic/neuronal functions with minimal adverse effects. Mainly, neurosteroids/endogenous hormones may demonstrate a neuroprotective action on the nervous system. These biological compounds diminish oxidative stress, inflammation, excitotoxicity, brain edema, and neurodegeneration [22]. Few endogenous hormones (estrogen, progesterone, and lipoic acid) are involved in neuroprotection [23]. Precisely, melatonin was found to exhibit a promising neuroprotective effect on HE [1]. Several patients with cirrhosis and HE have severely impaired melatonin metabolism [24], with altered melatonin secretion and circadian patterns. Melatonin is the sole endogenous hormone linked with HE. However, interpreting their pathophysiology remains finite, and their therapy is challenging. Here, we aim to recapitulate the multiple effects of melatonin in the HE brain.
2. Melatonin in the CNS
Melatonin (N-Acetyl-5-methoxytryptamine), a neurohormone, is known as an “internal synchronizer” involved in circadian rhythms [25]. Melatonin is synthesized in the pinealocytes and its derivatives are produced by the retina, astrocytes, kidney, lymphocytes, platelets, and skin [26]. Tryptophan is the vital precursor during melatonin synthesis, which is dependent on the light and dark cycle [27]. Hydroxylation and decarboxylation of tryptophan results in serotonin, and the acetylation of serotonin forms N-acetyl serotonin (NAS) by N-acetyltransferase [27]. Hydroxyindole-O-methyltransferase (HIOMT)/acetylserotonin methyltransferase then converts NAS to melatonin [27]. Melatonin synthesis merely relies on the precursors, enzyme availability, and seasonal and circadian rhythms [27]. Melatonin is a chronobiotic molecule that is not merely confined to circulation and augments to enable direct impacts in the central nervous system (CNS) [28]. Melatonin also acts as a circadian pacemaker, and this pleiotropic controller has numerous physiological roles, including in the sleep–wake cycle, neuro-immuno endocrine, and circadian rhythm [29,30]. Melatonin initiates the signaling pathway by binding to melatonin receptors (MT (1,2,3)) [31]. Both G-coupled transmembrane MT1 and MT2 receptors are predominantly located in the brain and other extra pineal tissues (liver, bone, and retina). MT3 is identified in the liver, kidneys, heart, adipose tissue, and brain. The activated MT receptors trigger various signaling and transcriptional pathways and act as a neuroprotective agent in various CNS disorders. These receptors are also involved in the pathology and chief drug target for CNS disorders. Melatonin can cross the BBB and protect against brain injury (neurodegenerative diseases, trauma, hypoxia, and HE) [32,33] by acting as a potent anti-inflammatory [34,35], anti-apoptotic [36], antioxidative [37,38], anti-tumor [30], anti-diabetic, anti-obese, neuroprotective, cardioprotective, and mood-stabilizing agent [28]. Collectively, melatonin has various potentials for treating both systemic pathology and neuropathology based on their characters (Figure 2).
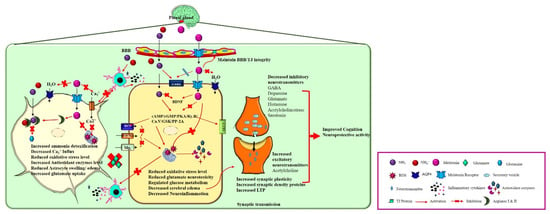
Figure 2.
Neuroprotective action of melatonin on HE brain. In the brain, melatonin is synthesized and released from pinealocytes of the pineal gland. Melatonin binds to its receptors and activates various physiological functions such as 1. In astrocytes: Melatonin detoxifies the excess ammonia by activating the Arginase I and II enzyme that prevents glutamine synthesis and glutamate accumulation in extracellular fluid. Furthermore, melatonin prevents neuroinflammation and astrocyte swelling by decreasing the Ca2+ influx and inhibiting water channel activation. 2. In a neuron, melatonin inhibits the cAMP/cGMP/PKA/Ry.R/Ca.V/GSK/PP-2A signaling pathway leading to the decreased oxidative stress level, inhibits the microglial activation, and reduces the inhibitory neurotransmitter synthesis and release. Moreover, melatonin regulates glucose metabolism by acting on insulin/GLUT receptors, facilitating synaptic plasticity, LTP, cognition by increasing the synaptic density proteins expression, and increasing the excitatory neurotransmitter release.
3. Hepatic Encephalopathy (HE) and Melatonin (Hyperammonemia)
The neuropathogenesis of HE remains unclear. The complications of HE include glutamine, chronic infections, and profuse gastrointestinal bleeding, and causes elevated ammonia levels in the blood and CNS [9]. According to the prevailing hypothesis of HE, gut-derived nitrogenous toxins of ammonia can cross the BBB and induce neurological symptoms [2]. The biochemical analysis in numerous clinical and experimental studies has confirmed increased circulatory ammonia levels in HE [13]. HE experimental models can also be created by increasing the ammonia level in blood circulation [39,40].
In this review, ammonia is defined as the concentration of both ammonia (NH3) and ammonium ion (NH4+). NH3 is a lipophilic compound that can cross the plasma membrane, while NH4+ is transported through ionic channels [41]. Ammonia is derived from all amino acids, nucleic acids, and renal glutamine. Ammonia is also produced by normal floral bacterial enzymes within the gastrointestinal tract (3–4 mg/day) [41] and is metabolized by bacterial enzymes in the gastrointestinal tract and in the liver via the urea cycle. A high level of ammonia crosses the BBB, which leads to oxidative stress, alters glucose and neurotransmitter metabolism, and disrupts of neuronal functions and structure, such as astrocyte swelling in HE [42].
Based on different studies, melatonin is a potent hepato-neuroprotector against hyperammonemia (Table 1). The liver is the principle organ involved in nitrogen homeostasis. Hepatic disease leads to impaired urea cycle, ammonia trafficking, and hyperammonemia [43]. In the urea cycle, ammonia is detoxified by five enzymes (arginase, argininosuccinate synthetase, argininosuccinate lyase, carbamyl phosphate synthetase I (CPS-I), and ornithine carbamyl transferase) [44]. Arginase is the final process enzyme that converts L-arginine to l-ornithine/urea to degrade the nitrogenous toxin of ammonia [45]. There are two types of arginase in mammals: 1. cytosolic arginase I, which is expressed in the liver (>98%); and 2. mitochondrial arginase II, which located in extrahepatic tissues (2%) (renal, brain, lung, intestine, and breast) [45]. According to Aydogdu et al., melatonin enhances arginase (I and II) expression and reduces the level of nitric oxide (NO) [46]. Another study revealed that melatonin reduces the metabolite accumulation end products, such as ornithine (Orn), homocitrulline (Hcit), and ammonia, in the urea cycle, owing to its antioxidant defense in hyperornithinemia–hyperammonemia–homocitrullinuria syndrome (HHH) [47]. Studies demonstrated that a high ammonia (>500 μM) level generates the free radical production in the cellular level [48]. However, hyperammonemia was found to alter mitochondrial functions by increasing the free radical production (LPO)/reactive oxygen species (ROS), decreasing adenosine triphosphate (ATP) synthesis [49] and disturbing cellular pH by reducing a-ketoglutarate [50].

Table 1.
Effects of melatonin on hyperammonemia.
Numerous in vivo and in vitro studies confirmed the antioxidant activity of melatonin on oxidative stress-induced damage [54]. In HE, the melatonin demonstrated antioxidative properties by inhibiting ammonia-induced free radical production [1,55,56]. In various oxidative stress markers, 3-nitrotyrosine is the main oxidative stress diagnostic marker (90% sensitivity and specificity) for MHE [57]. Hence, various scientific reports mentioned that melatonin inhibits the 3-nitrotyrosine generation induced by oxidative stress models [58,59]. Moreover, melatonin inhibits NO production by converting into NAS to reduce the oxidative stress [60]. As a result, these functions of melatonin on ammonia metabolism-related enzyme arginase and metabolites leads to the reduction of ammonia accumulation. Melatonin reduces the oxidative stress induced by hyperammonemia by generating antioxidants, and instantly scavenging ROS.
4. HE and Melatonin (Neuroinflammation and BBB Disruption)
HE is known to affect astrocyte dysfunction by making hyperammonemia toxicity [61]. In this review, we explain the paradigm of neuroglial communication, which is reconstructed by melatonin in HE. Several studies have affirmed that the accumulation of toxic metabolites alters cell signaling by facilitating the activation of microglia, neuroinflammation, and Alzheimer Type II astrocytosis, and plays an important key role in HE [61,62].
In the CNS, astrocytes impact the formation and maintenance of the BBB [63], and regulate cerebral blood flow, water channel expression [64], neurotransmitter release, and reuptake [65]. Microglia are immune cells that act as housekeeping factors and modulators of neuroinflammation [66]. Under physiological conditions, microglia monitor myelin homeostasis [67], synaptic activity, pathogen entry, and injury. However, under pathological conditions, microglia triggers neuroinflammation by increasing cytokines and chemokines [68]. Ammonia has multiple toxic impacts on cellular metabolisms, such as the production of free radicals by tricarboxylic acid (TCA) cycle enzymes, malate-aspartate shuttle, mitochondrial respiratory chain inhibition, and increase in glutamine to induce cell swelling [69]. These toxic attributes will be discussed later.
Astrocytes is a vital element of the BBB and regulates the arachidonic acid-dependent pathway to maintain cerebral blood flow (CBF) [70]. Astrocytes can also uptake and metabolize 7% of arterial ammonia [10]. Ammonia (NH3) crosses the BBB via passive diffusion to astrocytes [71]. In HE, the levels of blood ammonia, cytokines, transforming growth factor-beta (TGFβ1), tumor necrosis factor (TNF), matrix metalloproteinase 9 (MMP-9), and bile acids are increased [72]. Elevated MMP-9, TNF level, and bile acids impair the BBB’s tight junction (TJ) proteins, such as occludin and claudin-5 [72]. Damaged TJ allows the influx of ammonia. The accumulation of ammonia and bilirubin also reduces the BBB’s breast cancer resistance protein (BCRP) expression, which protects the brain from the toxin [72]. Astrocytes catalyze the glutamine formed from ammonia, which is converted to glutamate and NH4+ by glutamine synthetase [73]. In contrast, glutaminase converts glutamine to glutamate and stores it as a neurotransmitter in neurons for reuptake by astrocytes [71,74]. The osmolyte property of glutamine increases oxidative stress by activating mitochondrial pore transition in the mitochondria [75], and these factors are the main reason for astrocyte swelling and cerebral edema. Hyperammonemia over-activates the Na-K-2Cl cotransporter (NCCa-ATP) channel [76], increases ionic influx into the astrocyte, alters the water concentration gradient, and activates aquaporin 4 (AQP4) water channels [77], causing astrocyte swelling and brain edema. On the contrary, ammonia activates tryptophan metabolites, and induces ROS production, Ca2+ influx, NADPH oxidase, and mitochondrial pore transition [78] caused by oxidative stress. Finally, these mechanisms increase ROS and astrocyte senescence. ROS generation initiates p53 phosphorylation at serine 392 through mitogen-activated protein kinases (p38MAPK) [17,61]. Several studies confirmed that hyperammonemia activates the secretion of inflammatory cytokines (interleukin-6 (IL-6), interleukin-1beta (IL-1β), interferon gamma (IFN-γ), and tumor necrosis factor α (TNFα) in ammonia-induced astrocyte cultures [50]. These cytokines further activate nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) [79]. Similar to increased nitric oxide synthase (iNOS), IL-1β and hemoxygenase-1 (HO-1) were found to increase in ammonia-induced astrocyte cultures [80]. These findings indicate a direct connection between inflammatory cytokines, ROS, and ammonia in HE associated with astrocyte swelling and cerebral edema [61]. Thus, studies proved that astrocytes are crucial glial cells that link ammonia and inflammation by unlocking the BBB via an arachidonic acid-dependent mechanism [81]. Astrocytes demonstrated the pattern of Alzheimer’s Type II astrocytosis to have prominent nucleoli and large pale nuclei characters, whichare found in white and gray matter in the HE brain [62].
In astrocytes, lactate dehydrogenase (LDH)-1 and LDH-5 expression levels are markedly enhanced due to hyperammonemia [82]. Further, reduced glucose utilization causes ATP depletion and TCA cycle enzyme (α-ketoglutarate dehydrogenase) inhibition [9]. Therefore, excess deposition of lactate induces cytotoxic edema known as astrocyte swelling. A recent study hypothesized that the swelling of astrocytes is caused by glial fibrillary acidic protein (GFAP) in HE [65]. Langer et al. reported that the protein expression of GFAP is reduced in the ALF rat cortex [83]. GFAP reduction alters the visco-elastic nature of astrocytes, causing astrocyte swelling and brain edema. Other studies also reported that astrocyte swelling and brain edema are caused by the reduction of protein and gene expression of a water channel (aquaporin II), glucose transporter 1 (GLUT-1) [84], and GFAP [85] in HE.
Microglial activation is the second key factor for neuroinflammation in HE. In AHE/chronic hepatic disease, the increased level of ammonia, TCA, TGFβ1, and TNF interacts with neuronal receptors and increases C-C Motif Chemokine Ligand 2 (CCL2) production, which is followed by microglial activation [1]. Activated microglia can release proinflammatory cytokines (TNFα, IL-1α, IL-1β, and IL-6), other inflammatory markers (Toll-like receptor 4 (TLR4), OX-42, OX-46, CD11b), and numerous inflammatory signaling pathway factors (NF-kB, mitogen-activated protein kinase (MAPK) p53, and NO/cGMP pathway) [40], which are involved in neuropathogenesis-induced HE [61,86]. Alternatively, neuroinflammation can also be triggered by hyperammonia-induced oxidative stress within astrocytes and neurons [50]. Oxidative stress and cerebral edema alter the physiological functions of astrocytes [87] and inhibit neuronal-glial cell communication, leading to symptoms of HE [88].
Melatonin is a potent immunomodulator with diverse functions. Melatonin has a conceivable function in inhibiting the activation of the pro-inflammatory cytokines in the MAPK and NF-κB pathways [89]. An injection of melatonin reduces BBB permeability and brain edema in an in vivo and in vitro model [90]. Based on the cell requirement, melatonin acts as anti/pro-inflammatory agent and regulates immunological responses [91]. Melatonin exerts its anti-inflammatory activity by blocking iNOS, cyclooxygenase -2 (COX-2), and NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) expression [92,93]. According to Permpoonputtana et al., melatonin inhibits TNFα mRNA expression, phosphorylated p65 NF-κB, and nuclear factor erythroid-2-related factor 2 (Nrf2) in dopamine SH-SY5Y cell lines [94]. Melatonin also maintains the BBB integrity mediated via the TLR4/NF-κB-signaling pathway [95]. To sum up, melatonin acts as potent anti-inflammatory agent and maintains the BBB integrity by inhibiting the neuroinflammatory pathways (TLR4/NF-κB, MAPK pathways) and microglial activation, as well as maintaining the tight junction proteins’ integrity and inhibiting the astrocyte swelling brain edema by inactivating the ammonia-induced AQP4 channels.
5. HE and Melatonin (Neurotransmitters)
As there is evidence of glial activation and neuroinflammation in HE, neurotransmitters should be reviewed to understand the pathologies of HE. The major excitatory neurotransmitter related to HE is glutamate [13]. Previously, we explained the synthesis and metabolism of glutamate in astrocytes. Experimental HE studies revealed that glutamate’s release is increased in extracellular fluid and leads to hyperammonemia [96]. Ammonia directly influences glutamatergic neurotransmission [97]. Further, studies have suggested that hyperammonemia boosted the secretion of glutamate from astrocytes [98]. Astrocyte swelling has an impact on the release of glutamate by regulating a pH and Ca2+-dependent mechanism [99]. Ammonia affects the expression of N-methyl-D-aspartate (NMDA) receptors, and controls α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor-mediated currents [100]. Ammonia also decreases the depolarization caused by NMDA and AMPA receptors and reduces the production of inositol-3-phosphate (IP3) [101]. These findings indicate that ammonia has a direct impact on glutamatergic transmission in the neuronal cell synapse. The neuroprotective effect of melatonin against glutamate neurotoxicity has been demonstrated in various clinical and experimental studies via reduced NO production, decreased Ca2+ influx [102], and antioxidative signaling [103,104].
The level of gamma-aminobutyric acid (GABA) as an inhibitory neurotransmitter is increased in HE [100]. In fact, the increased GABAergic tone is the principal neuropathology of HE [100]. In HE, hyperammonemia increases GABA release and activates the peripheral benzodiazepine (PTBR) receptors [105]. PTBR and diazepam binding inhibitor (DBI) are increased in astrocytes and CSF [106]. Furthermore, the clinical report of HE comatose patients revealed that the upregulation of PTBR receptors increased astrocyte swelling [107,108] (II astrocytosis) [109]. Activated PTBR initiates the de novo synthesis of the neurosteroid/neuroinhibitor 3a-5a-tetrahydro-progesterone (Allopregnanolone) [110]. In the brain of the deceased HE coma patients’ brain, increased allopregnanolone was found, which increased the GABA-recruited chloride currents [62]. In contrast, acetylcholine inhibits the GABA receptor-mediated inhibitory effects. In HE, acetylcholinesterase levels are high, thereby catalyzing acetylcholine in the synaptic cleft [111,112]. To confirm these findings, one study has demonstrated that the administration of acetylcholine reverses is beneficial on the coma in HE patients [113]. The antagonist of these neurosteroids is the potential target for HE [22]. Numerous studies reported that melatonin inhibits the expression of GABA and acetylcholinestrase [114,115]. Claudia et al. revealed that melatonin receptors modulate the GABAergic system by inhibiting increased calcium accumulation, which activates GABA in xenopus tectal cells [116]. Cheng et al. reported that melatonin modulates the rat hippocampal GABAergic responses via benzodiazepine (BZ) receptors [117]. Huang et al. found that melatonin inhibited lateral hypothalamic GABAergic neurons via the inhibition of HCN ion channels [118]. Fernandez-Bachiller et al. mentioned that the melatonin hybrid interacts with the peripheral anionic site (PAS) of acetylcholinesterase, which modulates the acetylcholinesterase activity on acetylcholine [119].
Next, another vital neuromodulator in neuropathology is adenosine. Adenosine inhibits the release of postsynaptic neurotransmitters (glutamate, GABA, serotonin, and dopamine), and modulates neuronal excitability [120]. Studies confirmed that the adenosinergic mechanism is disturbed in HE compared to the normal brain [15]. Adenosine receptors (A1, A2A, A3) are downregulated in severe HE [15,100]. A1 receptor downregulation causes increased level of glutamate, leading to glutamate neurotoxicity [121]. The downregulation of A2 receptors increases GABA release, which is known as an increased GABAergic tone [122].
Furthermore, some reports have been published on the roles of monoamines in the HE brain [100]. The main monoamine involved in HE neuropathology is a serotonin. A disturbed serotonergic system is observed in many clinical HE conditions [123,124]. Research reports have revealed that melatonin increases serotonin synthesis, and increased serotonin facilitates for melatonin production [125,126]. In contrast, Agrawal et al. reported that melatonin inhibits the serotonin transporter function in the epithelial cells of the intestine. Thus, the melatonin and serotonin interconnection might be confusing the action on melatonin on the HE brain [127].
According previous studies, the levels of cerebral dopamine and its metabolite, homovanillinic acid, increases the HE brain [128,129,130,131]. Melatonin is used as an antidopaminergic agent in Parkinson’s disease (PD) [132]. Dopamine release is inhibited by melatonin, and is demonstrated in diverse brain regions such as the hippocampus, hypothalamus, pons-medulla, and retina [133]. Zisapal et al. mentioned that the dopaminergic pathway is modulated by melatonin and subsequently affects antioxidant responses and mitochondrial activity in PD patients [132]. Striatal BTZ receptors play a major role in controlling dopamine-related neuropathology [134]. A study suggests that melatonin alleviates PD symptoms such as dyskinesia by allosterically interacting with BZ and GABAA receptors [135]. Moreover, melatonin inhibits the cAMP production and significantly regulates the neuropathology induced by the D1 or D2 dopamine receptor agonists [136].
Melatonin inhibits the responses of postsynaptic NMDA-receptors to glutamate that modulates long-term potentiation (LTP) [132,133]. Based on another theory, melatonin receptors mediate dopamine/cAMP signaling, which modulates dopaminergic neurotransmission [137].
In addition, the activation of histamine and its precursors increased in HE [124,138]. These are the main causative factors for depression and sleep disturbances [100]. The link between histamine and melatonin regulates hormonal, neuronal, and behavioral activities [100].
Investigators have proposed the H1HR/CaV1.3/RyR and H1HR/Gβγ/cAMP/PKA/CFTR pathways, which mediate histamine and melatonin [139]. Silva et al. also confirmed that histamine-induced NO generation in endothelial cells was inhibited by melatonin [140]. From the above mentioned, melatonin regulates the neurotransmitter synthesis and secretion by acting on ionic channels (HCN, Ca2+, Mg2+, and GLUT), acting on receptor (PAS/NMDA/GABA/glutamate), and modulating the signaling pathways (cAMP/cGMP/PKA/Ry.R/GSK/PP-2A).
6. HE and Melatonin (Insulin Resistance)
In the CNS, the brain is the insulin-sensitive organ [141] that contains remarkable amounts of insulin binding receptors, especially in cerebral cortex, hypothalamus, and hippocampal post synaptic densities [142,143]. Insulin and glucose uptake regulate various neurophysiological functions, including neurogenesis, synaptic plasticity, and cognition [144]. Impaired insulin and glucose regulation lead to cognitive decline and the development of neurodegenerative diseases [145,146,147]. The liver, which is a metabolic organ, is involved in glucose metabolism (gluconeogenesis, glycogenesis, and glycolysis) [3]. Therefore, altered liver functions or liver diseases impair the glucose metabolism, and might cause glucose metabolic diseases such as diabetes mellitus. Diabetes and insulin resistance are interrelated with HE. In fact, studies have demonstrated that 96% of cirrhotic patients displayed glucose intolerance and 30% had type 2 diabetes (T2DM) [148].
Inconsistent scientific reports suggest that hyperammonemia impairs blood glucose and insulin secretion [149,150,151]. Elevated ammonia increased intermediate metabolites, such as nonesterified fatty acids, glucose, pyruvate, and a-ketoglutarate, and decreased glucose phosphate [152]. A generally known theory is that augmented glutamate dampens the TCA cycle of a-ketoglutarate to interrupt ATP and energy metabolism [153].
According to numerous reports, melatonin regulates insulin secretion by acting on carbohydrate and glucose metabolism [154,155]. Melatonin is also used as an anti-hyperglycemic agent for T2DM [154] and can maintain insulin secretion by acting on three signaling pathways in pancreatic beta cells: 1. MT1 receptor-mediated cAMP/PKA/Ca2+ pathway [155], 2. MT2 receptor-mediated cGMP/PKG/Ca2+ pathway [156], and 3. MT2 receptor-mediated PLC/IP3/ER/SR/Ca2+ or PLC/DAG/PKC/Ca2+ pathway [157,158]. Insulin binds to an extracellular insulin receptor (InsR), leading to intracellular β subunit autophosphorylation followed by the activation and phosphorylation of InsR substrate (IRS-1) elements [159]. Liu et al. revealed that 10 mg/kg/day of melatonin from the day of embryonic administration reduced the neural tube defects in embryos (e.g., exencephaly) by activating neural stem cell proliferation and inhibiting apoptosis regulated via the extracellular signal-regulated kinase (ERK) pathway [160]. Furthermore, a various genome-wide association study (GWAS) demonstrated that melatonin’s single nucleotide polymorphism (MTNR1B) is associated with hyperglycemia and T2DM [161,162,163].
These findings strongly suggest that melatonin regulates insulin and glucose metabolism, and may serve as the reason for reduced hyperglycemia-induced hyperammonemia in HE by modulating signaling pathways (cAMP/PKA/Ca2+/cGMP/PKC/Ca2+ pathway/PLC/DAG/PKC/Ca2+ pathway/ERK), and regulating glucose metabolism by acting on its respective receptors (GLUT/InsR).
Given these consequences, melatonin could improve insulin sensitivity and brain function in HE.
7. HE and Melatonin (Cognitive Function)
Several studies revealed that the main neuropathological symptoms of HE is a cognitive decline [4,61]. Melatonin has been found to promote cognition in both clinical and experimental studies (Table 2). Astrocyte senescence is intensely linked with oxidative stress and cognitive decline and is observed in HE [61]. Many studies revealed that ammonia inhibits astrocyte growth via arrest in the S-phase of the cell cycle [17]. Ammonia mainly upregulates SA-β-Gal, which is the diagnostic marker for senescence [164].
Table 2.
Effects of melatonin on HE with cognitive decline.
The mechanisms of astrocyte senescence have not been clearly elucidated; however, based on the main hypothesis, astrocyte senescence decreases synaptic connections [17]. Ammonia-induced astrocyte cultures demonstrated reduced synaptic connections, and are linked with a decreased level of the brain-derived neurotrophic factor (BDNF) and thrombospondins (TSP) [168]. Structural and functional alterations in astrocyte synapses are primarily due to BDNF-induced TrkBT-dependent (Tyrosine Receptor Kinase B) signaling [169]. However, reduced BDNF-actin polymerization induction was found in ammonia-induced astrocyte cultures [17]. Ephrins (Eph)/Ephrin-Receptors (EphR) and BDNF- TrkBT signaling interaction with astrocyte tripartite synapses and neurons intensify the synaptic contacts [170]. Another study revealed the inhibition of Eph/EphR signaling in ammonia-induced astrocyte cultures from the HE patient’s brain [170]. Hence, hyperammonia-induced astrocyte senescence is linked with disturbing synaptic stability/connectivity via the BDNF inhibition, blocking TrkBT-dependent and ephrin/ephrin receptor signaling in the brain [97]. Thus, defective astrocyte senescence and neuronal/glial transmission can lead to persistent morphological alterations in the HE brain, which may proceed for the resolution of overt HE [17].
To support these findings, Gorg et al. reported that astrocyte senescence in an in vitro HE model activated by hyperammonia-induced glutamine synthesis-dependent O-GlcNAcylation in an in vitro study [171]. Moreover, ammonia-induced oxidative stress activates the astrocyte senescence by triggering the p53 dependent transcription inhibitory genes (p21, GADD45α) [168]. Therefore, astrocyte senescence is an important key factor that activates neuroinflammation, aging of neuro-glial cells, and causes cognitive decline [172].
Synaptic connection is another hypothesis related to learning and memory [173]. Synapses are a specialized intercellular (functional) approximation between neurons, and synaptic plasticity denotes learning and memory [174]. Synaptic function and synaptic signal transduction are regulated by postsynaptic density (PSD) [173]. In various PSD types, PSD-95 is a vital protein that regulates and integrates synaptic signals, and is linked with cerebral diseases [175]. PSD-95 mediates the learning and memory process by aggregating the N-methyl-D-aspartate receptor (NMDAR) to generate LTP [176,177]. PSD-95 can also transmit neurotoxic signals via NMDAR overexpression [178].
Fawad et al. demonstrated the cognitive enhancement activity of melatonin administration (5 mg/kg) in middle cerebral artery occlusion (MCAO) rat models. In this study, melatonin facilitates the NR2a/PSD-95 complex association/PI3K/Akt/GSK3β pathway. Moreover, melatonin boosts the neuroprotective factor of γ-enolase expression and conserves the synaptophysin and SNAP25 presynaptic protein expression and p-GluR1845 postsynaptic protein expression [179]. Furthermore, in HE, the accumulation of ROS, increased glutamate, altered synaptic contacts/morphology, and the LTP leads to cognitive decline [180].
Numerous clinical/experimental studies and meta-analyses demonstrated the neurocognitive effect of melatonin in cognitive decline models [181,182,183]. Melatonin exerts neuroprotection against the cholinergic/serotonergic system and promotes GABAergic neurotransmission [184]. Guermonprez et al. reported that melatonin facilitates the choline and choline acetyltransferase functions of synaptosome/synaptic vesicles [185]. Melatonin administration was found to inhibit GSK-3/PKA/PP-2A activation in the rodent brain [186,187,188]. By using melatonin-treated glutamate-exposed neuronal cultures, Wei et al. demonstrated that MMP-9, PSD-95, and growth-associated protein 43 (GAP-43) proteins were not only upregulated, but facilitated neuronal plasticity in the rodent stroke model [189]. Melatonin was also found to increase BDNF expression via the PLC pathway [190].
These studies suggest that melatonin plays an important neurocognitive role in addressing HE-induced cognitive decline. Melatonin is a potent neurocognitive agent by increasing synaptic connectivity, synaptic density proteins, and increasing LTP, inhibiting GSK3β/PKA/PP-2A signaling pathways, decreasing inhibitory neurotransmitter synthesis and release. Melatonin’s neurocognitive effect was confirmed to be due to its antioxidant, anti-inflammatory, and anti-apoptotic properties on HE.
In summary, this review mainly focused on how melatonin communicates with the HE brain (Figure 2). A hallmark of HE is loss of neuro-glial function, which in turn leads to cognitive decline. Researchers have extensively studied the pathogenesis of HE and the treatments available. However, there are no studies examining how melatonin influences HE. In this review, we have described the precise molecular mechanism of HE and how melatonin protects against HE.
8. Conclusions and Future Prospects
HE is a severe neuropsychiatric hepatic disease that triggers various neuropathological alterations. Here, we summarized the functions of melatonin in HE neuropathology.
In HE, melatonin exhibited neuroprotective effects by increasing the enzyme activity involved in ammonia detoxification, by controlling liver enzymes, and by inhibiting ammonia’s entry into the brain by maintaining BBB integrity.
In the astrocyte, melatonin inhibits the conversion glutamate to glutamine by activating the ammonia detoxify enzymes and increasing the antioxidant enzymes’ level, ultimately decreasing the Ca2+ influx by melatonin, which leads to astrocyte swelling and brain edema. In the neuron, melatonin inhibits glutamine synthesis, proinflammatory cytokines, and inflammatory signaling pathways by activating free radical scavengers. This leads to decreased neuro-glial inflammation, insulin resistance, and the increased synaptic plasticity that is involving in cognitive function. Moreover, melatonin demonstrated potent hepatoprotective activity by regulating liver enzymes, reducing oxidative stress by increasing the antioxidant level, and decreasing inflammation in the HE liver. Here, we suggest the therapeutic potential of melatonin in the HE brain. Based on recent evidences, melatonin is involved in multiple neuroprotective responses in HE brains, including enhancing insulin sensitivity, modifying abnormal neurotransmitter and neuromodulator secretion, and reducing inflammatory responses and inhibiting BBB disruptions.
Although a limited number of studies have been attempted to investigate the effects of melatonin on HE, there are still few studies on the regulatory mechanisms of melatonin on neurotransmitters, cognition, and insulin regulation mechanisms in hepatic encephalopathy.
As part of this review, we described clinical and experimental studies conducted on melatonin and liver failure (Table 2), which increased the level of the antioxidant enzymes, reduced hyperammonemia, and hepato-neurotoxicity [1,191,192]. Numerous clinical studies demonstrated that liver diseases are associated with altered circulatory melatonin levels [25,193]. Mina Bahram et al. reported that administration of 6 mg melatonin had a refinement impact on non-alcoholic fatty liver disease (NAFLD) features such as imbalance anthropometric measurements, high blood pressure, abnormal liver enzymes, high sensitive C-reactive protein (hs-CRP), and abnormal leptin levels [194]. Moreover, melatonin is used as a therapeutic agent against obesity [195,196,197], obesity-induced leptin resistance [198,199], diabetes mellitus [200], hepatic steatosis [201], and myocardial injury [202].
Additionally, numerous studies have demonstrated the neuroprotective role of melatonin on liver diseases in vitro and in vivo models (Table 2). Various experimental results have demonstrated that melatonin exhibits antioxidative [203], anti-inflammatory [204], anti-hyperglycemic, and anti-apoptotic properties [205].
Given these clinical and experimental evidences, melatonin may be a new challenge for the treatment of HE neuropathology.
Further studies and clinical studies are needed to apply the appropriate melatonin therapy for brain damage following the progression of HE. Additionally, the monitoring serum melatonin level could be used as a predictive indicator of brain damage due to HE.
Hence, we suggest the possibility of using melatonin in combination with the existing drug treatment for HE and the melatonin alone treatment effect, and we expect to improve the quality of the life of patients with HE.
Author Contributions
Writing, A.A., Y.D.J. and J.S.; figures, A.A. and D.K.S.; manuscript revision, Y.D.J. and J.S.; manuscript finalization, J.S. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported and funded by the grant 2022R1A2C1006125 (Juhyun Song) from the National Research Foundation of Korea (NRF), Republic of Korea. Moreover, this study was supported by the Basic Science Research Program grant through the National Research Foundation of Korea funded by the Ministry of Education, Science, and Technology grant 2018R1D1A1B07049918 (Young Do Jung).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
Ach, acetylcholine; ALF, acute liver failure; ALP, alkaline phosphatase; ALT, alanine aminotransferase; AMPA, α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; AST, aspartate aminotransferase; ATP, adenosine triphosphate; AQP4, aquaporin 4; BBB, alood–brain barrier; BCRP, breast cancer resistance protein; BDNF, brain-derived growth factor; BD, block design; BDL, bile duct ligation; BNT, Boston naming test; BUN, blood urea nitrogen; cAMP, adenosine 3′,5′-cyclic monophosphate; CBF, cerebral blood flow; CCL2, C-C motif chemokine ligand 2; CCl4, carbon tetrachloride; Cd11b, Macrophage-1 antigen/Complement receptor; CFTR, cystic fibrosis transmembrane conductance regulator; cGMP, cyclic guanosine monophosphate; CNS, central nervous system; COX-2, Cycloxygenase-2; CSF, cerebrospinal fluid; CPS-1, carbamyl phosphate synthetase I; CVLT, California Verbal Learning Test; DAG, diacyl glycerol; DBI, Diazepam binding inhibitor; DMSO, Dimethylsulfoxide; DST, Digit Symbol Test; Eph/EphR, Ephrins/Ephrin-Receptor; ERK, extracellular signal-regulated kinase; FC, functional connectivity; GABA, gamma-aminobutyric acid; GAP-43, growth-associated protein-43; Gβγ, G protein–coupled receptors (βγ); GFAP, glial fibrillary acidic protein; GSH, glutathione; GSH/GSSG, reduced glutathione/oxidized glutathione ratio; GSK-3, glycogen synthase kinase-3; GWAS, genome-wide association studies; Hcit, homocitrulline; HCN, hyperpolarization-activated cyclic nucleotide-gated ion channel; HE, hepatic encephalopathy; H1HR, H1 histamine receptor; HHH, hyperornithinemia-hyperammonemia-homocitrullinuria syndrome; HI, hippocampus; HIMOT, Hydroxyindole-O-methyltransferase; HO-1, Hemoxygenase-1; ICT, inhibitory control test; IFN-γ, Interferon-γ; IHC, immunohistochemistry; IL, interleukin; InsR, insulin receptor; iNOS, nitric oxide synthase; IP3, Inositol-3-phosphate; IP, intraperitoneally; IRS-1, InsR substrate; LDH, lactate dehydrogenase; LF, liver fibrosis; LPO, lipid peroxidation; LTT, line tracing test; LTP, long-term potentiation; MAPK, mitogen-activated protein kinase; MDA, malondialdehyde; MHE, minimal hepatic encephalopathy; MMP-9, matrix metalloproteinase 9; MMSE, mini-mental state examination; MRI, magnetic resonance imaging; MT, melatonin receptors; MTNR1B, melatonin single nucleotide polymorphism receptor 2; NCT-A, number connecting test A; NADPH, nicotinamide adenine dinucleotide phosphate oxidase; Nrf2, Nuclear factor erythroid 2-related factor 2; NAS, N-acetyl serotonin; NCCa-ATP, Na-K-2Cl cotransporter; NF-κB, nuclear factor kappa B; NLRP3, nucleotide-binding domain (NOD)-like receptor protein 3; NMDA, N-methyl-D-aspartate; No, nitric oxide; Orn, ornithine; OS, oxidative stress; PD, Parkinson’s disease; PFC, Prefrontal cortex; PHES, Psychometric hepatic encephalopathy score; PKA, protein kinase A; PKG, protein kinase G; PLC, phospholipase; PP-2A, protein phosphatease-2A; PSD, postsynaptic density; PSE, porto-systemic encephalopathy; PTBR, peripheral benzodiazepine receptor; ROS, reactive oxygen species; RyR, ryanodine receptor; SA-β-Gal, senescence-associated β-d-galactosidase; SDT, serial dotting test; SIP, sickness impact profile; TAA, thioacetamide; TAVEC, Test de Aprendizaje Verbal España Complutense; TCA, tricarboxylic acid cycle; T2DM, Type 2 diabetic mellitus; TGFβ1, transforming growth factor β1; TJ, tight junction; TLR4, toll-like receptor 4; TNF, tumor necrosis factor; TrkBT, tyrosine receptor kinase B; WAIS, Wechsler adult intelligence scale.
References
- Zhang, J.J.; Meng, X.; Li, Y.; Zhou, Y.; Xu, D.P.; Li, S.; Li, H.B. Effects of Melatonin on Liver Injuries and Diseases. Int. J. Mol. Sci. 2017, 18, 673. [Google Scholar] [CrossRef] [PubMed]
- Ciecko-Michalska, I.; Szczepanek, M.; Slowik, A.; Mach, T. Pathogenesis of hepatic encephalopathy. Gastroenterol. Res. Pract. 2012, 2012, 642108. [Google Scholar] [CrossRef] [PubMed]
- Cheon, S.Y.; Song, J. The Association between Hepatic Encephalopathy and Diabetic Encephalopathy: The Brain-Liver Axis. Int. J. Mol. Sci. 2021, 22, 463. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, R.F.; Norenberg, M.D.; Felipo, V.; Ferenci, P.; Albrecht, J.; Blei, A.T.; ISHEN Commission on Experimental Models of HE. Experimental models of hepatic encephalopathy: ISHEN guidelines. Liver Int. 2009, 29, 783–788. [Google Scholar] [CrossRef] [PubMed]
- Ferenci, P. Hepatic encephalopathy. Gastroenterol. Rep. 2017, 5, 138–147. [Google Scholar] [CrossRef]
- Stinton, L.M.; Jayakumar, S. Minimal hepatic encephalopathy. Can J. Gastroenterol. 2013, 27, 572–574. [Google Scholar] [CrossRef]
- Khungar, V.; Poordad, F. Management of overt hepatic encephalopathy. Clin. Liver Dis. 2012, 16, 73–89. [Google Scholar] [CrossRef]
- Garcia-Garcia, R.; Cruz-Gomez, A.J.; Urios, A.; Mangas-Losada, A.; Forn, C.; Escudero-Garcia, D.; Kosenko, E.; Torregrosa, I.; Tosca, J.; Giner-Duran, R.; et al. Learning and Memory Impairments in Patients with Minimal Hepatic Encephalopathy are Associated with Structural and Functional Connectivity Alterations in Hippocampus. Sci. Rep. 2018, 8, 9664. [Google Scholar] [CrossRef]
- Felipo, V.; Butterworth, R.F. Neurobiology of ammonia. Prog. Neurobiol. 2002, 67, 259–279. [Google Scholar] [CrossRef]
- Levitt, D.G.; Levitt, M.D. A model of blood-ammonia homeostasis based on a quantitative analysis of nitrogen metabolism in the multiple organs involved in the production, catabolism, and excretion of ammonia in humans. Clin. Exp. Gastroenterol. 2018, 11, 193–215. [Google Scholar] [CrossRef]
- Olde Damink, S.W.; Deutz, N.E.; Dejong, C.H.; Soeters, P.B.; Jalan, R. Interorgan ammonia metabolism in liver failure. Neurochem. Int. 2002, 41, 177–188. [Google Scholar] [CrossRef]
- Sorensen, M. Update on cerebral uptake of blood ammonia. Metab. Brain Dis. 2013, 28, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Lemberg, A.; Fernandez, M.A. Hepatic encephalopathy, ammonia, glutamate, glutamine and oxidative stress. Ann. Hepatol. 2009, 8, 95–102. [Google Scholar] [CrossRef]
- Montoliu, C.; Piedrafita, B.; Serra, M.A.; del Olmo, J.A.; Urios, A.; Rodrigo, J.M.; Felipo, V. IL-6 and IL-18 in blood may discriminate cirrhotic patients with and without minimal hepatic encephalopathy. J. Clin. Gastroenterol. 2009, 43, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Palomero-Gallagher, N.; Bidmon, H.J.; Cremer, M.; Schleicher, A.; Kircheis, G.; Reifenberger, G.; Kostopoulos, G.; Haussinger, D.; Zilles, K. Neurotransmitter receptor imbalances in motor cortex and basal ganglia in hepatic encephalopathy. Cell Physiol. Biochem. 2009, 24, 291–306. [Google Scholar] [CrossRef] [PubMed]
- Rama Rao, K.V.; Norenberg, M.D. Brain energy metabolism and mitochondrial dysfunction in acute and chronic hepatic encephalopathy. Neurochem. Int. 2012, 60, 697–706. [Google Scholar] [CrossRef]
- Gorg, B.; Karababa, A.; Haussinger, D. Hepatic Encephalopathy and Astrocyte Senescence. J. Clin. Exp. Hepatol. 2018, 8, 294–300. [Google Scholar] [CrossRef]
- Bosoi, C.R.; Zwingmann, C.; Marin, H.; Parent-Robitaille, C.; Huynh, J.; Tremblay, M.; Rose, C.F. Increased brain lactate is central to the development of brain edema in rats with chronic liver disease. J. Hepatol. 2014, 60, 554–560. [Google Scholar] [CrossRef]
- Patidar, K.R.; Bajaj, J.S. Antibiotics for the treatment of hepatic encephalopathy. Metab. Brain Dis. 2013, 28, 307–312. [Google Scholar] [CrossRef]
- Bemeur, C.; Desjardins, P.; Butterworth, R.F. Role of nutrition in the management of hepatic encephalopathy in end-stage liver failure. J. Nutr. Metab. 2010, 2010, 489823. [Google Scholar] [CrossRef]
- Zoratti, C.; Moretti, R.; Rebuzzi, L.; Albergati, I.V.; Di Somma, A.; Decorti, G.; Di Bella, S.; Croce, L.S.; Giuffre, M. Antibiotics and Liver Cirrhosis: What the Physicians Need to Know. Antibiotics 2021, 11, 31. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, R.F. Neurosteroids in hepatic encephalopathy: Novel insights and new therapeutic opportunities. J. Steroid Biochem. Mol. Biol. 2016, 160, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Naseem, M.; Parvez, S. Role of melatonin in traumatic brain injury and spinal cord injury. Sci. World J. 2014, 2014, 586270. [Google Scholar] [CrossRef] [PubMed]
- Sherlock, S.; Summerskill, W.H.; White, L.P.; Phear, E.A. Portal-systemic encephalopathy; neurological complications of liver disease. Lancet 1954, 267, 454–457. [Google Scholar] [CrossRef]
- Velissaris, D.; Karamouzos, V.; Polychronopoulos, P.; Karanikolas, M. Chronotypology and melatonin alterations in minimal hepatic encephalopathy. J. Circadian Rhythm. 2009, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.D.F.; Mellanby, R.J.; Gow, A.G. Serum melatonin in dogs with congenital portosystemic shunting, with and without hepatic encephalopathy. Vet. Rec. 2020, 187, e23. [Google Scholar] [CrossRef]
- Amaral, F.G.D.; Cipolla-Neto, J. A brief review about melatonin, a pineal hormone. Arch. Endocrinol. Metab. 2018, 62, 472–479. [Google Scholar] [CrossRef]
- Gurunathan, S.; Kang, M.H.; Kim, J.H. Role and Therapeutic Potential of Melatonin in the Central Nervous System and Cancers. Cancers 2020, 12, 1567. [Google Scholar] [CrossRef]
- Claustrat, B.; Brun, J.; Chazot, G. The basic physiology and pathophysiology of melatonin. Sleep Med. Rev. 2005, 9, 11–24. [Google Scholar] [CrossRef]
- Yu, X.; Li, Z.; Zheng, H.; Ho, J.; Chan, M.T.; Wu, W.K. Protective roles of melatonin in central nervous system diseases by regulation of neural stem cells. Cell Prolif. 2017, 50, e12323. [Google Scholar] [CrossRef]
- Dubocovich, M.L.; Delagrange, P.; Krause, D.N.; Sugden, D.; Cardinali, D.P.; Olcese, J. International Union of Basic and Clinical Pharmacology. LXXV. Nomenclature, classification, and pharmacology of G protein-coupled melatonin receptors. Pharmacol. Rev. 2010, 62, 343–380. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.H.; Chen, Y.C.; Sheen, J.M.; Li, S.W.; Huang, L.T. Melatonin prevented spatial deficits and increases in brain asymmetric dimethylarginine in young bile duct ligation rats. Neuroreport 2018, 29, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.T.; Tiao, M.M.; Tain, Y.L.; Chen, C.C.; Hsieh, C.S. Melatonin ameliorates bile duct ligation-induced systemic oxidative stress and spatial memory deficits in developing rats. Pediatr. Res. 2009, 65, 176–180. [Google Scholar] [CrossRef]
- Zhao, L.; An, R.; Yang, Y.; Yang, X.; Liu, H.; Yue, L.; Li, X.; Lin, Y.; Reiter, R.J.; Qu, Y. Melatonin alleviates brain injury in mice subjected to cecal ligation and puncture via attenuating inflammation, apoptosis, and oxidative stress: The role of SIRT1 signaling. J. Pineal. Res. 2015, 59, 230–239. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, H.; Yue, L.; Zhang, J.; Li, X.; Wang, B.; Lin, Y.; Qu, Y. Melatonin Attenuates Early Brain Injury via the Melatonin Receptor/Sirt1/NF-kappaB Signaling Pathway Following Subarachnoid Hemorrhage in Mice. Mol. Neurobiol. 2017, 54, 1612–1621. [Google Scholar] [CrossRef] [PubMed]
- Babaee, A.; Eftekhar-Vaghefi, S.H.; Asadi-Shekaari, M.; Shahrokhi, N.; Soltani, S.D.; Malekpour-Afshar, R.; Basiri, M. Melatonin treatment reduces astrogliosis and apoptosis in rats with traumatic brain injury. Iran J. Basic Med. Sci. 2015, 18, 867–872. [Google Scholar] [PubMed]
- Sinha, B.; Wu, Q.; Li, W.; Tu, Y.; Sirianni, A.C.; Chen, Y.; Jiang, J.; Zhang, X.; Chen, W.; Zhou, S.; et al. Protection of melatonin in experimental models of newborn hypoxic-ischemic brain injury through MT1 receptor. J. Pineal. Res. 2018, 64, e12443. [Google Scholar] [CrossRef]
- Das, A.; Belagodu, A.; Reiter, R.J.; Ray, S.K.; Banik, N.L. Cytoprotective effects of melatonin on C6 astroglial cells exposed to glutamate excitotoxicity and oxidative stress. J. Pineal. Res. 2008, 45, 117–124. [Google Scholar] [CrossRef]
- Khan, A.; Ayub, M.; Khan, W.M. Hyperammonemia Is Associated with Increasing Severity of Both Liver Cirrhosis and Hepatic Encephalopathy. Int. J. Hepatol. 2016, 2016, 6741754. [Google Scholar] [CrossRef]
- Jayakumar, A.R.; Norenberg, M.D. Hyperammonemia in Hepatic Encephalopathy. J. Clin. Exp. Hepatol. 2018, 8, 272–280. [Google Scholar] [CrossRef]
- Mohiuddin, S.S.; Khattar, D. Biochemistry, Ammonia. In StatPearls; StatPearls Publisher: Treasure Island, FL, USA, 2022. [Google Scholar]
- Choi, J.M.; Kim, Y.H.; Roh, S.Y. Acute hepatic encephalopathy presenting as cortical laminar necrosis: Case report. Korean J. Radiol. 2013, 14, 324–328. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Olde Damink, S.W.; Jalan, R.; Redhead, D.N.; Hayes, P.C.; Deutz, N.E.; Soeters, P.B. Interorgan ammonia and amino acid metabolism in metabolically stable patients with cirrhosis and a TIPSS. Hepatology 2002, 36, 1163–1171. [Google Scholar] [CrossRef]
- Matsuda, I.; Matsuura, T.; Hoshide, R.; Uchino, T.; Matsubasa, T. Molecular basis of urea cycle disorders. Nihon Rinsho. 1993, 51, 520–524. [Google Scholar]
- Caldwell, R.W.; Rodriguez, P.C.; Toque, H.A.; Narayanan, S.P.; Caldwell, R.B. Arginase: A Multifaceted Enzyme Important in Health and Disease. Physiol. Rev. 2018, 98, 641–665. [Google Scholar] [CrossRef] [PubMed]
- Aydogdu, N.; Erbas, H.; Atmaca, G.; Erten, O.; Kaymak, K. Melatonin reduces nitric oxide via increasing arginase in rhabdomyolysis-induced acute renal failure in rats. Ren. Fail. 2006, 28, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Zanatta, A.; Viegas, C.M.; Tonin, A.M.; Busanello, E.N.; Grings, M.; Moura, A.P.; Leipnitz, G.; Wajner, M. Disturbance of redox homeostasis by ornithine and homocitrulline in rat cerebellum: A possible mechanism of cerebellar dysfunction in HHH syndrome. Life Sci. 2013, 93, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Bosoi, C.R.; Rose, C.F. Oxidative stress: A systemic factor implicated in the pathogenesis of hepatic encephalopathy. Metab. Brain Dis. 2013, 28, 175–178. [Google Scholar] [CrossRef]
- Tunez, I.; Munoz, M.C.; Villavicencio, M.A.; Medina, F.J.; de Prado, E.P.; Espejo, I.; Barcos, M.; Salcedo, M.; Feijoo, M.; Montilla, P. Hepato- and neurotoxicity induced by thioacetamide: Protective effects of melatonin and dimethylsulfoxide. Pharmacol. Res. 2005, 52, 223–228. [Google Scholar] [CrossRef]
- Ochoa-Sanchez, R.; Rose, C.F. Pathogenesis of Hepatic Encephalopathy in Chronic Liver Disease. J. Clin. Exp. Hepatol. 2018, 8, 262–271. [Google Scholar] [CrossRef]
- Tunez, I.; Munoz, M.C.; Medina, F.J.; Salcedo, M.; Feijoo, M.; Montilla, P. Comparison of melatonin, vitamin E and L-carnitine in the treatment of neuro- and hepatotoxicity induced by thioacetamide. Cell Biochem. Funct. 2007, 25, 119–127. [Google Scholar] [CrossRef]
- Lena, P.J.; Subramanian, P. Effects of melatonin on the levels of antioxidants and lipid peroxidation products in rats treated with ammonium acetate. Pharmazie 2004, 59, 636–639. [Google Scholar] [PubMed]
- Lena, P.J.; Subramanian, P. Evaluation of the antiperoxidative effects of melatonin in ammonium acetate-treated Wistar rats. Pol. J. Pharmacol. 2003, 55, 1031–1036. [Google Scholar]
- Morvaridzadeh, M.; Sadeghi, E.; Agah, S.; Nachvak, S.M.; Fazelian, S.; Moradi, F.; Persad, E.; Heshmati, J. Effect of melatonin supplementation on oxidative stress parameters: A systematic review and meta-analysis. Pharmacol. Res. 2020, 161, 105210. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Yang, Q.; Liu, Y.; Zhou, S.; Jiang, J.; Reiter, R.J.; Bhattacharya, P.; Cui, Y.; Yang, H.; Ma, H.; et al. The multiple protective roles and molecular mechanisms of melatonin and its precursor N-acetylserotonin in targeting brain injury and liver damage and in maintaining bone health. Free Radic. Biol. Med. 2019, 130, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R.; Cardinali, D.P.; Brown, G.M.; Pandi-Perumal, S.R. Melatonin and brain inflammaging. Prog. Neurobiol. 2015, 127–128, 46–63. [Google Scholar] [CrossRef] [PubMed]
- Montoliu, C.; Cauli, O.; Urios, A.; ElMlili, N.; Serra, M.A.; Giner-Duran, R.; Gonzalez-Lopez, O.; Del Olmo, J.A.; Wassel, A.; Rodrigo, J.M.; et al. 3-nitro-tyrosine as a peripheral biomarker of minimal hepatic encephalopathy in patients with liver cirrhosis. Am. J. Gastroenterol. 2011, 106, 1629–1637. [Google Scholar] [CrossRef]
- Cimen, B.; Turkozkan, N.; Unlu, A.; Erbil, M.K. Effects of melatonin on 3-nitrotyrosine formation and energy charge ratio in guinea pig kidney in LPS-induced stress. Cell Biochem. Funct. 2005, 23, 273–277. [Google Scholar] [CrossRef]
- Yin, J.; Liu, Y.H.; Xu, Y.F.; Zhang, Y.J.; Chen, J.G.; Shu, B.H.; Wang, J.Z. Melatonin arrests peroxynitrite-induced tau hyperphosphorylation and the overactivation of protein kinases in rat brain. J. Pineal. Res. 2006, 41, 124–129. [Google Scholar] [CrossRef]
- Hardeland, R. Melatonin metabolism in the central nervous system. Curr. Neuropharmacol. 2010, 8, 168–181. [Google Scholar] [CrossRef]
- Jayakumar, A.R.; Rama Rao, K.V.; Norenberg, M.D. Neuroinflammation in hepatic encephalopathy: Mechanistic aspects. J. Clin. Exp. Hepatol. 2015, 5, S21–S28. [Google Scholar] [CrossRef]
- Butterworth, R.F. Altered glial-neuronal crosstalk: Cornerstone in the pathogenesis of hepatic encephalopathy. Neurochem. Int. 2010, 57, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Zhou, J.; Kong, H.; Hua, X.; Xiao, M.; Ding, J.; Hu, G. Altered blood-brain barrier integrity in adult aquaporin-4 knockout mice. Neuroreport 2008, 19, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed]
- Nayak, D.; Roth, T.L.; McGavern, D.B. Microglia development and function. Annu. Rev. Immunol. 2014, 32, 367–402. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Sheeler, C.; Rosa, J.G.; Ferro, A.; McAdams, B.; Borgenheimer, E.; Cvetanovic, M. Glia in Neurodegeneration: The Housekeeper, the Defender and the Perpetrator. Int. J. Mol. Sci. 2020, 21, 9188. [Google Scholar] [CrossRef]
- Adlimoghaddam, A.; Sabbir, M.G.; Albensi, B.C. Ammonia as a Potential Neurotoxic Factor in Alzheimer’s Disease. Front. Mol. Neurosci. 2016, 9, 57. [Google Scholar] [CrossRef]
- Takano, T.; Tian, G.F.; Peng, W.; Lou, N.; Libionka, W.; Han, X.; Nedergaard, M. Astrocyte-mediated control of cerebral blood flow. Nat. Neurosci. 2006, 9, 260–267. [Google Scholar] [CrossRef]
- Ott, P.; Larsen, F.S. Blood-brain barrier permeability to ammonia in liver failure: A critical reappraisal. Neurochem. Int. 2004, 44, 185–198. [Google Scholar] [CrossRef]
- Claeys, W.; Van Hoecke, L.; Lefere, S.; Geerts, A.; Verhelst, X.; Van Vlierberghe, H.; Degroote, H.; Devisscher, L.; Vandenbroucke, R.E.; Van Steenkiste, C. The neurogliovascular unit in hepatic encephalopathy. JHEP Rep. 2021, 3, 100352. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J. The role of glutamine synthetase and glutamate dehydrogenase in cerebral ammonia homeostasis. Neurochem. Res. 2012, 37, 2439–2455. [Google Scholar] [CrossRef] [PubMed]
- Goldbecker, A.; Buchert, R.; Berding, G.; Bokemeyer, M.; Lichtinghagen, R.; Wilke, F.; Ahl, B.; Weissenborn, K. Blood-brain barrier permeability for ammonia in patients with different grades of liver fibrosis is not different from healthy controls. J. Cereb. Blood Flow Metab. 2010, 30, 1384–1393. [Google Scholar] [CrossRef]
- Rama Rao, K.V.; Norenberg, M.D. Glutamine in the pathogenesis of hepatic encephalopathy: The trojan horse hypothesis revisited. Neurochem. Res. 2014, 39, 593–598. [Google Scholar] [CrossRef]
- Jayakumar, A.R.; Liu, M.; Moriyama, M.; Ramakrishnan, R.; Forbush, B., 3rd; Reddy, P.V.; Norenberg, M.D. Na-K-Cl Cotransporter-1 in the mechanism of ammonia-induced astrocyte swelling. J. Biol. Chem. 2008, 283, 33874–33882. [Google Scholar] [CrossRef] [PubMed]
- Stokum, J.A.; Kurland, D.B.; Gerzanich, V.; Simard, J.M. Mechanisms of astrocyte-mediated cerebral edema. Neurochem. Res. 2015, 40, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Reinehr, R.; Gorg, B.; Becker, S.; Qvartskhava, N.; Bidmon, H.J.; Selbach, O.; Haas, H.L.; Schliess, F.; Haussinger, D. Hypoosmotic swelling and ammonia increase oxidative stress by NADPH oxidase in cultured astrocytes and vital brain slices. Glia 2007, 55, 758–771. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Jayakumar, A.R.; Tong, X.Y.; Ospel, J.; Norenberg, M.D. Role of cerebral endothelial cells in the astrocyte swelling and brain edema associated with acute hepatic encephalopathy. Neuroscience 2012, 218, 305–316. [Google Scholar] [CrossRef]
- Wright, G.; Jalan, R. Ammonia and inflammation in the pathogenesis of hepatic encephalopathy: Pandora’s box? Hepatology 2007, 46, 291–294. [Google Scholar] [CrossRef]
- Mehrotra, A.; Trigun, S.K. Moderate grade hyperammonemia activates lactate dehydrogenase-4 and 6-phosphofructo-2-kinase to support increased lactate turnover in the brain slices. Mol. Cell Biochem. 2013, 381, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Belanger, M.; Desjardins, P.; Chatauret, N.; Butterworth, R.F. Loss of expression of glial fibrillary acidic protein in acute hyperammonemia. Neurochem. Int. 2002, 41, 155–160. [Google Scholar] [CrossRef]
- Belanger, M.; Desjardins, P.; Chatauret, N.; Butterworth, R.F. Selectively increased expression of the astrocytic/endothelial glucose transporter protein GLUT1 in acute liver failure. Glia 2006, 53, 557–562. [Google Scholar] [CrossRef]
- Rama Rao, K.V.; Norenberg, M.D. Aquaporin-4 in hepatic encephalopathy. Metab. Brain Dis. 2007, 22, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Rabaza, V.; Cabrera-Pastor, A.; Taoro-Gonzalez, L.; Malaguarnera, M.; Agusti, A.; Llansola, M.; Felipo, V. Hyperammonemia induces glial activation, neuroinflammation and alters neurotransmitter receptors in hippocampus, impairing spatial learning: Reversal by sulforaphane. J. Neuroinflamm. 2016, 13, 41. [Google Scholar] [CrossRef]
- Gorg, B.; Schliess, F.; Haussinger, D. Osmotic and oxidative/nitrosative stress in ammonia toxicity and hepatic encephalopathy. Arch. Biochem. Biophys. 2013, 536, 158–163. [Google Scholar] [CrossRef]
- Pierzchala, K.; Simicic, D.; Sienkiewicz, A.; Sessa, D.; Mitrea, S.; Braissant, O.; McLin, V.A.; Gruetter, R.; Cudalbu, C. Central nervous system and systemic oxidative stress interplay with inflammation in a bile duct ligation rat model of type C hepatic encephalopathy. Free Radic. Biol. Med. 2022, 178, 295–307. [Google Scholar] [CrossRef]
- Genovese, T.; Mazzon, E.; Muia, C.; Bramanti, P.; De Sarro, A.; Cuzzocrea, S. Attenuation in the evolution of experimental spinal cord trauma by treatment with melatonin. J. Pineal. Res. 2005, 38, 198–208. [Google Scholar] [CrossRef]
- Alluri, H.; Wilson, R.L.; Anasooya Shaji, C.; Wiggins-Dohlvik, K.; Patel, S.; Liu, Y.; Peng, X.; Beeram, M.R.; Davis, M.L.; Huang, J.H.; et al. Melatonin Preserves Blood-Brain Barrier Integrity and Permeability via Matrix Metalloproteinase-9 Inhibition. PLoS ONE 2016, 11, e0154427. [Google Scholar] [CrossRef]
- Hardeland, R. Aging, Melatonin, and the Pro- and Anti-Inflammatory Networks. Int. J. Mol. Sci. 2019, 20, 1223. [Google Scholar] [CrossRef]
- Ortiz, F.; Acuna-Castroviejo, D.; Doerrier, C.; Dayoub, J.C.; Lopez, L.C.; Venegas, C.; Garcia, J.A.; Lopez, A.; Volt, H.; Luna-Sanchez, M.; et al. Melatonin blunts the mitochondrial/NLRP3 connection and protects against radiation-induced oral mucositis. J. Pineal. Res. 2015, 58, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Esposito, E.; Cuzzocrea, S. Antiinflammatory activity of melatonin in central nervous system. Curr. Neuropharmacol. 2010, 8, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Permpoonputtana, K.; Govitrapong, P. The anti-inflammatory effect of melatonin on methamphetamine-induced proinflammatory mediators in human neuroblastoma dopamine SH-SY5Y cell lines. Neurotox. Res. 2013, 23, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wang, Z.; Pan, S.; Zhang, H.; Fang, M.; Jiang, H.; Zhang, H.; Gao, Z.; Xu, K.; Li, Z.; et al. Melatonin protects against blood-brain barrier damage by inhibiting the TLR4/ NF-kappaB signaling pathway after LPS treatment in neonatal rats. Oncotarget 2017, 8, 31638–31654. [Google Scholar] [CrossRef] [PubMed]
- Gorg, B.; Qvartskhava, N.; Keitel, V.; Bidmon, H.J.; Selbach, O.; Schliess, F.; Haussinger, D. Ammonia induces RNA oxidation in cultured astrocytes and brain in vivo. Hepatology 2008, 48, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Galland, F.; Negri, E.; Da Re, C.; Froes, F.; Strapazzon, L.; Guerra, M.C.; Tortorelli, L.S.; Goncalves, C.A.; Leite, M.C. Hyperammonemia compromises glutamate metabolism and reduces BDNF in the rat hippocampus. Neurotoxicology 2017, 62, 46–55. [Google Scholar] [CrossRef]
- Rose, C. Increased extracellular brain glutamate in acute liver failure: Decreased uptake or increased release? Metab. Brain Dis. 2002, 17, 251–261. [Google Scholar] [CrossRef]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8, 184. [Google Scholar] [CrossRef]
- Palomero-Gallagher, N.; Zilles, K. Neurotransmitter receptor alterations in hepatic encephalopathy: A review. Arch. Biochem. Biophys. 2013, 536, 109–121. [Google Scholar] [CrossRef]
- Vaquero, J.; Butterworth, R.F. The brain glutamate system in liver failure. J. Neurochem. 2006, 98, 661–669. [Google Scholar] [CrossRef]
- Escames, G.; Macias, M.; Leon, J.; Garcia, J.; Khaldy, H.; Martin, M.; Vives, F.; Acuna-Castroviejo, D. Calcium-dependent effects of melatonin inhibition of glutamatergic response in rat striatum. J. Neuroendocrinol. 2001, 13, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; McDowell, M.; Pava, M.J.; Smith, J.A.; Reiter, R.J.; Woodward, J.J.; Varma, A.K.; Ray, S.K.; Banik, N.L. The inhibition of apoptosis by melatonin in VSC4.1 motoneurons exposed to oxidative stress, glutamate excitotoxicity, or TNF-alpha toxicity involves membrane melatonin receptors. J. Pineal. Res. 2010, 48, 157–169. [Google Scholar] [CrossRef]
- Vishnoi, S.; Raisuddin, S.; Parvez, S. Glutamate Excitotoxicity and Oxidative Stress in Epilepsy: Modulatory Role of Melatonin. J. Environ. Pathol. Toxicol. Oncol. 2016, 35, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Casellas, P.; Galiegue, S.; Basile, A.S. Peripheral benzodiazepine receptors and mitochondrial function. Neurochem. Int. 2002, 40, 475–486. [Google Scholar] [CrossRef]
- Yanase, H.; Shimizu, H.; Yamada, K.; Iwanaga, T. Cellular localization of the diazepam binding inhibitor in glial cells with special reference to its coexistence with brain-type fatty acid binding protein. Arch. Histol. Cytol. 2002, 65, 27–36. [Google Scholar] [CrossRef]
- Butterworth, R.F. Hepatic encephalopathy: A central neuroinflammatory disorder? Hepatology 2011, 53, 1372–1376. [Google Scholar] [CrossRef]
- Hazell, A.S.; Normandin, L.; Nguyen, B.; Kennedy, G. Upregulation of ‘peripheral-type’ benzodiazepine receptors in the globus pallidus in a sub-acute rat model of manganese neurotoxicity. Neurosci. Lett. 2003, 349, 13–16. [Google Scholar] [CrossRef]
- Haussinger, D. Low grade cerebral edema and the pathogenesis of hepatic encephalopathy in cirrhosis. Hepatology 2006, 43, 1187–1190. [Google Scholar] [CrossRef]
- Reddy, D.S. Neurosteroids: Endogenous role in the human brain and therapeutic potentials. Prog. Brain Res. 2010, 186, 113–137. [Google Scholar] [CrossRef]
- Garcia-Ayllon, M.S.; Cauli, O.; Silveyra, M.X.; Rodrigo, R.; Candela, A.; Compan, A.; Jover, R.; Perez-Mateo, M.; Martinez, S.; Felipo, V.; et al. Brain cholinergic impairment in liver failure. Brain 2008, 131, 2946–2956. [Google Scholar] [CrossRef]
- Li, F.; Endo, T.; Isa, T. Presynaptic muscarinic acetylcholine receptors suppress GABAergic synaptic transmission in the intermediate grey layer of mouse superior colliculus. Eur. J. Neurosci. 2004, 20, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Pakala, R.S.; Brown, K.N.; Preuss, C.V. Cholinergic Medications. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Kalsbeek, A.; Garidou, M.L.; Palm, I.F.; Van Der Vliet, J.; Simonneaux, V.; Pevet, P.; Buijs, R.M. Melatonin sees the light: Blocking GABA-ergic transmission in the paraventricular nucleus induces daytime secretion of melatonin. Eur. J. Neurosci. 2000, 12, 3146–3154. [Google Scholar] [CrossRef]
- Rosenstein, R.E.; Cardinali, D.P. Central gabaergic mechanisms as targets for melatonin activity in brain. Neurochem. Int. 1990, 17, 373–379. [Google Scholar] [CrossRef]
- Prada, C.; Udin, S.B.; Wiechmann, A.F.; Zhdanova, I.V. Stimulation of melatonin receptors decreases calcium levels in xenopus tectal cells by activating GABA(C) receptors. J. Neurophysiol. 2005, 94, 968–978. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.P.; Sun, H.; Ye, Z.Y.; Zhou, J.N. Melatonin modulates the GABAergic response in cultured rat hippocampal neurons. J. Pharmacol. Sci. 2012, 119, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, Y.; Leng, Z. Melatonin inhibits GABAergic neurons in the hypothalamus consistent with a reduction in wakefulness. Neuroreport 2020, 31, 92–98. [Google Scholar] [CrossRef]
- Fernandez-Bachiller, M.I.; Perez, C.; Campillo, N.E.; Paez, J.A.; Gonzalez-Munoz, G.C.; Usan, P.; Garcia-Palomero, E.; Lopez, M.G.; Villarroya, M.; Garcia, A.G.; et al. Tacrine-melatonin hybrids as multifunctional agents for Alzheimer’s disease, with cholinergic, antioxidant, and neuroprotective properties. ChemMedChem 2009, 4, 828–841. [Google Scholar] [CrossRef]
- Sperlagh, B.; Vizi, E.S. The role of extracellular adenosine in chemical neurotransmission in the hippocampus and Basal Ganglia: Pharmacological and clinical aspects. Curr. Top. Med. Chem. 2011, 11, 1034–1046. [Google Scholar] [CrossRef]
- Ribeiro, J.A.; Sebastiao, A.M.; de Mendonca, A. Adenosine receptors in the nervous system: Pathophysiological implications. Prog. Neurobiol. 2002, 68, 377–392. [Google Scholar] [CrossRef]
- Schiffmann, S.N.; Fisone, G.; Moresco, R.; Cunha, R.A.; Ferre, S. Adenosine A2A receptors and basal ganglia physiology. Prog. Neurobiol. 2007, 83, 277–292. [Google Scholar] [CrossRef]
- al Mardini, H.; Harrison, E.J.; Ince, P.G.; Bartlett, K.; Record, C.O. Brain indoles in human hepatic encephalopathy. Hepatology 1993, 17, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Lozeva, V.; Montgomery, J.A.; Tuomisto, L.; Rocheleau, B.; Pannunzio, M.; Huet, P.M.; Butterworth, R.F. Increased brain serotonin turnover correlates with the degree of shunting and hyperammonemia in rats following variable portal vein stenosis. J. Hepatol. 2004, 40, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.; Caride, A.; Pereiro, N.; Lafuente, A. Modulatory effects of melatonin on cadmium-induced changes in biogenic amines in rat hypothalamus. Neurotox. Res. 2011, 20, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Miguez, J.M.; Martin, F.J.; Aldegunde, M. Effects of single doses and daily melatonin treatments on serotonin metabolism in rat brain regions. J. Pineal. Res. 1994, 17, 170–176. [Google Scholar] [CrossRef]
- Agrawal, R.; Tyagi, E.; Shukla, R.; Nath, C. Effect of insulin and melatonin on acetylcholinesterase activity in the brain of amnesic mice. Behav. Brain Res. 2008, 189, 381–386. [Google Scholar] [CrossRef]
- Als-Nielsen, B.; Gluud, L.L.; Gluud, C. Dopaminergic agonists for hepatic encephalopathy. Cochrane Database Syst. Rev. 2004, CD003047. [Google Scholar] [CrossRef]
- Dhanda, S.; Sandhir, R. Role of dopaminergic and serotonergic neurotransmitters in behavioral alterations observed in rodent model of hepatic encephalopathy. Behav. Brain Res. 2015, 286, 222–235. [Google Scholar] [CrossRef]
- Chen, B.; Yang, Y.; Li, S.; Zhu, X.; Qi, Y.; Hong, F. The critical role of hippocampal dopamine in the pathogenesis of hepatic encephalopathy. Physiol. Res. 2021, 70, 101–110. [Google Scholar] [CrossRef]
- Junker, A.E.; Als-Nielsen, B.; Gluud, C.; Gluud, L.L. Dopamine agents for hepatic encephalopathy. Cochrane Database Syst. Rev. 2014, CD003047. [Google Scholar] [CrossRef]
- Zisapel, N. Melatonin-dopamine interactions: From basic neurochemistry to a clinical setting. Cell Mol. Neurobiol. 2001, 21, 605–616. [Google Scholar] [CrossRef]
- Alexiuk, N.A.; Vriend, J.P. Melatonin reduces dopamine content in the neurointermediate lobe of male Syrian hamsters. Brain Res. Bull. 1993, 32, 433–436. [Google Scholar] [CrossRef]
- Roberts, B.M.; Lopes, E.F.; Cragg, S.J. Axonal Modulation of Striatal Dopamine Release by Local gamma-Aminobutyric Acid (GABA) Signalling. Cells 2021, 10, 709. [Google Scholar] [CrossRef] [PubMed]
- Tenn, C.C.; Niles, L.P. Mechanisms underlying the antidopaminergic effect of clonazepam and melatonin in striatum. Neuropharmacology 1997, 36, 1659–1663. [Google Scholar] [CrossRef]
- Tenn, C.C.; Niles, L.P. Central-type benzodiazepine receptors mediate the antidopaminergic effect of clonazepam and melatonin in 6-hydroxydopamine lesioned rats: Involvement of a GABAergic mechanism. J. Pharmacol. Exp. Ther. 1995, 274, 84–89. [Google Scholar]
- Undieh, A.S. Pharmacology of signaling induced by dopamine D(1)-like receptor activation. Pharmacol. Ther. 2010, 128, 37–60. [Google Scholar] [CrossRef]
- Lozeva, V.; Tuomisto, L.; Tarhanen, J.; Butterworth, R.F. Increased concentrations of histamine and its metabolite, tele-methylhistamine and down-regulation of histamine H3 receptor sites in autopsied brain tissue from cirrhotic patients who died in hepatic coma. J. Hepatol. 2003, 39, 522–527. [Google Scholar] [CrossRef]
- Pham, L.; Baiocchi, L.; Kennedy, L.; Sato, K.; Meadows, V.; Meng, F.; Huang, C.K.; Kundu, D.; Zhou, T.; Chen, L.; et al. The interplay between mast cells, pineal gland, and circadian rhythm: Links between histamine, melatonin, and inflammatory mediators. J. Pineal. Res. 2021, 70, e12699. [Google Scholar] [CrossRef]
- Silva, C.L.; Tamura, E.K.; Macedo, S.M.; Cecon, E.; Bueno-Alves, L.; Farsky, S.H.; Ferreira, Z.S.; Markus, R.P. Melatonin inhibits nitric oxide production by microvascular endothelial cells in vivo and in vitro. Br. J. Pharmacol. 2007, 151, 195–205. [Google Scholar] [CrossRef]
- Seaquist, E.R.; Damberg, G.S.; Tkac, I.; Gruetter, R. The effect of insulin on in vivo cerebral glucose concentrations and rates of glucose transport/metabolism in humans. Diabetes 2001, 50, 2203–2209. [Google Scholar] [CrossRef]
- Plum, L.; Belgardt, B.F.; Bruning, J.C. Central insulin action in energy and glucose homeostasis. J. Clin. Investig. 2006, 116, 1761–1766. [Google Scholar] [CrossRef]
- McNay, E.C.; Ong, C.T.; McCrimmon, R.J.; Cresswell, J.; Bogan, J.S.; Sherwin, R.S. Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol. Learn. Mem. 2010, 93, 546–553. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M. Insulin resistance and Alzheimer’s disease. BMB Rep. 2009, 42, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Cholerton, B.; Baker, L.D.; Craft, S. Insulin, cognition, and dementia. Eur. J. Pharmacol. 2013, 719, 170–179. [Google Scholar] [CrossRef]
- Morris, J.K.; Vidoni, E.D.; Honea, R.A.; Burns, J.M.; Alzheimer’s Disease Neuroimaging, I. Impaired glycemia increases disease progression in mild cognitive impairment. Neurobiol. Aging 2014, 35, 585–589. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M.; Wands, J.R. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef]
- Ampuero, J.; Ranchal, I.; del Mar Diaz-Herrero, M.; del Campo, J.A.; Bautista, J.D.; Romero-Gomez, M. Role of diabetes mellitus on hepatic encephalopathy. Metab. Brain Dis. 2013, 28, 277–279. [Google Scholar] [CrossRef]
- Machado, M.C.; Pinheiro da Silva, F. Hyperammonemia due to urea cycle disorders: A potentially fatal condition in the intensive care setting. J. Intensiv. Care 2014, 2, 22. [Google Scholar] [CrossRef]
- Alfadhel, M.; Mutairi, F.A.; Makhseed, N.; Jasmi, F.A.; Al-Thihli, K.; Al-Jishi, E.; AlSayed, M.; Al-Hassnan, Z.N.; Al-Murshedi, F.; Haberle, J.; et al. Guidelines for acute management of hyperammonemia in the Middle East region. Ther. Clin. Risk Manag. 2016, 12, 479–487. [Google Scholar] [CrossRef]
- Kelly, A.; Ng, D.; Ferry, R.J., Jr.; Grimberg, A.; Koo-McCoy, S.; Thornton, P.S.; Stanley, C.A. Acute insulin responses to leucine in children with the hyperinsulinism/hyperammonemia syndrome. J. Clin. Endocrinol. Metab. 2001, 86, 3724–3728. [Google Scholar] [CrossRef]
- Visek, W.J. Ammonia: Its effects on biological systems, metabolic hormones, and reproduction. J. Dairy Sci. 1984, 67, 481–498. [Google Scholar] [CrossRef]
- Ivanovski, I.; Jesic, M.; Ivanovski, A.; Garavelli, L.; Ivanovski, P. Metabolically based liver damage pathophysiology in patients with urea cycle disorders—A new hypothesis. World J. Gastroenterol. 2017, 23, 7930–7938. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Singh, H.; Ahmad, N.; Mishra, P.; Tiwari, A. The role of melatonin in diabetes: Therapeutic implications. Arch. Endocrinol. Metab. 2015, 59, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Peschke, E.; Muhlbauer, E.; Musshoff, U.; Csernus, V.J.; Chankiewitz, E.; Peschke, D. Receptor (MT(1)) mediated influence of melatonin on cAMP concentration and insulin secretion of rat insulinoma cells INS-1. J. Pineal. Res. 2002, 33, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, I.; Bazwinsky, I.; Peschke, E. Modulation of the cGMP signaling pathway by melatonin in pancreatic beta-cells. J. Pineal. Res. 2009, 46, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Bach, A.G.; Wolgast, S.; Muhlbauer, E.; Peschke, E. Melatonin stimulates inositol-1,4,5-trisphosphate and Ca2+ release from INS1 insulinoma cells. J. Pineal. Res. 2005, 39, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Peschke, E.; Bach, A.G.; Muhlbauer, E. Parallel signaling pathways of melatonin in the pancreatic beta-cell. J. Pineal. Res. 2006, 40, 184–191. [Google Scholar] [CrossRef]
- Kim, B.; Feldman, E.L. Insulin resistance in the nervous system. Trends Endocrinol. Metab. 2012, 23, 133–141. [Google Scholar] [CrossRef]
- Liu, S.; Guo, Y.; Yuan, Q.; Pan, Y.; Wang, L.; Liu, Q.; Wang, F.; Wang, J.; Hao, A. Melatonin prevents neural tube defects in the offspring of diabetic pregnancy. J. Pineal. Res. 2015, 59, 508–517. [Google Scholar] [CrossRef]
- Ronn, T.; Wen, J.; Yang, Z.; Lu, B.; Du, Y.; Groop, L.; Hu, R.; Ling, C. A common variant in MTNR1B, encoding melatonin receptor 1B, is associated with type 2 diabetes and fasting plasma glucose in Han Chinese individuals. Diabetologia 2009, 52, 830–833. [Google Scholar] [CrossRef]
- Staiger, H.; Machicao, F.; Schafer, S.A.; Kirchhoff, K.; Kantartzis, K.; Guthoff, M.; Silbernagel, G.; Stefan, N.; Haring, H.U.; Fritsche, A. Polymorphisms within the novel type 2 diabetes risk locus MTNR1B determine beta-cell function. PLoS ONE 2008, 3, e3962. [Google Scholar] [CrossRef]
- Prokopenko, I.; Langenberg, C.; Florez, J.C.; Saxena, R.; Soranzo, N.; Thorleifsson, G.; Loos, R.J.; Manning, A.K.; Jackson, A.U.; Aulchenko, Y.; et al. Variants in MTNR1B influence fasting glucose levels. Nat. Genet. 2009, 41, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Nagelhus, E.A.; Amiry-Moghaddam, M.; Bergersen, L.H.; Bjaalie, J.G.; Eriksson, J.; Gundersen, V.; Leergaard, T.B.; Morth, J.P.; Storm-Mathisen, J.; Torp, R.; et al. The glia doctrine: Addressing the role of glial cells in healthy brain ageing. Mech. Ageing Dev. 2013, 134, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Haeger, P.; Bouchet, A.; Ossandon, C.; Bresky, G. Treatment with Melatonin Improves Cognitive Behavior and Motor Skills in a Rat Model of Liver Fibrosis. Ann. Hepatol. 2019, 18, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Brodersen, C.; Koen, E.; Ponte, A.; Sanchez, S.; Segal, E.; Chiapella, A.; Fernandez, M.; Torres, M.; Tripodi, V.; Lemberg, A. Cognitive function in patients with alcoholic and nonalcoholic chronic liver disease. J. Neuropsychiatry Clin. Neurosci. 2014, 26, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Schubert, C.M.; Heuman, D.M.; Wade, J.B.; Gibson, D.P.; Topaz, A.; Saeian, K.; Hafeezullah, M.; Bell, D.E.; Sterling, R.K.; et al. Persistence of cognitive impairment after resolution of overt hepatic encephalopathy. Gastroenterology 2010, 138, 2332–2340. [Google Scholar] [CrossRef] [PubMed]
- Gorg, B.; Karababa, A.; Shafigullina, A.; Bidmon, H.J.; Haussinger, D. Ammonia-induced senescence in cultured rat astrocytes and in human cerebral cortex in hepatic encephalopathy. Glia 2015, 63, 37–50. [Google Scholar] [CrossRef]
- Saba, J.; Turati, J.; Ramirez, D.; Carniglia, L.; Durand, D.; Lasaga, M.; Caruso, C. Astrocyte truncated tropomyosin receptor kinase B mediates brain-derived neurotrophic factor anti-apoptotic effect leading to neuroprotection. J. Neurochem. 2018, 146, 686–702. [Google Scholar] [CrossRef]
- Sobczyk, K.; Jordens, M.S.; Karababa, A.; Gorg, B.; Haussinger, D. Ephrin/Ephrin receptor expression in ammonia-treated rat astrocytes and in human cerebral cortex in hepatic encephalopathy. Neurochem. Res. 2015, 40, 274–283. [Google Scholar] [CrossRef]
- Gorg, B.; Karababa, A.; Schutz, E.; Paluschinski, M.; Schrimpf, A.; Shafigullina, A.; Castoldi, M.; Bidmon, H.J.; Haussinger, D. O-GlcNAcylation-dependent upregulation of HO1 triggers ammonia-induced oxidative stress and senescence in hepatic encephalopathy. J. Hepatol. 2019, 71, 930–941. [Google Scholar] [CrossRef]
- Csipo, T.; Lipecz, A.; Ashpole, N.M.; Balasubramanian, P.; Tarantini, S. Astrocyte senescence contributes to cognitive decline. Geroscience 2020, 42, 51–55. [Google Scholar] [CrossRef]
- Kennedy, M.B. Synaptic Signaling in Learning and Memory. Cold Spring Harb. Perspect. Biol. 2013, 8, a016824. [Google Scholar] [CrossRef] [PubMed]
- Grossberg, S. From brain synapses to systems for learning and memory: Object recognition, spatial navigation, timed conditioning, and movement control. Brain Res. 2015, 1621, 270–293. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, L.I.; Tayler, H.M.; Love, S. Synaptic protein levels altered in vascular dementia. Neuropathol. Appl. Neurobiol. 2015, 41, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Terashima, A.; Pelkey, K.A.; Rah, J.C.; Suh, Y.H.; Roche, K.W.; Collingridge, G.L.; McBain, C.J.; Isaac, J.T. An essential role for PICK1 in NMDA receptor-dependent bidirectional synaptic plasticity. Neuron 2008, 57, 872–882. [Google Scholar] [CrossRef]
- Yuen, E.Y.; Ren, Y.; Yan, Z. Postsynaptic density-95 (PSD-95) and calcineurin control the sensitivity of N-methyl-D-aspartate receptors to calpain cleavage in cortical neurons. Mol. Pharmacol. 2008, 74, 360–370. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, T.X.; Hallett, P.J.; Watanabe, M.; Grant, S.G.; Isacson, O.; Yao, W.D. PSD-95 uncouples dopamine-glutamate interaction in the D1/PSD-95/NMDA receptor complex. J. Neurosci. 2009, 29, 2948–2960. [Google Scholar] [CrossRef]
- Shah, F.A.; Liu, G.; Al Kury, L.T.; Zeb, A.; Abbas, M.; Li, T.; Yang, X.; Liu, F.; Jiang, Y.; Li, S.; et al. Melatonin Protects MCAO-Induced Neuronal Loss via NR2A Mediated Prosurvival Pathways. Front. Pharmacol. 2019, 10, 297. [Google Scholar] [CrossRef]
- Hu, B.R.; Park, M.; Martone, M.E.; Fischer, W.H.; Ellisman, M.H.; Zivin, J.A. Assembly of proteins to postsynaptic densities after transient cerebral ischemia. J. Neurosci. 1998, 18, 625–633. [Google Scholar] [CrossRef]
- Lin, L.; Huang, Q.X.; Yang, S.S.; Chu, J.; Wang, J.Z.; Tian, Q. Melatonin in Alzheimer’s disease. Int. J. Mol. Sci. 2013, 14, 14575–14593. [Google Scholar] [CrossRef]
- Sumsuzzman, D.M.; Choi, J.; Jin, Y.; Hong, Y. Neurocognitive effects of melatonin treatment in healthy adults and individuals with Alzheimer’s disease and insomnia: A systematic review and meta-analysis of randomized controlled trials. Neurosci. Biobehav. Rev. 2021, 127, 459–473. [Google Scholar] [CrossRef]
- Cardinali, D.P.; Vigo, D.E.; Olivar, N.; Vidal, M.F.; Furio, A.M.; Brusco, L.I. Therapeutic application of melatonin in mild cognitive impairment. Am. J. Neurodegener. Dis. 2012, 1, 280–291. [Google Scholar] [PubMed]
- Cardinali, D.P.; Srinivasan, V.; Brzezinski, A.; Brown, G.M. Melatonin and its analogs in insomnia and depression. J. Pineal. Res. 2012, 52, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Guermonprez, L.; Ducrocq, C.; Gaudry-Talarmain, Y.M. Inhibition of acetylcholine synthesis and tyrosine nitration induced by peroxynitrite are differentially prevented by antioxidants. Mol. Pharmacol. 2001, 60, 838–846. [Google Scholar] [PubMed]
- Liu, S.J.; Wang, J.Z. Alzheimer-like tau phosphorylation induced by wortmannin in vivo and its attenuation by melatonin. Acta Pharmacol. Sin. 2002, 23, 183–187. [Google Scholar] [PubMed]
- Wang, D.L.; Ling, Z.Q.; Cao, F.Y.; Zhu, L.Q.; Wang, J.Z. Melatonin attenuates isoproterenol-induced protein kinase A overactivation and tau hyperphosphorylation in rat brain. J. Pineal. Res. 2004, 37, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.C.; Zhang, J.; Yu, X.; Han, L.; Zhou, Z.T.; Zhang, Y.; Wang, J.Z. Prevention of isoproterenol-induced tau hyperphosphorylation by melatonin in the rat. Sheng Li Xue Bao 2005, 57, 7–12. [Google Scholar]
- Juan, W.S.; Huang, S.Y.; Chang, C.C.; Hung, Y.C.; Lin, Y.W.; Chen, T.Y.; Lee, A.H.; Lee, A.C.; Wu, T.S.; Lee, E.J. Melatonin improves neuroplasticity by upregulating the growth-associated protein-43 (GAP-43) and NMDAR postsynaptic density-95 (PSD-95) proteins in cultured neurons exposed to glutamate excitotoxicity and in rats subjected to transient focal cerebral ischemia even during a long-term recovery period. J. Pineal. Res. 2014, 56, 213–223. [Google Scholar] [CrossRef]
- Luo, Y.; Peng, M.; Wei, H. Melatonin Promotes Brain-Derived Neurotrophic Factor (BDNF) Expression and Anti-Apoptotic Effects in Neonatal Hemolytic Hyperbilirubinemia via a Phospholipase (PLC)-Mediated Mechanism. Med. Sci. Monit. 2017, 23, 5951–5959. [Google Scholar] [CrossRef]
- Sato, K.; Meng, F.; Francis, H.; Wu, N.; Chen, L.; Kennedy, L.; Zhou, T.; Franchitto, A.; Onori, P.; Gaudio, E.; et al. Melatonin and circadian rhythms in liver diseases: Functional roles and potential therapies. J. Pineal. Res. 2020, 68, e12639. [Google Scholar] [CrossRef]
- Chojnacki, C.; Blonska, A.; Chojnacki, J. The Effects of Melatonin on Elevated Liver Enzymes during Statin Treatment. Biomed. Res. Int. 2017, 2017, 3204504. [Google Scholar] [CrossRef]
- Velissaris, D.; Karanikolas, M.; Kalogeropoulos, A.; Solomou, E.; Polychronopoulos, P.; Thomopoulos, K.; Labropoulou-Karatza, C. Pituitary hormone circadian rhythm alterations in cirrhosis patients with subclinical hepatic encephalopathy. World J. Gastroenterol. 2008, 14, 4190–4195. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, M.; Cheraghpour, M.; Jafarirad, S.; Alavinejad, P.; Asadi, F.; Hekmatdoost, A.; Mohammadi, M.; Yari, Z. The effect of melatonin on treatment of patients with non-alcoholic fatty liver disease: A randomized double blind clinical trial. Complement. Ther. Med. 2020, 52, 102452. [Google Scholar] [CrossRef] [PubMed]
- Genario, R.; Cipolla-Neto, J.; Bueno, A.A.; Santos, H.O. Melatonin supplementation in the management of obesity and obesity-associated disorders: A review of physiological mechanisms and clinical applications. Pharmacol. Res. 2021, 163, 105254. [Google Scholar] [CrossRef] [PubMed]
- de Farias, T.; Cruz, M.M.; de Sa, R.; Severi, I.; Perugini, J.; Senzacqua, M.; Cerutti, S.M.; Giordano, A.; Cinti, S.; Alonso-Vale, M.I.C. Melatonin Supplementation Decreases Hypertrophic Obesity and Inflammation Induced by High-Fat Diet in Mice. Front. Endocrinol. 2019, 10, 750. [Google Scholar] [CrossRef] [PubMed]
- Overberg, J.; Kalveram, L.; Keller, T.; Krude, H.; Kuhnen, P.; Wiegand, S. Interactions between nocturnal melatonin secretion, metabolism, and sleeping behavior in adolescents with obesity. Int. J. Obes. 2022. [Google Scholar] [CrossRef]
- Delpino, F.M.; Figueiredo, L.M. Melatonin supplementation and anthropometric indicators of obesity: A systematic review and meta-analysis. Nutrition 2021, 91–92, 111399. [Google Scholar] [CrossRef]
- Suriagandhi, V.; Nachiappan, V. Protective Effects of Melatonin against Obesity-Induced by Leptin Resistance. Behav. Brain Res. 2022, 417, 113598. [Google Scholar] [CrossRef]
- Patel, R.; Parmar, N.; Pramanik Palit, S.; Rathwa, N.; Ramachandran, A.V.; Begum, R. Diabetes mellitus and melatonin: Where are we? Biochimie 2022, in press. [Google Scholar] [CrossRef]
- Sun, H.; Huang, F.F.; Qu, S. Melatonin: A potential intervention for hepatic steatosis. Lipids Health Dis. 2015, 14, 75. [Google Scholar] [CrossRef]
- Naaz, S.; Mishra, S.; Pal, P.K.; Chattopadhyay, A.; Das, A.R.; Bandyopadhyay, D. Activation of SIRT1/PGC 1alpha/SIRT3 pathway by melatonin provides protection against mitochondrial dysfunction in isoproterenol induced myocardial injury. Heliyon 2020, 6, e05159. [Google Scholar] [CrossRef]
- Ling, L.; Alattar, A.; Tan, Z.; Shah, F.A.; Ali, T.; Alshaman, R.; Koh, P.O.; Li, S. A Potent Antioxidant Endogenous Neurohormone Melatonin, Rescued MCAO by Attenuating Oxidative Stress-Associated Neuroinflammation. Front. Pharmacol. 2020, 11, 1220. [Google Scholar] [CrossRef] [PubMed]
- Tarocco, A.; Caroccia, N.; Morciano, G.; Wieckowski, M.R.; Ancora, G.; Garani, G.; Pinton, P. Melatonin as a master regulator of cell death and inflammation: Molecular mechanisms and clinical implications for newborn care. Cell Death Dis. 2019, 10, 317. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. The antiapoptotic activity of melatonin in neurodegenerative diseases. CNS Neurosci. Ther. 2009, 15, 345–357. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).