
Electrophysiological and Behavioral Evidence for Hyper- and Hyposensitivity in Rare Genetic Syndromes Associated with Autism

 , , and
, , and
Abstract
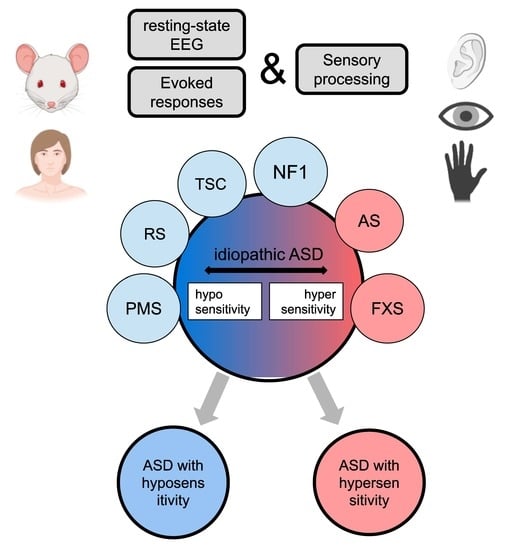
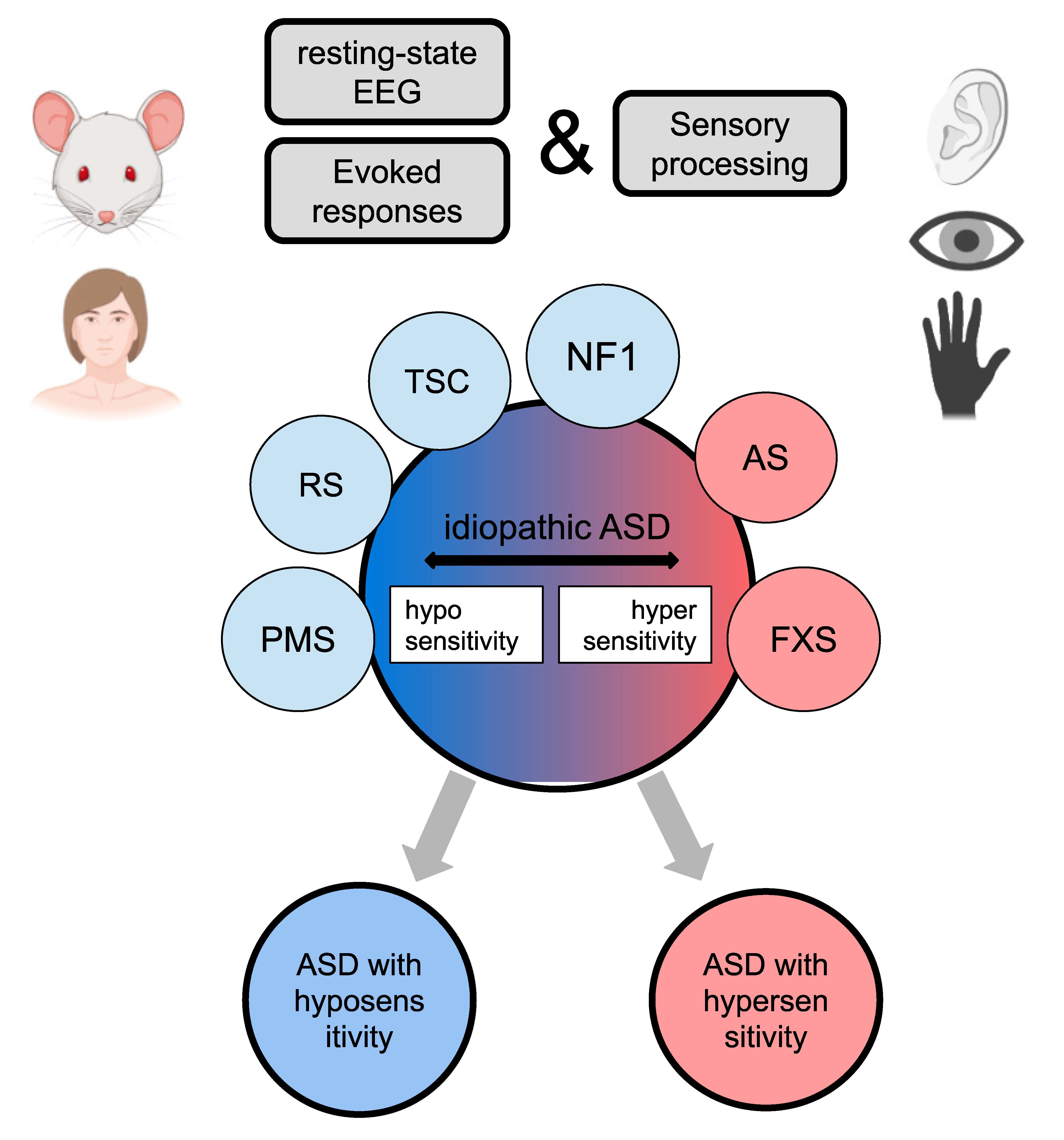
1. Introduction
2. Hypersensitive Syndromes
2.1. Fragile X Syndrome
2.1.1. General Info
2.1.2. Sensory Processing
2.1.3. EEG: Resting State
2.1.4. EEG: Evoked Responses
2.2. Angelman Syndrome
2.2.1. General Info
2.2.2. Sensory Processing
2.2.3. EEG: Resting State
2.2.4. EEG: Evoked Responses
3. Hyposensitive Syndromes
3.1. Phelan–McDermid Syndrome
3.1.1. General Info
3.1.2. Sensory Processing
3.1.3. EEG: Resting State
3.1.4. EEG: Evoked Responses
3.2. Rett Syndrome
3.2.1. General Info
3.2.2. Sensory Processing
3.2.3. EEG: Resting State
3.2.4. EEG: Evoked Responses
3.3. Tuberous Sclerosis
3.3.1. General Info
3.3.2. Sensory Processing
3.3.3. EEG: Resting State
3.3.4. EEG: Evoked Responses
3.4. Neurofibromatosis Type 1
3.4.1. General Info
3.4.2. Sensory Processing
3.4.3. EEG: Resting State
3.4.4. EEG: Evoked Responses

{kind=link}
| Syndrome | Epilepsy, Onset | Resting-State EEG | Auditory EEG | Visual EEG | Somatosensory EEG |
|---|---|---|---|---|---|
| FXS | 10–20%, before 10 y.o. [80,81] | ↑ γ power [84,85] ↓ α power [101] ↑ θ power [86] | ↓ evoked γ power [88] ↑ N1, P2 amplitude [90,91,92,93] ↓ MMN, P3a, P3b amplitude [92,99] ↑↓ N2 amplitude [89] ↑↓ N1 habituation [88,92,94] ↑ N2 latency [54] | ↑ N1, N170, N2 amplitude [94,98,99] ↓ P3b amplitude [98] ↓ N170 habituation [99] | n/a |
| AS | 80%, before 3 y.o. [109,110] | ↑ Δ power [114,115,116,117] in patients with deletion: ↓ β power [114] ↑ θ power [114] | ↑ amplitude 200–500 ms for repeated non-words was associated with better communication skills [118,119] | n/a | in patients with deletion: ↑ N1m amplitude [120] ↓ P1m amplitude [120] Prolonged N1m, P1m component [120] |
| PMS | 63%, between 2–6 y.o. [133,134] | ↓ γ power [140] ↓ β power [140] ↓ α power [135,138,139] | ↓ 40Hz ASSR [142] ↓ P50, P2 amplitude [148,149] ↑ P2 habituation [148] ↑ N250 latency [150] | ↓ P60-N75, N75-P100 amplitude [140] | n/a |
| RS | 60–80%, between 2–4 y.o. [158] | ↓ β power [159] ↓ α power [159] ↑ θ power [159] ↑ Δ power [159,160] | ↓P2 and N2 amplitude [164] ↓ MMN amplitude [167,168] ↑ N1 and P2 latencies [164,165] ↑ MMN latency [167] | ↓ N1-P2 amplitude [169] ↑ P1, N2 latency [169,170] | ↑ amplitude of SEP [171,172] |
| TSC | 80–90%, before 2 y.o. [176] | ↓ α peak frequency [177] in TSC with ASD: ↑ Δ power [178] | ↓ P1-N1 amplitude [95,179] in TSC with ASD: ↓ MMN amplitude [179] ↑ N1, MMN latencies [179] | ↑ N170 and N290 latency in response to human faces [181,182] | n/a |
| NF1 | 4–7%, varies a lot [191] | ↑ θ power [191] ↑ α power [191] | ↑ latency in brainstem auditory evoked potential [194,195] ↑ latency in pitch-change detection response in infants [195] | ↑ α oscillations during visual stimulation [191] ↓ P1, P3 amplitude [194,198] ↓ N2 amplitude in incompatible trials [206] ↑ N450 amplitude in compatible trials [206] ↑ P1 latency [194,195,198] ↓ P3 latency in n-back task [204] | ↑ latency in SEP [194,195] |
4. Discussion
4.1. Oscillatory Activity
4.2. Approach for Idiopathic ASD Stratification
4.3. A possible Role of Inhibition in Sensory Abnormalities
4.4. Concluding Remarks/Open Questions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maenner, M.J. Prevalence of autism spectrum disorder among children aged 8 years—Autism and developmental disabilities monitoring network, 11 sites, united states, 2016. MMWR Surveill. Summ. 2020, 69, 503. [Google Scholar] [CrossRef] [PubMed]
- Marco, E.J.; Hinkley, L.B.N.; Hill, S.S.; Nagarajan, S.S. Sensory processing in autism: A review of neurophysiologic findings. Pediatr. Res. 2011, 69, 48R–54R. [Google Scholar] [CrossRef] [PubMed]
- Robertson, C.E.; Baron-Cohen, S. Sensory perception in autism. Nat. Rev. Neurosci. 2017, 18, 671–684. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013; ISBN 978-0-89042-555-8. [Google Scholar]
- Baranek, G.T.; David, F.J.; Poe, M.D.; Stone, W.L.; Watson, L.R. Sensory experiences questionnaire: Discriminating sensory features in young children with autism, developmental delays, and typical development. J. Child Psychol. Psychiatry 2006, 47, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, A.; Tillmann, J.; Cliquet, F.; Amsellem, F.; Maruani, A.; Leblond, C.; Beggiato, A.; Germanaud, D.; Amestoy, A.; Moal, M.L.-L.; et al. Hypo- and hyper- sensory processing heterogeneity in autism spectrum disorder. Res. Sq. 2021. submitted. [Google Scholar]
- Sztainberg, Y.; Zoghbi, H.Y. Lessons learned from studying syndromic autism spectrum disorders. Nat. Neurosci. 2016, 19, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Ziats, C.A.; Rennert, O.M.; Ziats, M.N. Toward a pathway-driven clinical-molecular framework for classifying autism spectrum disorders. Pediatr. Neurol. 2019, 98, 46–52. [Google Scholar] [CrossRef]
- Rylaarsdam, L.; Guemez-Gamboa, A. Genetic causes and modifiers of autism spectrum disorder. Front. Cell. Neurosci. 2019, 13, 385. [Google Scholar] [CrossRef]
- Chen, C.-H.; Huang, C.-C.; Cheng, M.-C.; Chiu, Y.-N.; Tsai, W.-C.; Wu, Y.-Y.; Liu, S.-K.; Gau, S.S.-F. Genetic analysis of GABRB3 as a Candidate gene of autism spectrum disorders. Mol. Autism 2014, 5, 36. [Google Scholar] [CrossRef]
- Stefansson, H.; Meyer-Lindenberg, A.; Steinberg, S.; Magnusdottir, B.; Morgen, K.; Arnarsdottir, S.; Bjornsdottir, G.; Walters, G.B.; Jonsdottir, G.A.; Doyle, O.M.; et al. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature 2014, 505, 361–366. [Google Scholar] [CrossRef]
- Leblond, C.S.; Nava, C.; Polge, A.; Gauthier, J.; Huguet, G.; Lumbroso, S.; Giuliano, F.; Stordeur, C.; Depienne, C.; Mouzat, K.; et al. Meta-analysis of SHANK mutations in autism spectrum disorders: A Gradient of Severity in Cognitive Impairments. PLoS Genet. 2014, 10, e1004580. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wang, X.; Li, X.-L.; Towers, A.; Cao, X.; Wang, P.; Bowman, R.; Yang, H.; Goldstein, J.; Li, Y.-J.; et al. Epigenetic dysregulation of SHANK3 in Brain tissues from individuals with autism spectrum disorders. Hum. Mol. Genet. 2014, 23, 1563–1578. [Google Scholar] [CrossRef]
- Lam, C.-W.; Yeung, W.-L.; Ko, C.-H.; Poon, P.M.K.; Tong, S.-F.; Chan, K.-Y.; Lo, I.F.M.; Chan, L.Y.S.; Hui, J.; Wong, V.; et al. Spectrum of mutations in the MECP2 gene in patients with infantile autism and rett syndrome. J. Med. Genet. 2000, 37, e41. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.S.; Blasi, F.; Bacchelli, E.; Klauck, S.M.; Maestrini, E.; Poustka, A. International molecular genetic study of autism consortium (IMGSAC) mutation analysis of the coding sequence of the MECP2 gene in infantile autism. Hum. Genet. 2002, 111, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Loat, C.S.; Curran, S.; Lewis, C.M.; Duvall, J.; Geschwind, D.; Bolton, P.; Craig, I.W. Methyl-CpG-Binding protein 2 polymorphisms and vulnerability to autism. Genes Brain Behav. 2008, 7, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, R.P.; Hogart, A.R.; Gwye, Y.; Martin, M.R.; LaSalle, J.M. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant mecp2 promoter methylation. Epigenetics 2006, 1, e1–e11. [Google Scholar] [CrossRef]
- Liu, C.; Wang, Y.; Deng, J.; Lin, J.; Hu, C.; Li, Q.; Xu, X. Social deficits and repetitive behaviors are improved by early postnatal low-dose VPA intervention in a novel shank3-deficient zebrafish model. Front. Neurosci. 2021, 15, 1125. [Google Scholar] [CrossRef]
- Wang, X.; Bey, A.; Chang, L.; Krystal, A.D.; Jiang, Y. Therapeutic approaches for shankopathies. Dev. Neurobiol. 2014, 74, 123–135. [Google Scholar] [CrossRef]
- Sur, S.; Sinha, V.K. Event-related potential: An overview. Ind. Psychiatry, J. 2009, 18, 70–73. [Google Scholar] [CrossRef]
- Rubenstein, J.L.R.; Merzenich, M.M. Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003, 2, 255–267. [Google Scholar] [CrossRef]
- Gonzalez-Burgos, G.; Cho, R.Y.; Lewis, D.A. Alterations in cortical network oscillations and parvalbumin neurons in schizophrenia. Biol. Psychiatry 2015, 77, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Whittington, M.A.; Traub, R.D.; Kopell, N.; Ermentrout, B.; Buhl, E.H. Inhibition-based rhythms: Experimental and mathematical observations on network dynamics. Int. J. Psychophysiol. 2000, 38, 315–336. [Google Scholar] [CrossRef]
- Huber, K.M.; Gallagher, S.M.; Warren, S.T.; Bear, M.F. Altered synaptic plasticity in a mouse model of fragile x mental retardation. Proc. Natl. Acad. Sci. USA 2002, 99, 7746–7750. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, F.; Heitz, D.; Biancalana, V.; Blumenfeld, S.; Kretz, C.; Boué, J.; Tommerup, N.; Hagen, C.V.D.; DeLozier-Blanchet, C.; Croquette, M.-F.; et al. Direct diagnosis by DNA analysis of the fragile X syndrome of mental retardation. N. Engl. J. Med. 1991, 325, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Rotschafer, S.; Razak, K. Auditory processing in fragile X syndrome. Front. Cell. Neurosci. 2014, 8, 19. [Google Scholar] [CrossRef]
- Heald, M.; Adams, D.; Oliver, C. Profiles of atypical sensory processing in angelman, cornelia de lange and fragile X syndromes. J. Intellect. Disabil. Res. JIDR 2020, 64, 117–130. [Google Scholar] [CrossRef]
- Rais, M.; Binder, D.K.; Razak, K.A.; Ethell, I.M. Sensory processing phenotypes in fragile X syndrome. ASN Neuro 2018, 10, 1759091418801092. [Google Scholar] [CrossRef]
- Sinclair, D.; Oranje, B.; Razak, K.A.; Siegel, S.J.; Schmid, S. Sensory processing in autism spectrum disorders and fragile X syndrome-from the clinic to animal models. Neurosci. Biobehav. Rev. 2017, 76, 235–253. [Google Scholar] [CrossRef]
- Finestack, L.H.; Richmond, E.K.; Abbeduto, L. Language development in individuals with fragile X syndrome. Top. Lang. Disord. 2009, 29, 133–148. [Google Scholar] [CrossRef]
- Bailey, D.B.; Raspa, M.; Olmsted, M.; Holiday, D.B. Co-occurring conditions associated with FMR1 gene variations: Findings from a national parent survey. Am. J. Med. Genet. A 2008, 146, 2060–2069. [Google Scholar] [CrossRef]
- Sullivan, K.; Hatton, D.; Hammer, J.; Sideris, J.; Hooper, S.; Ornstein, P.; Bailey, D. ADHD symptoms in children with FXS. Am. J. Med Genet. 2006, 140, 2275–2288. [Google Scholar] [CrossRef]
- Mabb, A.M.; Judson, M.C.; Zylka, M.J.; Philpot, B.D. Angelman syndrome: Insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 2011, 34, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Lossie, A.C.; Whitney, M.M.; Amidon, D.; Dong, H.J.; Chen, P.; Theriaque, D.; Hutson, A.; Nicholls, R.D.; Zori, R.T.; Williams, C.A.; et al. Distinct phenotypes distinguish the molecular classes of angelman syndrome. J. Med. Genet. 2001, 38, 834–845. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.; Jones, C.; Groves, L.; Moss, J.; Oliver, C. Prevalence of autism spectrum disorder phenomenology in genetic disorders: A systematic review and meta-analysis. Lancet Psychiatry 2015, 2, 909–916. [Google Scholar] [CrossRef]
- Clarke, D.J.; Marston, G. Problem behaviors associated with 15q- angelman syndrome. Am. J. Ment. Retard. AJMR 2000, 105, 25–31. [Google Scholar] [CrossRef]
- Buntinx, I.M.; Hennekam, R.C.; Brouwer, O.F.; Stroink, H.; Beuten, J.; Mangelschots, K.; Fryns, J.P. Clinical profile of angelman syndrome at different ages. Am. J. Med. Genet. 1995, 56, 176–183. [Google Scholar] [CrossRef]
- Williams, C.A. Neurological aspects of the angelman syndrome. Brain Dev. 2005, 27, 88–94. [Google Scholar] [CrossRef]
- Walz, N.C.; Baranek, G.T. Sensory processing patterns in persons with angelman syndrome. Am. J. Occup. Ther. Off. Publ. Am. Occup. Ther. Assoc. 2006, 60, 472–479. [Google Scholar] [CrossRef]
- Buiting, K.; Williams, C.; Horsthemke, B. Angelman syndrome—Insights into a rare neurogenetic disorder. Nat. Rev. Neurol. 2016, 12, 584–593. [Google Scholar] [CrossRef]
- Gentile, J.K.; Tan, W.-H.; Horowitz, L.T.; Bacino, C.A.; Skinner, S.A.; Barbieri-Welge, R.; Bauer-Carlin, A.; Beaudet, A.L.; Bichell, T.J.; Lee, H.-S.; et al. A neurodevelopmental survey of angelman syndrome with genotype-phenotype correlations. J. Dev. Behav. Pediatr. JDBP 2010, 31, 592–601. [Google Scholar] [CrossRef]
- Kalsner, L.; Chamberlain, S.J. Prader-willi, angelman, and 15q11-Q13 duplication syndromes. Pediatr. Clin. N. Am. 2015, 62, 587–606. [Google Scholar] [CrossRef] [PubMed]
- Barry, R.J.; Berry, R.J.; Leitner, R.P.; Clarke, A.R.; Einfeld, S.L. Behavioral aspects of angelman syndrome: A case control study. Am. J. Med. Genet. A 2005, 132, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Sarasua, S.M.; Dwivedi, A.; Boccuto, L.; Rollins, J.D.; Chen, C.-F.; Rogers, R.C.; Phelan, K.; DuPont, B.R.; Collins, J.S. Association between deletion size and important phenotypes expands the genomic region of interest in phelan-mcdermid syndrome (22q13 deletion syndrome). J. Med. Genet. 2011, 48, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Soorya, L.; Kolevzon, A.; Zweifach, J.; Lim, T.; Dobry, Y.; Schwartz, L.; Frank, Y.; Wang, A.T.; Cai, G.; Parkhomenko, E.; et al. Prospective investigation of autism and genotype-phenotype correlations in 22q13 deletion syndrome and SHANK3 deficiency. Mol. Autism 2013, 4, 18. [Google Scholar] [CrossRef]
- Sarasua, S.M.; Boccuto, L.; Sharp, J.L.; Dwivedi, A.; Chen, C.-F.; Rollins, J.D.; Rogers, R.C.; Phelan, K.; DuPont, B.R. Clinical and genomic evaluation of 201 patients with phelan–mcdermid syndrome. Hum. Genet. 2014, 133, 847–859. [Google Scholar] [CrossRef]
- Mieses, A.M.; Tavassoli, T.; Li, E.; Soorya, L.; Lurie, S.; Wang, A.T.; Siper, P.M.; Kolevzon, A. Brief report: Sensory reactivity in children with phelan-mcdermid syndrome. J. Autism Dev. Disord. 2016, 46, 2508–2513. [Google Scholar] [CrossRef]
- Droogmans, G.; Swillen, A.; Van Buggenhout, G. Deep phenotyping of development, communication and behaviour in phelan-mcdermid syndrome. Mol. Syndromol. 2020, 10, 294–305. [Google Scholar] [CrossRef]
- Tavassoli, T.; Layton, C.; Levy, T.; Rowe, M.; George-Jones, J.; Zweifach, J.; Lurie, S.; Buxbaum, J.D.; Kolevzon, A.; Siper, P.M. Sensory reactivity phenotype in phelan-mcdermid syndrome is distinct from idiopathic ASD. Genes 2021, 12, 977. [Google Scholar] [CrossRef]
- Srivastava, S.; Condy, E.; Carmody, E.; Filip-Dhima, R.; Kapur, K.; Bernstein, J.A.; Berry-Kravis, E.; Powell, C.M.; Soorya, L.; Thurm, A.; et al. Parent-reported measure of repetitive behavior in phelan-mcdermid syndrome. J. Neurodev. Disord. 2021, 13, 53. [Google Scholar] [CrossRef]
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [CrossRef]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.C.; Qin, J.; Zoghbi, H.Y. MeCP2, a key contributor to neurological disease, Activates and represses transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Devarakonda, K.M.; Lowthian, D.; Raghavendra, T. A case of rett syndrome with reduced pain sensitivity. Paediatr. Anaesth. 2009, 19, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Downs, J.; Géranton, S.M.; Bebbington, A.; Jacoby, P.; Bahi-Buisson, N.; Ravine, D.; Leonard, H. Linking MECP2 and pain sensitivity: The example of rett syndrome. Am. J. Med. Genet. A 2010, 152, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Cardoza, B.; Clarke, A.; Wilcox, J.; Gibbon, F.; Smith, P.E.M.; Archer, H.; Hryniewiecka-Jaworska, A.; Kerr, M. Epilepsy in rett syndrome: Association between phenotype and genotype, and implications for practice. Seizure 2011, 20, 646–649. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.L.; Veenstra, G.J.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsberger, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef]
- Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.S.; Schanen, N.C.; Zappella, M.; et al. Rett syndrome: Revised diagnostic criteria and nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef]
- Sarajlija, A.; Kisic-Tepavcevic, D.; Nikolic, Z.; Pavicevic, D.S.; Obradovic, S.; Djuric, M.; Pekmezovic, T. Epidemiology of rett syndrome in serbia: Prevalence, incidence and survival. Neuroepidemiology 2015, 44, 1–5. [Google Scholar] [CrossRef]
- Annear, N.M.P.; Appleton, R.E.; Bassi, Z.; Bhatt, R.; Bolton, P.F.; Crawford, P.; Crowe, A.; Tossi, M.; Elmslie, F.; Finlay, E.; et al. Tuberous sclerosis complex (TSC): Expert recommendations for provision of coordinated care. Front. Neurol. 2019, 10, 1116. [Google Scholar] [CrossRef]
- Numis, A.L.; Major, P.; Montenegro, M.A.; Muzykewicz, D.A.; Pulsifer, M.B.; Thiele, E.A. Identification of risk factors for autism spectrum disorders in tuberous sclerosis complex. Neurology 2011, 76, 981–987. [Google Scholar] [CrossRef]
- Smalley, S.L. Autism and tuberous sclerosis. J. Autism Dev. Disord. 1998, 28, 407–414. [Google Scholar] [CrossRef]
- Goh, S.; Kwiatkowski, D.J.; Dorer, D.J.; Thiele, E.A. Infantile spasms and intellectual outcomes in children with tuberous sclerosis complex. Neurology 2005, 65, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Prather, P.; de Vries, P.J. Behavioral and cognitive aspects of tuberous sclerosis complex. J. Child Neurol. 2004, 19, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Curatolo, P.; Cusmai, R.; Cortesi, F.; Chiron, C.; Jambaque, I.; Dulac, O. Neuropsychiatric aspects of tuberous sclerosis. Ann. N. Y. Acad. Sci. 1991, 615, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.A.; Friedman, J.M. NF1 gene and neurofibromatosis 1. Am. J. Epidemiol. 2000, 151, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Eijk, S.; Mous, S.E.; Dieleman, G.C.; Dierckx, B.; Rietman, A.B.; de Nijs, P.F.A.; ten Hoopen, L.W.; van Minkelen, R.; Elgersma, Y.; Catsman-Berrevoets, C.E.; et al. Autism spectrum disorder in an unselected cohort of children with neurofibromatosis type 1 (NF1). J. Autism Dev. Disord. 2018, 48, 2278–2285. [Google Scholar] [CrossRef]
- Jett, K.; Friedman, J.M. Clinical and genetic aspects of neurofibromatosis 1. Genet. Med. Off. J. Am. Coll. Med. Genet. 2010, 12, 1–11. [Google Scholar] [CrossRef]
- Legius, E.; Descheemaeker, M.J.; Spaepen, A.; Casaer, P.; Fryns, J.P. Neurofibromatosis type 1 in childhood: A study of the neuropsychological profile in 45 children. Genet. Couns. Geneva Switz. 1994, 5, 51–60. [Google Scholar]
- Lorenzo, J.; Barton, B.; Acosta, M.T.; North, K. Mental, motor, and language development of toddlers with neurofibromatosis type 1. J. Pediatr. 2011, 158, 660–665. [Google Scholar] [CrossRef]
- Hyman, S.L.; Shores, A.; North, K.N. The nature and frequency of cognitive deficits in children with neurofibromatosis type 1. Neurology 2005, 65, 1037–1044. [Google Scholar] [CrossRef]
- Butler, M.G. Fragile X syndrome: A major cause of x-linked mental retardation. Compr. Ther. 1988, 14, 3–7. [Google Scholar]
- D’Hulst, C.; De Geest, N.; Reeve, S.P.; Van Dam, D.; De Deyn, P.P.; Hassan, B.A.; Kooy, R.F. Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res. 2006, 1121, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Chuang, S.-H.; Reddy, D.S. Genetic and molecular regulation of extrasynaptic GABA-A receptors in the brain: Therapeutic insights for epilepsy. J. Pharmacol. Exp. Ther. 2018, 364, 180–197. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Peng, Z.; Tong, X.; Lindemeyer, A.K.; Cetina, Y.; Huang, C.S.; Olsen, R.W.; Otis, T.S.; Houser, C.R. Decreased surface expression of the δ subunit of the GABA a receptor contributes to reduced tonic inhibition in dentate granule cells in a mouse model of fragile X syndrome. Exp. Neurol. 2017, 297, 168–178. [Google Scholar] [CrossRef]
- Paluszkiewicz, S.M.; Martin, B.S.; Huntsman, M.M. Fragile X syndrome: The GABAergic system and circuit dysfunction. Dev. Neurosci. 2011, 33, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Felgerolle, C.; Hébert, B.; Ardourel, M.; Meyer-Dilhet, G.; Menuet, A.; Pinto-Morais, K.; Bizot, J.-C.; Pichon, J.; Briault, S.; Perche, O. Visual behavior impairments as an aberrant sensory processing in the mouse model of fragile X syndrome. Front. Behav. Neurosci. 2019, 13, 228. [Google Scholar] [CrossRef]
- Chen, L.; Toth, M. Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience 2001, 103, 1043–1050. [Google Scholar] [CrossRef]
- Paylor, R.; Yuva-Paylor, L.A.; Nelson, D.L.; Spencer, C.M. Reversal of sensorimotor gating abnormalities in fmr1 knockout mice carrying a human fmr1 transgene. Behav. Neurosci. 2008, 122, 1371–1377. [Google Scholar] [CrossRef]
- He, C.X.; Cantu, D.A.; Mantri, S.S.; Zeiger, W.A.; Goel, A.; Portera-Cailliau, C. Tactile defensiveness and impaired adaptation of neuronal activity in the fmr1 knock-out mouse model of autism. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 6475–6487. [Google Scholar] [CrossRef]
- Berry-Kravis, E. Epilepsy in fragile X syndrome. Dev. Med. Child Neurol. 2002, 44, 724–728. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Filipink, R.A.; Frye, R.E.; Golla, S.; Morris, S.M.; Andrews, H.; Choo, T.-H.; Kaufmann, W.E. The FORWARD consortium seizures in fragile X syndrome: Associations and longitudinal analysis of a large clinic-based cohort. Front. Pediatr. 2021, 9, 736255. [Google Scholar] [CrossRef]
- Hagerman, P.J.; Stafstrom, C.E. Origins of epilepsy in fragile X syndrome. Epilepsy Curr. 2009, 9, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Sohal, V.S.; Rubenstein, J.L.R. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol. Psychiatry 2019, 24, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Lovelace, J.W.; Ethell, I.M.; Binder, D.K.; Razak, K.A. Translation-relevant EEG phenotypes in a mouse model of fragile X syndrome. Neurobiol. Dis. 2018, 115, 39–48. [Google Scholar] [CrossRef]
- Wen, T.H.; Lovelace, J.W.; Ethell, I.M.; Binder, D.K.; Razak, K.A. Developmental changes in EEG phenotypes in a mouse model of fragile X syndrome. Neuroscience 2019, 398, 126–143. [Google Scholar] [CrossRef]
- Wang, J.; Ethridge, L.E.; Mosconi, M.W.; White, S.P.; Binder, D.K.; Pedapati, E.V.; Erickson, C.A.; Byerly, M.J.; Sweeney, J.A. A resting EEG study of neocortical hyperexcitability and altered functional connectivity in fragile X syndrome. J. Neurodev. Disord. 2017, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Mathewson, K.E.; Lleras, A.; Beck, D.M.; Fabiani, M.; Ro, T.; Gratton, G. Pulsed out of awareness: EEG alpha oscillations represent a pulsed-inhibition of ongoing cortical processing. Front. Psychol. 2011, 2, 99. [Google Scholar] [CrossRef]
- Jonak, C.R.; Lovelace, J.W.; Ethell, I.M.; Razak, K.A.; Binder, D.K. Multielectrode array analysis of EEG biomarkers in a mouse model of fragile X syndrome. Neurobiol. Dis. 2020, 138, 104794. [Google Scholar] [CrossRef]
- Castrén, M.; Pääkkönen, A.; Tarkka, I.M.; Ryynänen, M.; Partanen, J. Augmentation of auditory N1 in children with fragile X syndrome. Brain Topogr. 2003, 15, 165–171. [Google Scholar] [CrossRef]
- Rojas, D.C.; Benkers, T.L.; Rogers, S.J.; Teale, P.D.; Reite, M.L.; Hagerman, R.J. Auditory evoked magnetic fields in adults with fragile X syndrome. Neuroreport 2001, 12, 2573–2576. [Google Scholar] [CrossRef]
- Van der Molen, M.J.W.; Van der Molen, M.W.; Ridderinkhof, K.R.; Hamel, B.C.J.; Curfs, L.M.G.; Ramakers, G.J.A. Auditory change detection in fragile X syndrome males: A brain potential study. Clin. Neurophysiol. 2012, 123, 1309–1318. [Google Scholar] [CrossRef]
- Ethridge, L.E.; De Stefano, L.A.; Schmitt, L.M.; Woodruff, N.E.; Brown, K.L.; Tran, M.; Wang, J.; Pedapati, E.V.; Erickson, C.A.; Sweeney, J.A. Auditory EEG biomarkers in fragile X syndrome: Clinical relevance. Front. Integr. Neurosci. 2019, 13, 60. [Google Scholar] [CrossRef] [PubMed]
- Knoth, I.S.; Vannasing, P.; Major, P.; Michaud, J.L.; Lippé, S. Alterations of visual and auditory evoked potentials in fragile X syndrome. Int. J. Dev. Neurosci. 2014, 36, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Côté, V.; Lalancette, È.; Knoth, I.S.; Côté, L.; Agbogba, K.; Vannasing, P.; Major, P.; Barlaam, F.; Michaud, J.; Lippé, S. Distinct patterns of repetition suppression in fragile X syndrome, down syndrome, tuberous sclerosis complex and mutations in SYNGAP1. Brain Res. 2021, 1751, 147205. [Google Scholar] [CrossRef] [PubMed]
- May, P.J.C.; Tiitinen, H. Mismatch negativity (MMN), the deviance-elicited auditory deflection, explained. Psychophysiology 2010, 47, 66–122. [Google Scholar] [CrossRef] [PubMed]
- Näätänen, R.; Sussman, E.S.; Salisbury, D.; Shafer, V.L. Mismatch negativity (MMN) as an index of cognitive dysfunction. Brain Topogr. 2014, 27, 451–466. [Google Scholar] [CrossRef]
- Fishman, Y.I. The mechanisms and meaning of the mismatch negativity. Brain Topogr. 2014, 27, 500–526. [Google Scholar] [CrossRef] [PubMed]
- Van der Molen, M.J.W.; Van der Molen, M.W.; Ridderinkhof, K.R.; Hamel, B.C.J.; Curfs, L.M.G.; Ramakers, G.J.A. Auditory and visual cortical activity during selective attention in fragile X syndrome: A cascade of processing deficiencies. Clin. Neurophysiol. 2012, 123, 720–729. [Google Scholar] [CrossRef]
- Rigoulot, S.; Knoth, I.S.; Lafontaine, M.-P.; Vannasing, P.; Major, P.; Jacquemont, S.; Michaud, J.L.; Jerbi, K.; Lippé, S. Altered visual repetition suppression in fragile X syndrome: New evidence from ERPs and oscillatory activity. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 2017, 59, 52–59. [Google Scholar] [CrossRef]
- St Clair, D.M.; Blackwood, D.H.; Oliver, C.J.; Dickens, P. P3 Abnormality in fragile X syndrome. Biol. Psychiatry 1987, 22, 303–312. [Google Scholar] [CrossRef]
- Cassidy, S.B.; Schwartz, S. Prader-willi and angelman syndromes. disorders of genomic imprinting. Medicine 1998, 77, 140–151. [Google Scholar] [CrossRef]
- Hochstrasser, M. Evolution and function of ubiquitin-like protein-conjugation systems. Nat. Cell Biol. 2000, 2, E153–E157. [Google Scholar] [CrossRef]
- Vatsa, N.; Jana, N.R. UBE3A and its link with autism. Front. Mol. Neurosci. 2018, 11, 448. [Google Scholar] [CrossRef] [PubMed]
- Keute, M.; Miller, M.T.; Krishnan, M.L.; Sadhwani, A.; Chamberlain, S.; Thibert, R.L.; Tan, W.-H.; Bird, L.M.; Hipp, J.F. Angelman syndrome genotypes manifest varying degrees of clinical severity and developmental impairment. Mol. Psychiatry 2021, 26, 3625–3633. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.D.; Cadle, R.G. Adjunct diagnostic test for angelman syndrome: The tuning fork response. Am. J. Med. Genet. 2002, 112, 429. [Google Scholar] [CrossRef] [PubMed]
- Judson, M.C.; Wallace, M.L.; Sidorov, M.S.; Burette, A.C.; Gu, B.; van Woerden, G.M.; King, I.F.; Han, J.E.; Zylka, M.J.; Elgersma, Y.; et al. GABAergic neuron-specific loss of Ube3a causes angelman syndrome-like EEG abnormalities and enhances seizure susceptibility. Neuron 2016, 90, 56–69. [Google Scholar] [CrossRef]
- Sonzogni, M.; Wallaard, I.; Santos, S.S.; Kingma, J.; du Mee, D.; van Woerden, G.M.; Elgersma, Y. A behavioral test battery for mouse models of angelman syndrome: A powerful tool for testing drugs and novel Ube3a mutants. Mol. Autism 2018, 9, 47. [Google Scholar] [CrossRef]
- McCoy, E.S.; Taylor-Blake, B.; Aita, M.; Simon, J.M.; Philpot, B.D.; Zylka, M.J. Enhanced nociception in angelman syndrome model mice. J. Neurosci. 2017, 37, 10230–10239. [Google Scholar] [CrossRef]
- Pelc, K.; Cheron, G.; Dan, B. Behavior and neuropsychiatric manifestations in angelman syndrome. Neuropsychiatr. Dis. Treat. 2008, 4, 577–584. [Google Scholar]
- Thibert, R.L.; Larson, A.M.; Hsieh, D.T.; Raby, A.R.; Thiele, E.A. Neurologic manifestations of angelman syndrome. Pediatr. Neurol. 2013, 48, 271–279. [Google Scholar] [CrossRef]
- Dagli, A.; Buiting, K.; Williams, C.A. Molecular and clinical aspects of angelman syndrome. Mol. Syndromol. 2012, 2, 100–112. [Google Scholar] [CrossRef]
- Minassian, B.A.; DeLorey, T.M.; Olsen, R.W.; Philippart, M.; Bronstein, Y.; Zhang, Q.; Guerrini, R.; Van Ness, P.; Livet, M.O.; Delgado-Escueta, A.V. Angelman syndrome: Correlations between epilepsy phenotypes and genotypes. Ann. Neurol. 1998, 43, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Vendrame, M.; Loddenkemper, T.; Zarowski, M.; Gregas, M.; Shuhaiber, H.; Sarco, D.P.; Morales, A.; Nespeca, M.; Sharpe, C.; Haas, K.; et al. Analysis of EEG patterns and genotypes in patients with angelman syndrome. Epilepsy Behav. EB 2012, 23, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, J.; Miller, M.T.; Bird, L.M.; Garces, P.; Purtell, H.; Hoener, M.C.; Philpot, B.D.; Sidorov, M.S.; Tan, W.-H.; Hernandez, M.-C.; et al. Electrophysiological phenotype in angelman syndrome differs between genotypes. Biol. Psychiatry 2019, 85, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.; Zhu, M.; Glass, M.R.; Rougié, M.; Nikolova, V.D.; Moy, S.S.; Carney, P.R.; Philpot, B.D. Cannabidiol attenuates seizures and EEG abnormalities in angelman syndrome model mice. J. Clin. Invest 2019, 129, 5462–5467. [Google Scholar] [CrossRef]
- Sidorov, M.S.; Deck, G.M.; Dolatshahi, M.; Thibert, R.L.; Bird, L.M.; Chu, C.J.; Philpot, B.D. Delta rhythmicity is a reliable EEG biomarker in angelman syndrome: A parallel mouse and human analysis. J. Neurodev. Disord. 2017, 9, 17. [Google Scholar] [CrossRef]
- Hipp, J.F.; Frohlich, J.; Keute, M.; Tan, W.-H.; Bird, L.M. Electrophysiological abnormalities in angelman syndrome correlate with symptom severity. Biol. Psychiatry Glob. Open Sci. 2021, 1, 201–209. [Google Scholar] [CrossRef]
- Key, A.P.; Jones, D.; Peters, S.; Dold, C. Feasibility of using auditory event-related potentials to investigate learning and memory in nonverbal individuals with angelman syndrome. Brain Cogn. 2018, 128, 73–79. [Google Scholar] [CrossRef]
- Key, A.P.; Jones, D. Social-emotional processing in nonverbal individuals with angelman syndrome: Evidence from brain responses to known and novel names. J. Intellect. Disabil. Res. JIDR 2019, 63, 244–254. [Google Scholar] [CrossRef]
- Egawa, K.; Asahina, N.; Shiraishi, H.; Kamada, K.; Takeuchi, F.; Nakane, S.; Sudo, A.; Kohsaka, S.; Saitoh, S. Aberrant somatosensory-evoked responses imply GABAergic dysfunction in angelman syndrome. NeuroImage 2008, 39, 593–599. [Google Scholar] [CrossRef]
- Egawa, K.; Kitagawa, K.; Inoue, K.; Takayama, M.; Takayama, C.; Saitoh, S.; Kishino, T.; Kitagawa, M.; Fukuda, A. Decreased tonic inhibition in cerebellar granule cells causes motor dysfunction in a mouse model of angelman syndrome. Sci. Transl. Med. 2012, 4, 163ra157. [Google Scholar] [CrossRef]
- Guerrini, R.; Bonanni, P.; de Lorey, T.M.; Serratosa, J.M.; Moncla, A.; Malzac, P.; Dravet, C.; Igvet, M.O.; Bureau, M.; Genton, P.; et al. Cortical myoclonus in angelman syndrome. Ann. Neurol. 1996, 40, 39–48. [Google Scholar] [CrossRef]
- Voytek, B.; Kramer, M.A.; Case, J.; Lepage, K.Q.; Tempesta, Z.R.; Knight, R.T.; Gazzaley, A. Age-related changes in 1/f neural electrophysiological noise. J. Neurosci. 2015, 35, 13257–13265. [Google Scholar] [CrossRef]
- Phelan, K.; McDermid, H.E. The 22q13.3 deletion syndrome (Phelan-mcdermid syndrome). Mol. Syndromol. 2012, 2, 186–201. [Google Scholar] [CrossRef] [PubMed]
- Boeckers, T.M.; Bockmann, J.; Kreutz, M.R.; Gundelfinger, E.D. ProSAP/shank proteins—A family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease. J. Neurochem. 2002, 81, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Kim, E. The shank family of scaffold proteins. J. Cell Sci. 2000, 113 Pt 11, 1851–1856. [Google Scholar] [CrossRef] [PubMed]
- Filice, F.; Vörckel, K.J.; Sungur, A.Ö.; Wöhr, M.; Schwaller, B. Reduction in parvalbumin expression not loss of the parvalbumin-expressing GABA interneuron subpopulation in genetic parvalbumin and shank mouse models of autism. Mol. Brain 2016, 9, 10. [Google Scholar] [CrossRef]
- Vreugdenhil, M.; Jefferys, J.G.R.; Celio, M.R.; Schwaller, B. Parvalbumin-deficiency facilitates repetitive ipscs and gamma oscillations in the hippocampus. J. Neurophysiol. 2003, 89, 1414–1422. [Google Scholar] [CrossRef]
- Chen, Q.; Deister, C.A.; Gao, X.; Guo, B.; Lynn-Jones, T.; Chen, N.; Wells, M.F.; Liu, R.; Goard, M.J.; Dimidschstein, J.; et al. Dysfunction of cortical GABAergic neurons leads to sensory hyper-reactivity in a shank3 mouse model of ASD. Nat. Neurosci. 2020, 23, 520–532. [Google Scholar] [CrossRef]
- Yoo, T.; Cho, H.; Lee, J.; Park, H.; Yoo, Y.-E.; Yang, E.; Kim, J.Y.; Kim, H.; Kim, E. GABA neuronal deletion of shank3 exons 14-16 in mice suppresses striatal excitatory synaptic input and induces social and locomotor abnormalities. Front. Cell. Neurosci. 2018, 12, 341. [Google Scholar] [CrossRef]
- Delling, J.P.; Boeckers, T.M. Comparison of SHANK3 deficiency in animal models: Phenotypes, treatment strategies, and translational implications. J. Neurodev. Disord. 2021, 13, 55. [Google Scholar] [CrossRef]
- Drapeau, E.; Riad, M.; Kajiwara, Y.; Buxbaum, J.D. Behavioral phenotyping of an improved mouse model of phelan–Mcdermid syndrome with a complete deletion of the shank3 gene. eNeuro 2018, 5, 1–55. [Google Scholar] [CrossRef] [PubMed]
- Costales, J.; Kolevzon, A. Phelan–Mcdermid syndrome and SHANK3: Implications for treatment. Neurother. J. Am. Soc. Exp. Neurother. 2015, 12, 620–630. [Google Scholar] [CrossRef]
- Reierson, G.; Bernstein, J.; Froehlich-Santino, W.; Urban, A.; Purmann, C.; Berquist, S.; Jordan, J.; O’Hara, R.; Hallmayer, J. Characterizing regression in phelan mcdermid syndrome (22q13 deletion syndrome). J. Psychiatr. Res. 2017, 91, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Khan, O.I.; Zhou, X.; Leon, J.; Kessler, R.; Gaughan, T.; D’Souza, P.; Gropman, A.; Cohen, N.; Rennert, O.; Buckley, A.; et al. Prospective longitudinal overnight video-EEG evaluation in phelan-mcdermid syndrome. Epilepsy Behav. EB 2018, 80, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Gropman, A.; D’Souza, P.; Thurm, A.; Inati, S. Epilepsy and electroencephalographic features in patients with phelan mcdermid syndrome (P6.270). Neurology 2015, 84, P6.270. [Google Scholar]
- De Rubeis, S.; Siper, P.M.; Durkin, A.; Weissman, J.; Muratet, F.; Halpern, D.; Trelles, M.d.P.; Frank, Y.; Lozano, R.; Wang, A.T.; et al. Delineation of the genetic and clinical spectrum of phelan-mcdermid syndrome caused by SHANK3 point mutations. Mol. Autism 2018, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Holder, J.L.; Quach, M.M. The spectrum of epilepsy and electroencephalographic abnormalities due to SHANK3 loss-of-function mutations. Epilepsia 2016, 57, 1651–1659. [Google Scholar] [CrossRef] [PubMed]
- Mariscal, M.G.; Berry-Kravis, E.; Buxbaum, J.D.; Ethridge, L.E.; Filip-Dhima, R.; Foss-Feig, J.H.; Kolevzon, A.; Modi, M.E.; Mosconi, M.W.; Nelson, C.A.; et al. Shifted phase of EEG cross-frequency coupling in individuals with phelan-mcdermid syndrome. Mol. Autism 2021, 12, 29. [Google Scholar] [CrossRef]
- Siper, P.M.; Rowe, M.A.; Guillory, S.B.; Rouhandeh, A.A.; George-Jones, J.L.; Tavassoli, T.; Lurie, S.; Zweifach, J.; Weissman, J.; Foss-Feig, J.; et al. Visual evoked potential abnormalities in phelan-mcdermid syndrome. J. Am. Acad. Child Adolesc. Psychiatry 2021, 61, 565–574. [Google Scholar] [CrossRef]
- Grosman, H.; Guillory, S.; McLaughlin, C.; Britvan, B.; Isenstein, E.; Keller, K.; Jones, O.; Siper, P.M.; Kolevzon, A.; Foss-Feig, J.H. Examining Gamma Oscillatory Response as a Marker of Disrupted Excitatory/Inhibitory Balance in Autism Spectrum Disorder and Phelan-Mcdermid Syndrome; INSAR: Kansas, MO, USA, 2020. [Google Scholar]
- Ono, Y.; Kudoh, K.; Ikeda, T.; Takahashi, T.; Yoshimura, Y.; Minabe, Y.; Kikuchi, M. Auditory steady-state response at 20 Hz and 40 Hz in young typically developing children and children with autism spectrum disorder. Psychiatry Clin. Neurosci. 2020, 74, 354–361. [Google Scholar] [CrossRef]
- Stroganova, T.; Komarov, K.; Goiaeva, D.; Obukhova, T.; Ovsiannikova, T.; Prokofiev, A.; Orekhova, E. Left hemispheric deficit in the sustained neuromagnetic response to periodic click trains in children with ASD. Mol. Autism 2020, 11, 100. [Google Scholar] [CrossRef] [PubMed]
- Neklyudova, A.K.; Portnova, G.V.; Rebreikina, A.B.; Voinova, V.Y.; Vorsanova, S.G.; Iourov, I.Y.; Sysoeva, O.V. 40-Hz auditory steady-state response (ASSR) as a biomarker of genetic defects in the SHANK3 gene: A case report of 15-year-old girl with a rare partial SHANK3 Duplication. Int. J. Mol. Sci. 2021, 22, 1898. [Google Scholar] [CrossRef] [PubMed]
- Koshiyama, D.; Kirihara, K.; Tada, M.; Nagai, T.; Fujioka, M.; Ichikawa, E.; Ohta, K.; Tani, M.; Tsuchiya, M.; Kanehara, A.; et al. Electrophysiological evidence for abnormal glutamate-GABA association following psychosis onset. Transl. Psychiatry 2018, 8, 211. [Google Scholar] [CrossRef] [PubMed]
- Sivarao, D.V.; Chen, P.; Senapati, A.; Yang, Y.; Fernandes, A.; Benitex, Y.; Whiterock, V.; Li, Y.-W.; Ahlijanian, M.K. 40 Hz auditory steady-state response is a pharmacodynamic biomarker for cortical NMDA receptors. Neuropsychopharmacology 2016, 41, 2232–2240. [Google Scholar] [CrossRef]
- Isenstein, E.; Durkin, A.; Zhang, Y.; Feldman, E.; Servedio, N.; Harony-nicolas, H.; Buxbaum, J.; Kolevzon, A.; Siper, P.; Foss-Feig, J. Electrophysiological evidence of auditory habituation abnormalities in young adults with phelan-mcdermid syndrome. Biol. Psychiatry 2018, 83, S200. [Google Scholar] [CrossRef]
- Reese, M. Effects of Age, Gender, and Genotype on Auditory Processing in Phelan-Mcdermid Syndrome. Master Dissertation, University of Oklahoma, Norman, Oklahoma, 2019. [Google Scholar]
- Ponson, L.; Gomot, M.; Blanc, R.; Barthelemy, C.; Roux, S.; Munnich, A.; Romana, S.; Aguillon-Hernandez, N.; Malan, V.; Bonnet-Brilhault, F. 22q13 deletion syndrome: Communication disorder or autism? Evidence from a specific clinical and neurophysiological phenotype. Transl. Psychiatry 2018, 8, 146. [Google Scholar] [CrossRef]
- Vidal, J.; Bonnet-Brilhault, F.; Roux, S.; Bruneau, N. Auditory evoked potentials to tones and syllables in adults: Evidence of specific influence on N250 wave. Neurosci. Lett. 2005, 378, 145–149. [Google Scholar] [CrossRef]
- Engineer, C.T.; Rahebi, K.C.; Borland, M.S.; Buell, E.P.; Im, K.W.; Wilson, L.G.; Sharma, P.; Vanneste, S.; Harony-Nicolas, H.; Buxbaum, J.D.; et al. Shank3-deficient rats exhibit degraded cortical responses to sound. Autism Res. 2018, 11, 59–68. [Google Scholar] [CrossRef]
- Brittenham, C. Objective Measures of Electrophysiological Responses of Children with Idiopathic Autism Spectrum Disorder and Phelan-Mcdermid Syndrome to a Contrast-Reversing Checkerboard. Master’s Thesis, CUNY Hunter College, New York, NY, USA, 2017. [Google Scholar]
- Moretti, P.; Zoghbi, H.Y. MeCP2 dysfunction in rett syndrome and related disorders. Curr. Opin. Genet. Dev. 2006, 16, 276–281. [Google Scholar] [CrossRef]
- Friez, M.J.; Jones, J.R.; Clarkson, K.; Lubs, H.; Abuelo, D.; Bier, J.-A.B.; Pai, S.; Simensen, R.; Williams, C.; Giampietro, P.F.; et al. Recurrent infections, hypotonia, and mental retardation caused by duplication of MECP2 and adjacent region in Xq28. Pediatrics 2006, 118, e1687–e1695. [Google Scholar] [CrossRef]
- Erhardt, E.B.; Rachakonda, S.; Bedrick, E.J.; Allen, E.A.; Adali, T.; Calhoun, V.D. Comparison of multi-subject ICA methods for analysis of FMRI data. Hum. Brain Mapp. 2011, 32, 2075–2095. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.-T.; Chen, H.; Samaco, R.C.; Xue, M.; Chahrour, M.; Yoo, J.; Neul, J.L.; Gong, S.; Lu, H.-C.; Heintz, N.; et al. Dysfunction in GABA signalling mediates autism-like stereotypies and rett syndrome phenotypes. Nature 2010, 468, 263–269. [Google Scholar] [CrossRef]
- Bhattacherjee, A.; Mu, Y.; Winter, M.K.; Knapp, J.R.; Eggimann, L.S.; Gunewardena, S.S.; Kobayashi, K.; Kato, S.; Krizsan-Agbas, D.; Smith, P.G. Neuronal cytoskeletal gene dysregulation and mechanical hypersensitivity in a rat model of rett syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, E6952–E6961. [Google Scholar] [CrossRef] [PubMed]
- Tarquinio, D.C.; Hou, W.; Berg, A.; Kaufmann, W.E.; Lane, J.B.; Skinner, S.A.; Motil, K.J.; Neul, J.L.; Percy, A.K.; Glaze, D.G. Longitudinal course of epilepsy in rett syndrome and related disorders. Brain 2017, 140, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, K.; Stroganova, T.; Molholm, S.; Sysoeva, O. Reviewing evidence for the relationship of EEG abnormalities and RTT phenotype paralleled by insights from animal studies. Int. J. Mol. Sci. 2021, 22, 5308. [Google Scholar] [CrossRef]
- Roche, K.J.; LeBlanc, J.J.; Levin, A.R.; O’Leary, H.M.; Baczewski, L.M.; Nelson, C.A. Electroencephalographic spectral power as a marker of cortical function and disease severity in girls with rett syndrome. J. Neurodev. Disord. 2019, 11, 15. [Google Scholar] [CrossRef]
- Lang, M.; Wither, R.G.; Colic, S.; Wu, C.; Monnier, P.P.; Bardakjian, B.L.; Zhang, L.; Eubanks, J.H. Rescue of behavioral and EEG deficits in male and female Mecp2-deficient mice by delayed mecp2 gene reactivation. Hum. Mol. Genet. 2014, 23, 303–318. [Google Scholar] [CrossRef]
- Paterno, R.; Marafiga, J.R.; Ramsay, H.; Li, T.; Salvati, K.A.; Baraban, S.C. Hippocampal gamma and sharp-wave ripple oscillations are altered in a cntnap2 mouse model of autism spectrum disorder. Cell Rep. 2021, 37, 109970. [Google Scholar] [CrossRef]
- Sysoeva, O.V.; Smirnov, K.; Stroganova, T.A. Sensory evoked potentials in patients with rett syndrome through the lens of animal studies: Systematic review. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2020, 131, 213–224. [Google Scholar] [CrossRef]
- Sysoeva, O.V.; Molholm, S.; Djukic, A.; Frey, H.-P.; Foxe, J.J. Atypical processing of tones and phonemes in rett syndrome as biomarkers of disease progression. Transl. Psychiatry 2020, 10, 188. [Google Scholar] [CrossRef]
- Badr, G.G.; Witt-Engerström, I.; Hagberg, B. Brain stem and spinal cord impairment in rett syndrome: Somatosensory and auditory evoked responses investigations. Brain Dev. 1987, 9, 517–522. [Google Scholar] [CrossRef]
- Goffin, D.; Allen, M.; Zhang, L.; Amorim, M.; Wang, I.-T.J.; Reyes, A.-R.S.; Mercado-Berton, A.; Ong, C.; Cohen, S.; Hu, L.; et al. Rett syndrome mutation MeCP2 T158A disrupts DNA binding, protein stability and ERP responses. Nat. Neurosci. 2011, 15, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Foxe, J.J.; Burke, K.M.; Andrade, G.N.; Djukic, A.; Frey, H.-P.; Molholm, S. Automatic cortical representation of auditory pitch changes in rett syndrome. J. Neurodev. Disord. 2016, 8, 34. [Google Scholar] [CrossRef]
- Brima, T.; Molholm, S.; Molloy, C.J.; Sysoeva, O.V.; Nicholas, E.; Djukic, A.; Freedman, E.G.; Foxe, J.J. Auditory sensory memory span for duration is severely curtailed in females with rett syndrome. Transl. Psychiatry 2019, 9, 130. [Google Scholar] [CrossRef]
- LeBlanc, J.J.; DeGregorio, G.; Centofante, E.; Vogel-Farley, V.K.; Barnes, K.; Kaufmann, W.E.; Fagiolini, M.; Nelson, C.A. Visual evoked potentials detect cortical processing deficits in rett syndrome. Ann. Neurol. 2015, 78, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Saunders, K.J.; McCulloch, D.L.; Kerr, A.M. Visual function in rett syndrome. Dev. Med. Child Neurol. 1995, 37, 496–504. [Google Scholar] [CrossRef]
- Yamanouchi, H.; Kaga, M.; Arima, M. Abnormal cortical excitability in rett syndrome. Pediatr. Neurol. 1993, 9, 202–206. [Google Scholar] [CrossRef]
- Yoshikawa, H.; Kaga, M.; Suzuki, H.; Sakuragawa, N.; Arima, M. Giant somatosensory evoked potentials in the rett syndrome. Brain Dev. 1991, 13, 36–39. [Google Scholar] [CrossRef]
- Zhang, L.; Wither, R.G.; Lang, M.; Wu, C.; Sidorova-Darmos, E.; Netchev, H.; Matolcsy, C.B.; Snead, O.C.; Eubanks, J.H. A role for diminished GABA transporter activity in the cortical discharge phenotype of MeCP2-deficient mice. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2016, 41, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Brió, M.C.; Fazzina, M.; Chindi, M. Tuberous sclerosis complex associated with autism spectrum features and bumetanide as a pharmacological indication: A case report. Open, J. Psychiatry 2021, 11, 202–213. [Google Scholar] [CrossRef]
- Sato, A.; Kasai, S.; Kobayashi, T.; Takamatsu, Y.; Hino, O.; Ikeda, K.; Mizuguchi, M. Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat. Commun. 2012, 3, 1292. [Google Scholar] [CrossRef] [PubMed]
- Moavero, R.; Benvenuto, A.; Emberti Gialloreti, L.; Siracusano, M.; Kotulska, K.; Weschke, B.; Riney, K.; Jansen, F.E.; Feucht, M.; Krsek, P.; et al. Early clinical predictors of autism spectrum disorder in infants with tuberous sclerosis complex: Results from the EPISTOP study. J. Clin. Med. 2019, 8, 788. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, A.; Varcin, K.J.; Sahin, M.; Nelson, C.A.; Jeste, S.S. Early patterns of functional brain development associated with autism spectrum disorder in tuberous sclerosis complex. Autism Res. Off. J. Int. Soc. Autism Res. 2019, 12, 1758–1773. [Google Scholar] [CrossRef] [PubMed]
- De Ridder, J.; Lavanga, M.; Verhelle, B.; Vervisch, J.; Lemmens, K.; Kotulska, K.; Moavero, R.; Curatolo, P.; Weschke, B.; Riney, K.; et al. Prediction of neurodevelopment in infants with tuberous sclerosis complex using early EEG characteristics. Front. Neurol. 2020, 11, 582891. [Google Scholar] [CrossRef]
- Seri, S.; Cerquiglini, A.; Pisani, F.; Curatolo, P. Autism in tuberous sclerosis: Evoked potential evidence for a deficit in auditory sensory processing. Clin. Neurophysiol. 1999, 110, 1825–1830. [Google Scholar] [CrossRef]
- O’Brien, A.M.; Bayet, L.; Riley, K.; Nelson, C.A.; Sahin, M.; Modi, M.E. Auditory processing of speech and tones in children with tuberous sclerosis complex. Front. Integr. Neurosci. 2020, 14, 14. [Google Scholar] [CrossRef]
- Jeste, S.S.; Hirsch, S.; Vogel-Farley, V.; Norona, A.; Navalta, M.-C.; Gregas, M.C.; Prabhu, S.P.; Sahin, M.; Nelson, C.A. Atypical face processing in children with tuberous sclerosis complex. J. Child Neurol. 2013, 28, 1569–1576. [Google Scholar] [CrossRef]
- Tye, C.; Farroni, T.; Volein, Á.; Mercure, E.; Tucker, L.; Johnson, M.H.; Bolton, P.F. Autism diagnosis differentiates neurophysiological responses to faces in adults with tuberous sclerosis complex. J. Neurodev. Disord. 2015, 7, 33. [Google Scholar] [CrossRef][Green Version]
- Varcin, K.J.; Nelson, C.A.; Ko, J.; Sahin, M.; Wu, J.Y.; Jeste, S.S. Visual evoked potentials as a readout of cortical function in infants with tuberous sclerosis complex. J. Child Neurol. 2016, 31, 195–202. [Google Scholar] [CrossRef]
- Bateup, H.S.; Johnson, C.A.; Denefrio, C.L.; Saulnier, J.L.; Kornacker, K.; Sabatini, B.L. Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 2013, 78, 510–522. [Google Scholar] [CrossRef]
- Mori, K.; Mori, T.; Toda, Y.; Fujii, E.; Miyazaki, M.; Harada, M.; Kagami, S. Decreased benzodiazepine receptor and increased GABA level in cortical tubers in tuberous sclerosis complex. Brain Dev. 2012, 34, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Kallionpää, R.A.; Uusitalo, E.; Leppävirta, J.; Pöyhönen, M.; Peltonen, S.; Peltonen, J. Prevalence of neurofibromatosis type 1 in the finnish population. Genet. Med. 2018, 20, 1082–1086. [Google Scholar] [CrossRef] [PubMed]
- Ferner, R.E. Neurofibromatosis 1 and neurofibromatosis 2: A twenty first century perspective. Lancet Neurol. 2007, 6, 340–351. [Google Scholar] [CrossRef]
- Cui, Y.; Costa, R.M.; Murphy, G.G.; Elgersma, Y.; Zhu, Y.; Gutmann, D.H.; Parada, L.F.; Mody, I.; Silva, A.J. Neurofibromin regulation of ERK signaling modulates GABA release and learning. Cell 2008, 135, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Kolesnik, A.M.; Jones, E.J.H.; Garg, S.; Green, J.; Charman, T.; Johnson, M.H.; Baron-Cohen, S.; Begum, J.; Bolton, P.; Cheung, C.; et al. Early development of infants with neurofibromatosis type 1: A case series. Mol. Autism 2017, 8, 62. [Google Scholar] [CrossRef] [PubMed]
- Serdaroglu, E.; Konuskan, B.; Karli Oguz, K.; Gurler, G.; Yalnizoglu, D.; Anlar, B. Epilepsy in neurofibromatosis type 1: Diffuse cerebral dysfunction? Epilepsy Behav. 2019, 98, 6–9. [Google Scholar] [CrossRef]
- Ribeiro, M.J.; d’Almeida, O.C.; Ramos, F.; Saraiva, J.; Silva, E.D.; Castelo-Branco, M. Abnormal late visual responses and alpha oscillations in neurofibromatosis type 1: A link to visual and attention deficits. J. Neurodev. Disord. 2014, 6, 4. [Google Scholar] [CrossRef]
- North, K.; Joy, P.; Yuille, D.; Cocks, N.; Mobbs, E.; Hutchins, P.; McHugh, K.; Silva, M.d. Specific learning disability in children with neurofibromatosis type 1: Significance of MRI abnormalities. Neurology 1994, 44, 878. [Google Scholar] [CrossRef]
- Tonsgard, J.H. Clinical manifestations and management of neurofibromatosis type 1. Semin. Pediatr. Neurol. 2006, 13, 2–7. [Google Scholar] [CrossRef]
- Ammendola, A.; Ciccone, G.; Ammendola, E. Utility of multimodal evoked potentials study in neurofibromatosis type 1 of childhood. Pediatr. Neurol. 2006, 34, 276–280. [Google Scholar] [CrossRef]
- Yerdelen, D.; Koc, F.; Durdu, M.; Karakas, M. Electrophysiological findings in neurofibromatosis type 1. J. Neurol. Sci. 2011, 306, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Begum-Ali, J.; Kolesnik-Taylor, A.; Quiroz, I.; Mason, L.; Garg, S.; Green, J.; Johnson, M.H.; Jones, E.J.H.; Holman, R.; Kalwarowsky, S.; et al. Early differences in auditory processing relate to autism spectrum disorder traits in infants with neurofibromatosis type I. J. Neurodev. Disord. 2021, 13, 22. [Google Scholar] [CrossRef] [PubMed]
- Dilts, C.V.; Carey, J.C.; Kircher, J.C.; Hoffman, R.O.; Creel, D.; Ward, K.; Clark, E.; Leonard, C.O. Children and adolescents with neurofibromatosis 1: A behavioral phenotype. J. Dev. Behav. Pediatr. JDBP 1996, 17, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Iannaccone, A.; McCluney, R.A.; Brewer, V.R.; Spiegel, P.H.; Taylor, J.S.; Kerr, N.C.; Pivnick, E.K. Visual evoked potentials in children with neurofibromatosis type 1. Doc. Ophthalmol. Adv. Ophthalmol. 2002, 105, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Dijk, H.v.; Schoffelen, J.-M.; Oostenveld, R.; Jensen, O. Prestimulus oscillatory activity in the alpha band predicts visual discrimination ability. J. Neurosci. 2008, 28, 1816–1823. [Google Scholar] [CrossRef] [PubMed]
- Thut, G.; Nietzel, A.; Brandt, S.A.; Pascual-Leone, A. Alpha-band electroencephalographic activity over occipital cortex indexes visuospatial attention bias and predicts visual target detection. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 9494–9502. [Google Scholar] [CrossRef]
- Foxe, J.J.; Snyder, A.C. The role of alpha-band brain oscillations as a sensory suppression mechanism during selective attention. Front. Psychol. 2011, 2, 154. [Google Scholar] [CrossRef]
- Lidzba, K.; Granstroem, S.; Leark, R.A.; Kraegeloh-Mann, I.; Mautner, V.-F. Pharmacotherapy of attention deficit in neurofibromatosis type 1: Effects on cognition. Neuropediatrics 2014, 45, 240–246. [Google Scholar] [CrossRef]
- Ribeiro, M.J.; Violante, I.R.; Bernardino, I.; Edden, R.A.E.; Castelo-Branco, M. Abnormal relationship between GABA, neurophysiology and impulsive behavior in neurofibromatosis type 1. Cortex, J. Devoted Study Nerv. Syst. Behav. 2015, 64, 194–208. [Google Scholar] [CrossRef]
- Huster, R.J.; Enriquez-Geppert, S.; Lavallee, C.F.; Falkenstein, M.; Herrmann, C.S. Electroencephalography of response inhibition tasks: Functional networks and cognitive contributions. Int. J. Psychophysiol. Off. J. Int. Organ. Psychophysiol. 2013, 87, 217–233. [Google Scholar] [CrossRef]
- Violante, I.R.; Patricio, M.; Bernardino, I.; Rebola, J.; Abrunhosa, A.J.; Ferreira, N.; Castelo-Branco, M. GABA deficiency in NF1: A multimodal [11C]-flumazenil and spectroscopy study. Neurology 2016, 87, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Bluschke, A.; von der Hagen, M.; Papenhagen, K.; Roessner, V.; Beste, C. Conflict processing in juvenile patients with neurofibromatosis type 1 (NF1) and healthy controls—Two pathways to success. NeuroImage Clin. 2017, 14, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Pobric, G.; Taylor, J.R.; Ramalingam, H.M.; Pye, E.; Robinson, L.; Vassallo, G.; Jung, J.; Bhandary, M.; Szumanska-Ryt, K.; Theodosiou, L.; et al. Cognitive and electrophysiological correlates of working memory impairments in neurofibromatosis type 1. J. Autism Dev. Disord. 2021, 52, 1478–1494. [Google Scholar] [CrossRef] [PubMed]
- Polich, J. Updating P300: An integrative theory of P3a and P3b. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2007, 118, 2128–2148. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Barstein, J.; Ethridge, L.E.; Mosconi, M.W.; Takarae, Y.; Sweeney, J.A. Resting state EEG abnormalities in autism spectrum disorders. J. Neurodev. Disord. 2013, 5, 24. [Google Scholar] [CrossRef]
- Clarke, A.R.; Barry, R.J.; Johnstone, S. Resting state EEG power research in attention-deficit/hyperactivity disorder: A review update. Clin. Neurophysiol. 2020, 131, 1463–1479. [Google Scholar] [CrossRef]
- Hamburg, S.; Bush, D.; Strydom, A.; Startin, C.M. Comparison of resting-state EEG between adults with down syndrome and typically developing controls. J. Neurodev. Disord. 2021, 13, 48. [Google Scholar] [CrossRef]
- Mason, L.M.; Barry, R.J.; Clarke, A.R. Age-related changes in the EEG in an Eyes-open condition: I. normal development. Int. J. Psychophysiol. 2022, 172, 40–45. [Google Scholar] [CrossRef]
- Wilkinson, C.L.; Nelson, C.A. Increased aperiodic gamma power in young boys with fragile X syndrome is associated with better language ability. Mol. Autism 2021, 12, 17. [Google Scholar] [CrossRef]
- Kepecs, A.; Fishell, G. Interneuron cell types: Fit to form and formed to fit. Nature 2014, 505, 318–326. [Google Scholar] [CrossRef]
- van Diessen, E.; Senders, J.; Jansen, F.E.; Boersma, M.; Bruining, H. Increased power of resting-state gamma oscillations in autism spectrum disorder detected by routine electroencephalography. Eur. Arch. Psychiatry Clin. Neurosci. 2015, 265, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, C.R.; Villalobos, M.E.; Schultz, R.T.; Herpertz-Dahlmann, B.; Konrad, K.; Kohls, G. Atypical laterality of resting gamma oscillations in autism spectrum disorders. J. Autism Dev. Disord. 2015, 45, 292–297. [Google Scholar] [CrossRef]
- Williams, Z.J.; Abdelmessih, P.G.; Key, A.P.; Woynaroski, T.G. Cortical auditory processing of simple stimuli is altered in autism: A meta-analysis of auditory evoked responses. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2021, 6, 767–781. [Google Scholar] [CrossRef]
- Kovarski, K.; Malvy, J.; Khanna, R.K.; Arsène, S.; Batty, M.; Latinus, M. Reduced visual evoked potential amplitude in autism spectrum disorder, a variability effect? Transl. Psychiatry 2019, 9, 293. [Google Scholar] [CrossRef]
- Pei, F.; Baldassi, S.; Norcia, A.M. Electrophysiological measures of low-level vision reveal spatial processing deficits and hemispheric asymmetry in autism spectrum disorder. J. Vis. 2014, 14, 3. [Google Scholar] [CrossRef]
- Roberts, T.P.L.; Matsuzaki, J.; Blaskey, L.; Bloy, L.; Edgar, J.C.; Kim, M.; Ku, M.; Kuschner, E.S.; Embick, D. Delayed M50/M100 evoked response component latency in minimally verbal/nonverbal children who have autism spectrum disorder. Mol. Autism 2019, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, J.; Kagitani-Shimono, K.; Goto, T.; Sanefuji, W.; Yamamoto, T.; Sakai, S.; Uchida, H.; Hirata, M.; Mohri, I.; Yorifuji, S.; et al. Differential responses of primary auditory cortex in autistic spectrum disorder with auditory hypersensitivity. NeuroReport 2012, 23, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wang, S.; Huang, D.; Wu, X.; Zhang, Y. Role of inter-trial phase coherence in atypical auditory evoked potentials to speech and nonspeech stimuli in children with autism. Clin. Neurophysiol. 2018, 129, 1374–1382. [Google Scholar] [CrossRef] [PubMed]
| Genetic Cause | Autism | Sensory Deficit | Intellectual Disability (ID) | Language Problem | Other Deficits | |
|---|---|---|---|---|---|---|
| FXS | FMR1 in chromosome X [24] | 22% [25,26] | auditory, tactile, and visual defensiveness or avoidance [27,28,29] | ID varies from mild to moderate degree [25] | expressive and receptive skills are mildly delayed [30] | hypotonia, ADHD 60–80% [31,32] |
| AS | UBE3A (+GABRB3, GABRA5, GABRG3) in chromosome 15 [33,34] | 25–40% [35] | hypo and hyperresponsiveness, hyporesponsiveness decreases with age; heat sensitivity, water attraction, sensory seeking [27,36,37,38,39] | 100%, severity depends on types of mutation (moderate to severe ID [40,41] | 80% delay in expressive language with minimal or no use of words [42] | hypotonia, attention deficit [36,40,41,43] |
| PMS | SHANK3 in chromosome 22 [44] | 50–75% [45,46] | high sensory threshold, hyporeactivity [47,48,49] | 53% profound ID, 23% severe ID, 10% moderate ID, 10% mild ID [45] | 100% delay, 50% had no expressive speech [45,50] | hypotonia, bipolar disorder—54% psychosis—12% irritability—36% [45,50] |
| RS | MECP2 in chromosome X [51,52] | 60% [35] | reduced pain threshold [53,54,55] | 100% severe ID [56] | 80–90% language in regression period, could say few or no words [56] | hypotonia motor stereotypes [57,58] |
| TSC | TSC1 and TSC2 in chromosome 9 [59] | 36% [60,61] | n/a | 64% severe ID [62] | 24–66% abnormal language: difficulties in expressive vocabulary and semantic-grammatical abilities, abstract language skills [60,62] | ADHD, anxiety, sleep disorders [63] |
| NF1 | NF1 in chromosome 17 [64] | 10–40% [65,66] | n/a | 5–33% mild ID [67,68] | 70% mild difficulties in articulation, naming, receptive and expressive vocabulary [67] | ADHD in 38% [69,70] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neklyudova, A.; Smirnov, K.; Rebreikina, A.; Martynova, O.; Sysoeva, O. Electrophysiological and Behavioral Evidence for Hyper- and Hyposensitivity in Rare Genetic Syndromes Associated with Autism. Genes 2022, 13, 671. https://doi.org/10.3390/genes13040671
Neklyudova A, Smirnov K, Rebreikina A, Martynova O, Sysoeva O. Electrophysiological and Behavioral Evidence for Hyper- and Hyposensitivity in Rare Genetic Syndromes Associated with Autism. Genes. 2022; 13(4):671. https://doi.org/10.3390/genes13040671
Chicago/Turabian StyleNeklyudova, Anastasia, Kirill Smirnov, Anna Rebreikina, Olga Martynova, and Olga Sysoeva. 2022. "Electrophysiological and Behavioral Evidence for Hyper- and Hyposensitivity in Rare Genetic Syndromes Associated with Autism" Genes 13, no. 4: 671. https://doi.org/10.3390/genes13040671
APA StyleNeklyudova, A., Smirnov, K., Rebreikina, A., Martynova, O., & Sysoeva, O. (2022). Electrophysiological and Behavioral Evidence for Hyper- and Hyposensitivity in Rare Genetic Syndromes Associated with Autism. Genes, 13(4), 671. https://doi.org/10.3390/genes13040671