
Parathyroid Hormone Induces Human Valvular Endothelial Cells Dysfunction That Impacts the Osteogenic Phenotype of Valvular Interstitial Cells

 , ,
, ,
Abstract
:1. Introduction
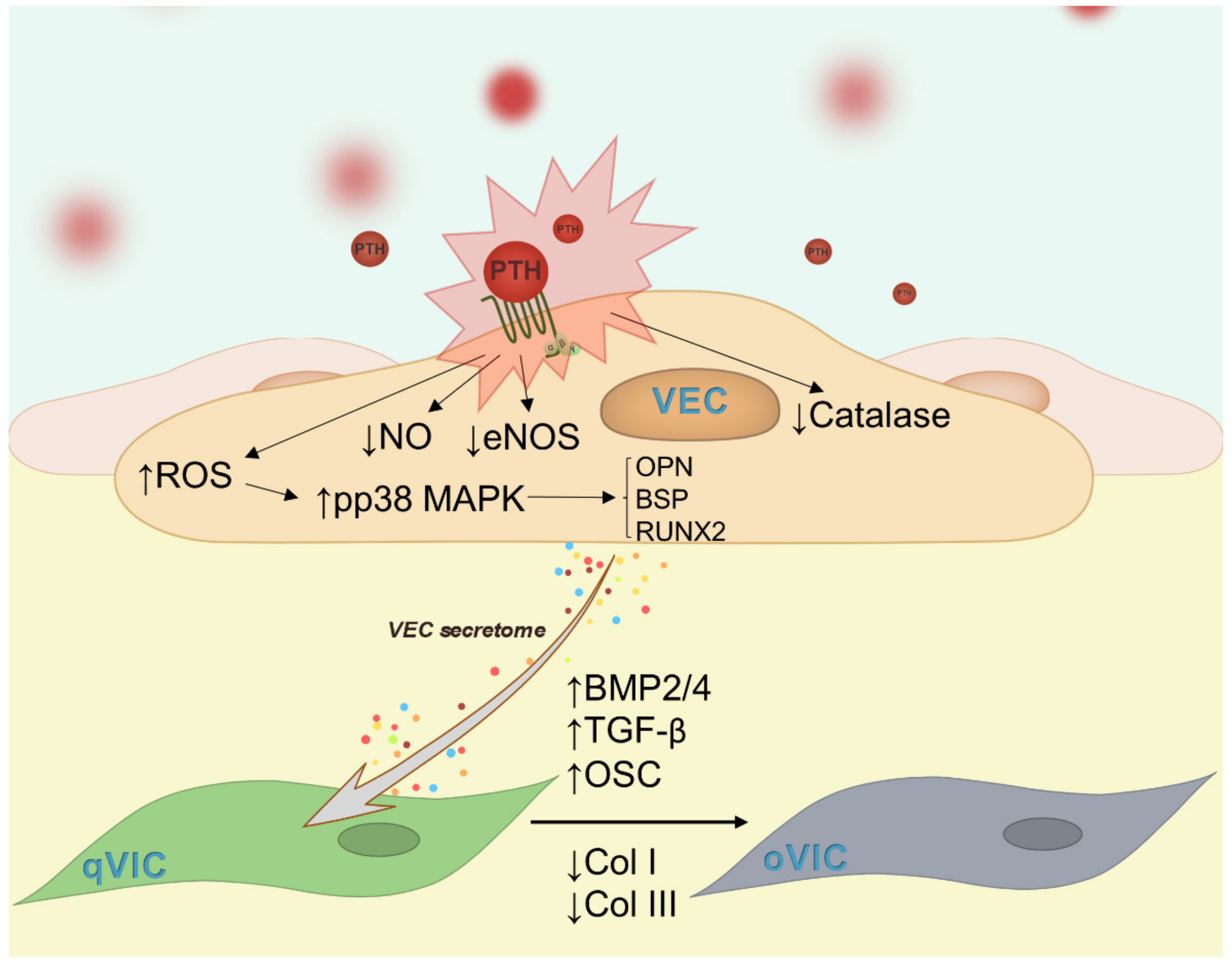
2. Results
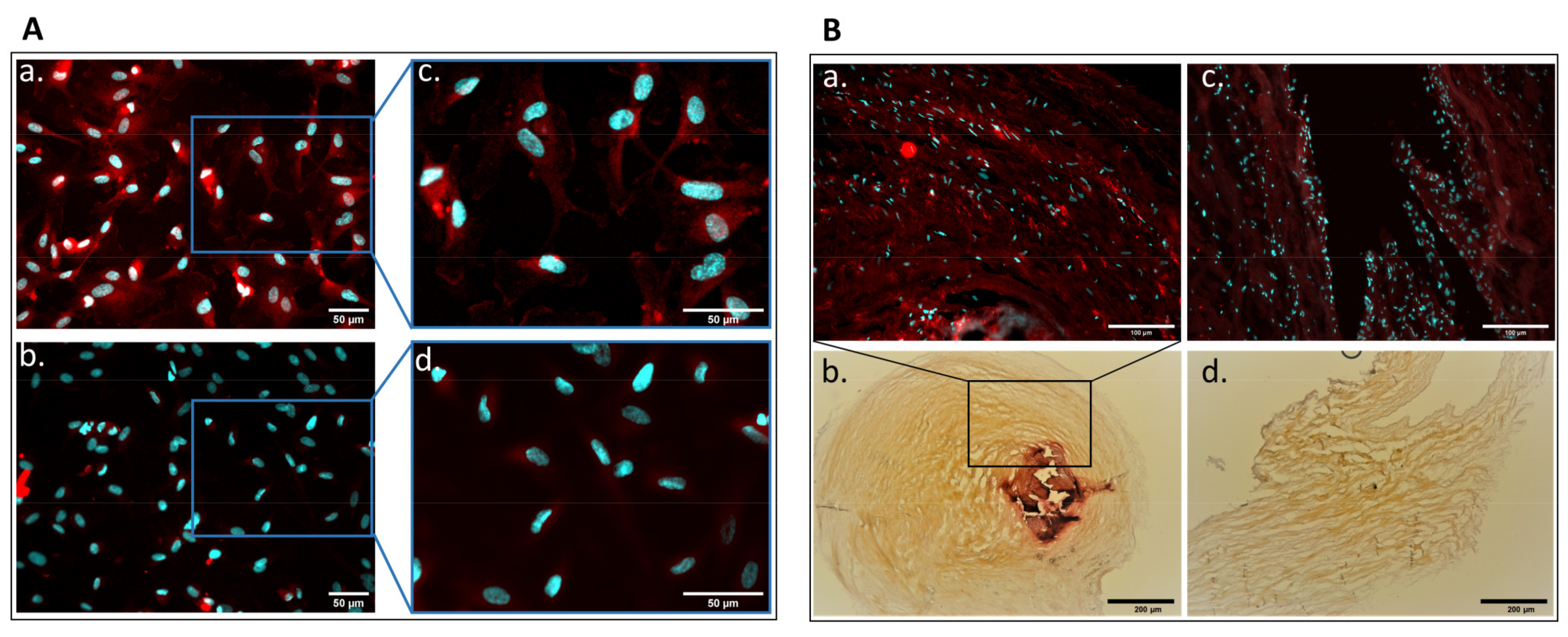
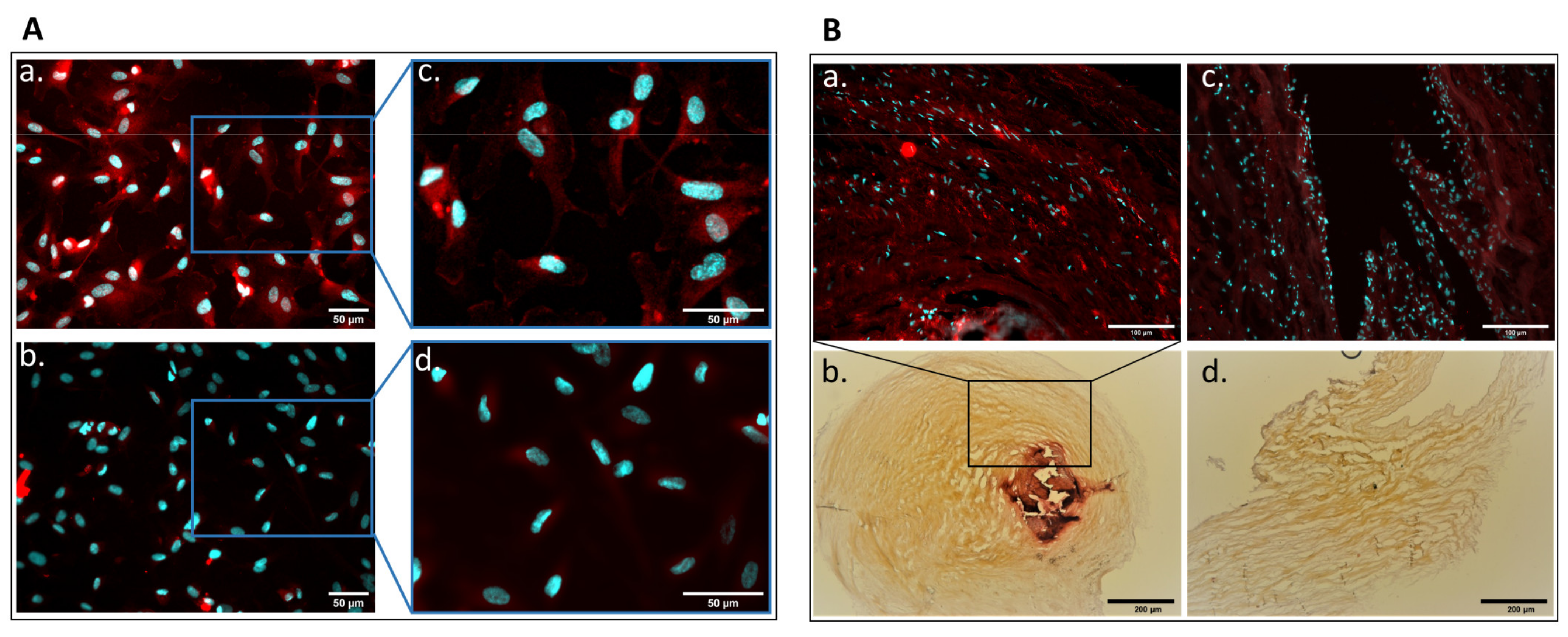
2.1. Detection of Parathyroid Hormone Receptor Expression in Human Aortic Valve and Cultured Valvular Endothelial Cells
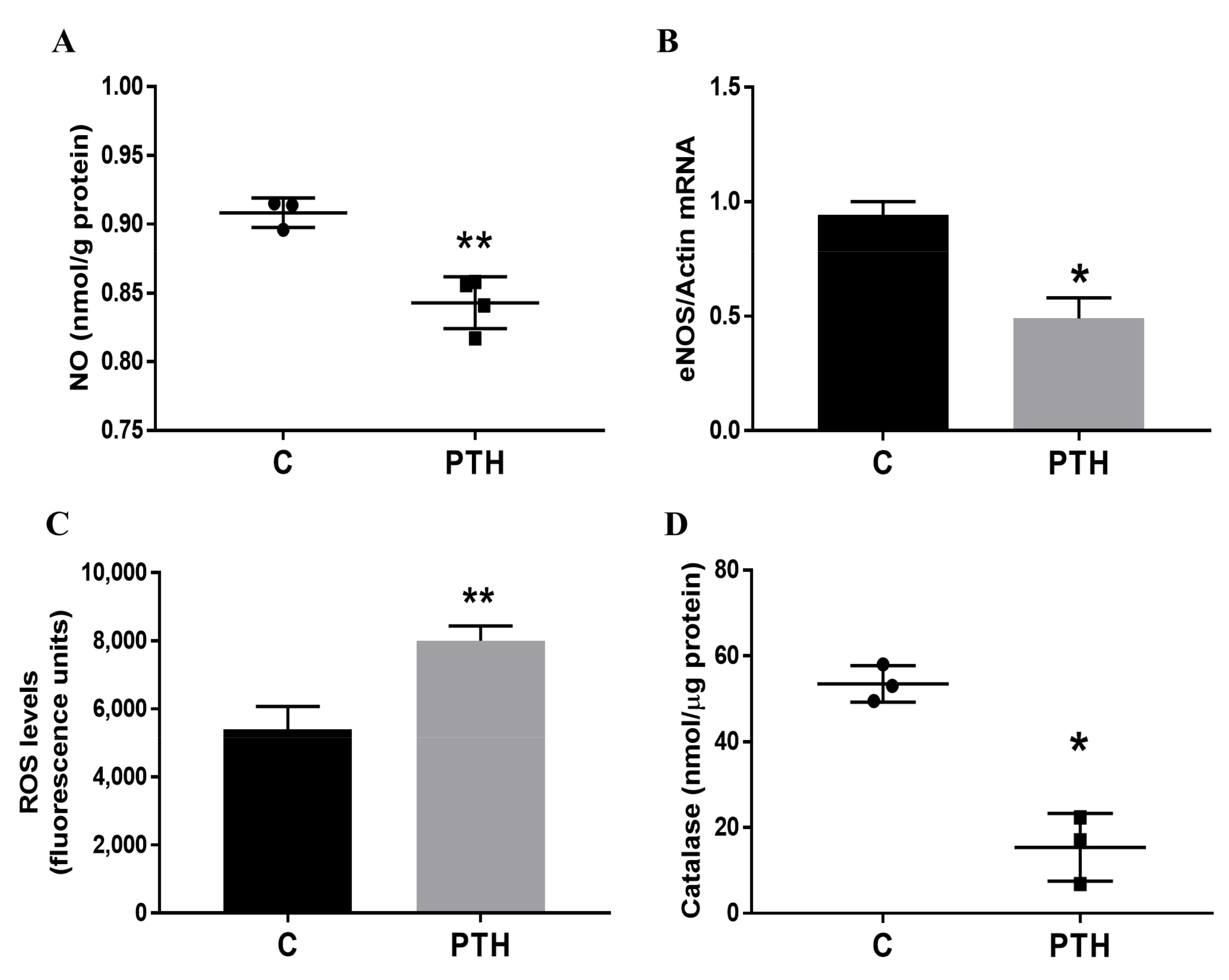
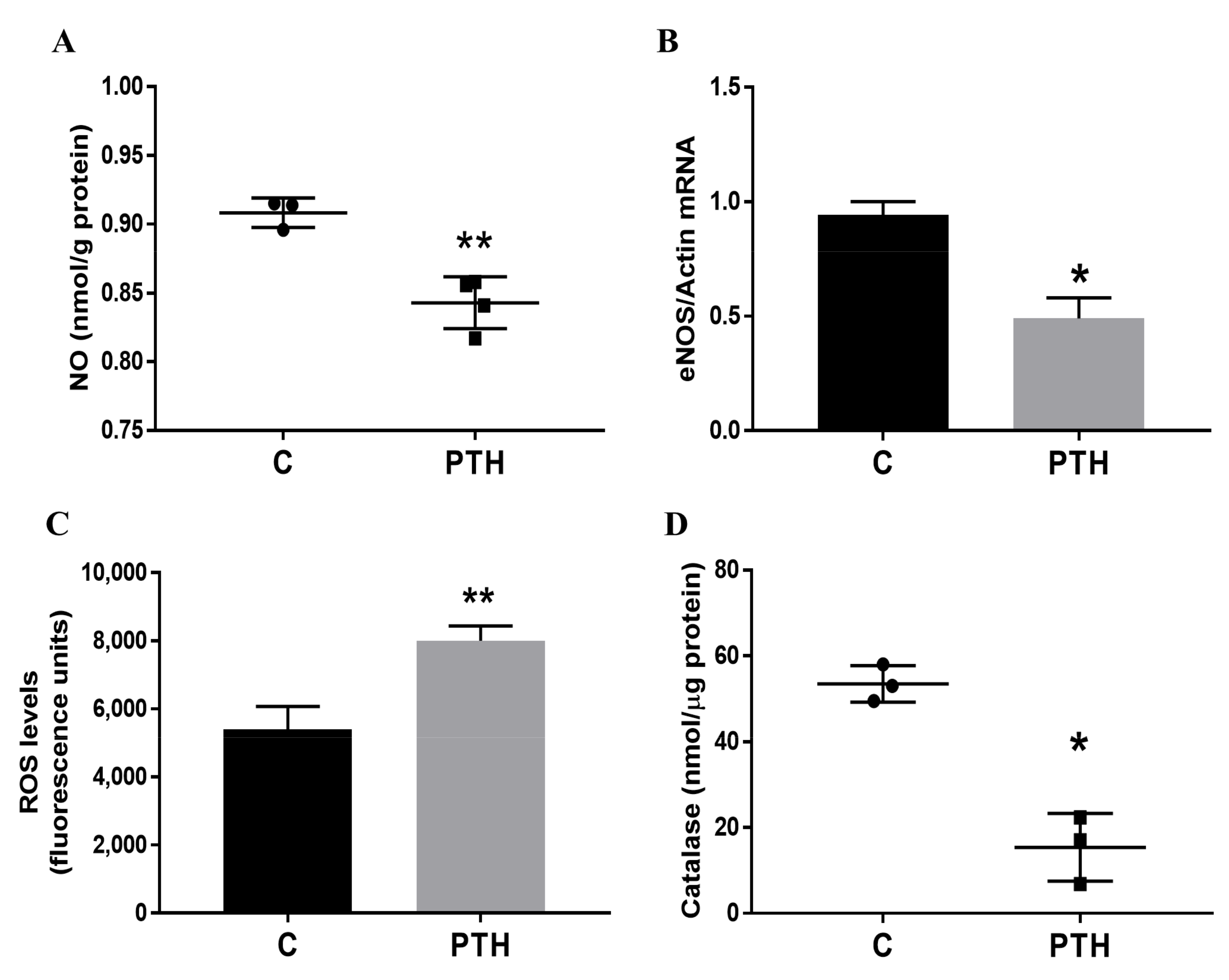
2.2. PTH Induces Dysfunction of Human Valvular Endothelial Cells
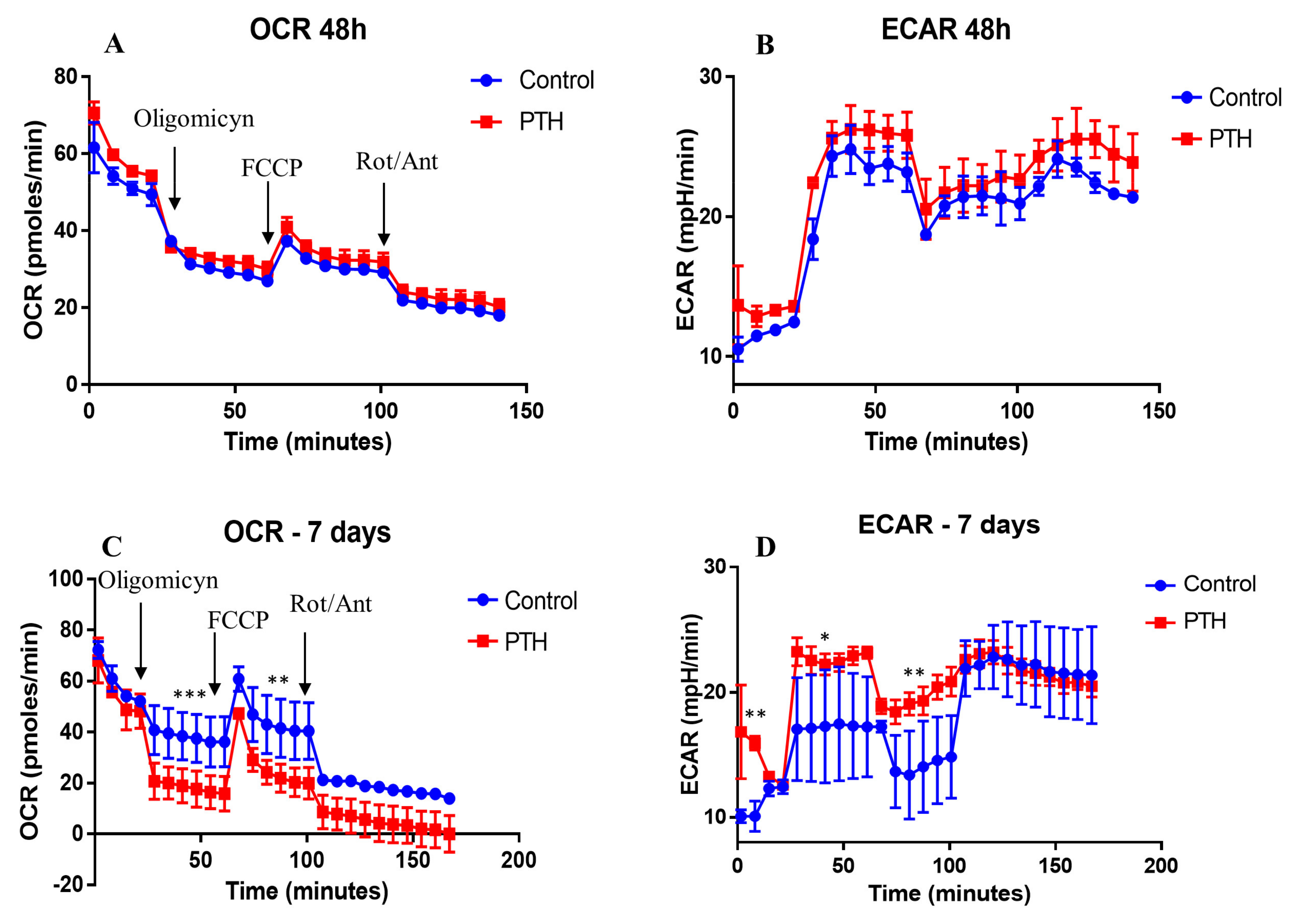
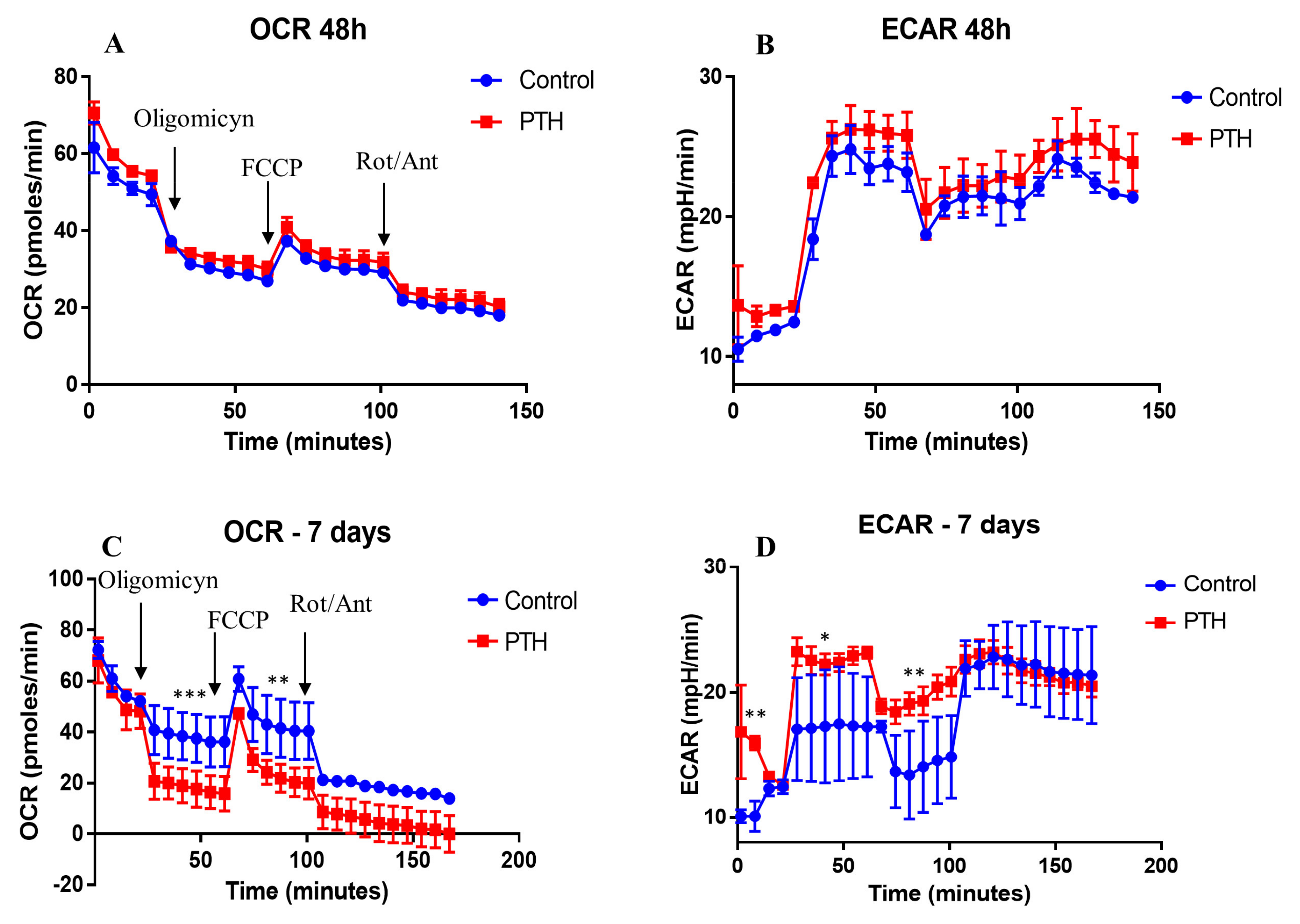
2.3. Energetic Metabolism in VEC Is Modified by PTH
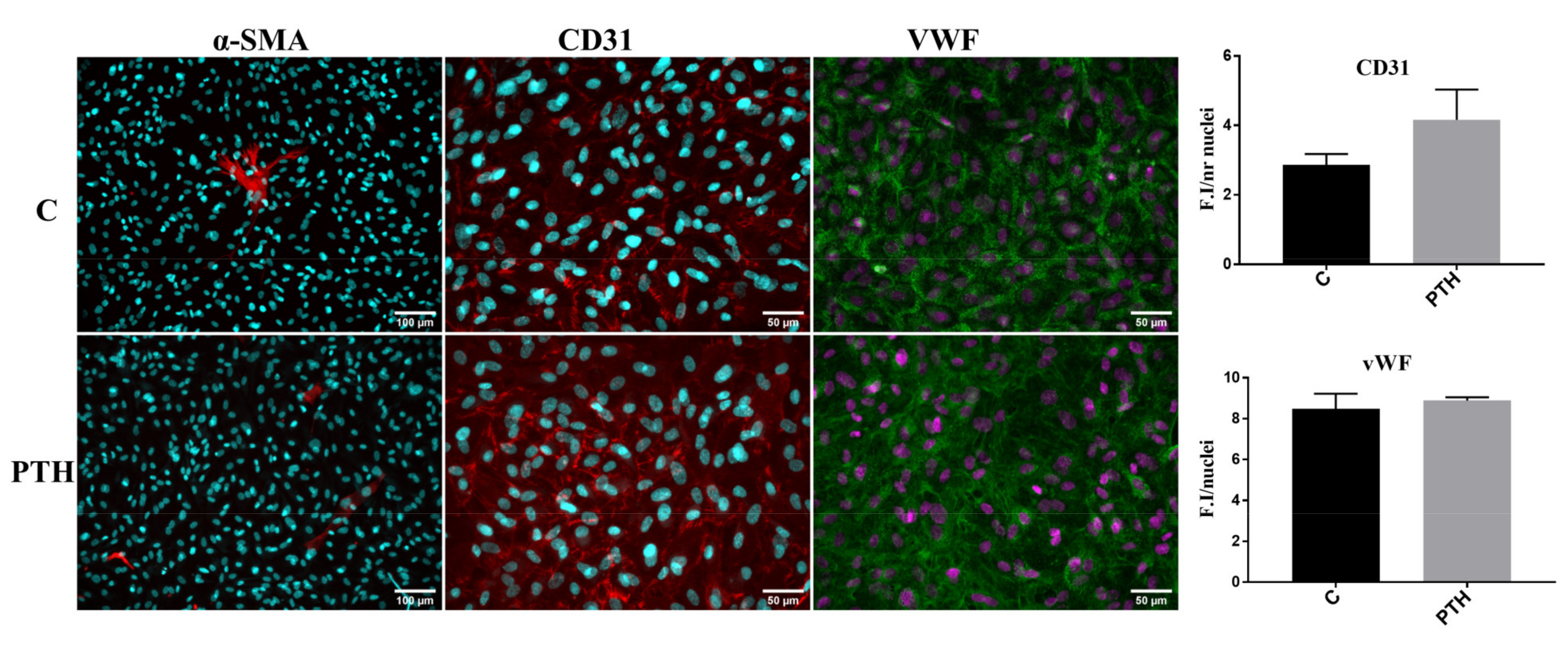
2.4. PTH Does Not Modify the Expression of Endothelial and Mesenchymal Markers: CD31, vWF and α-SMA in VEC
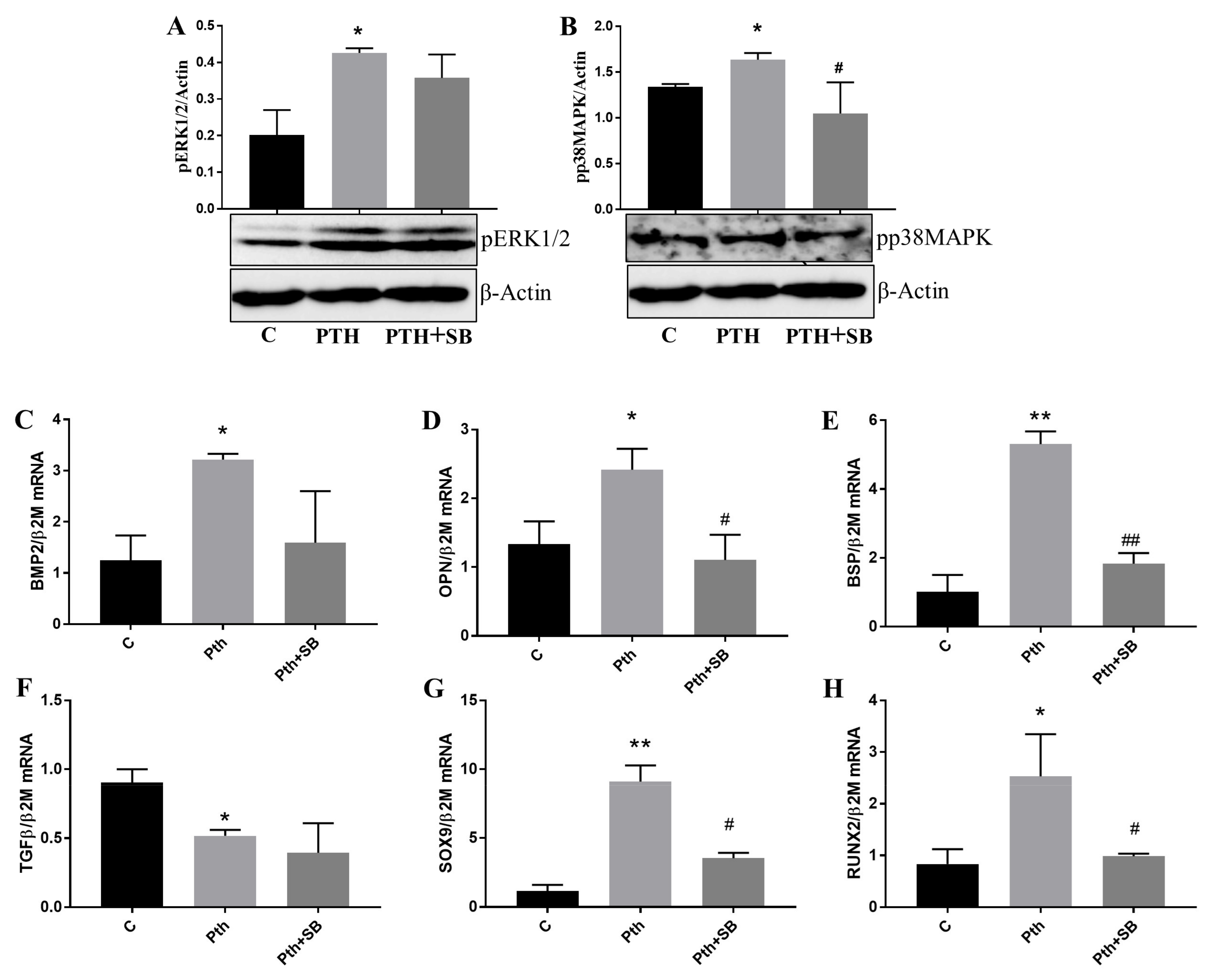
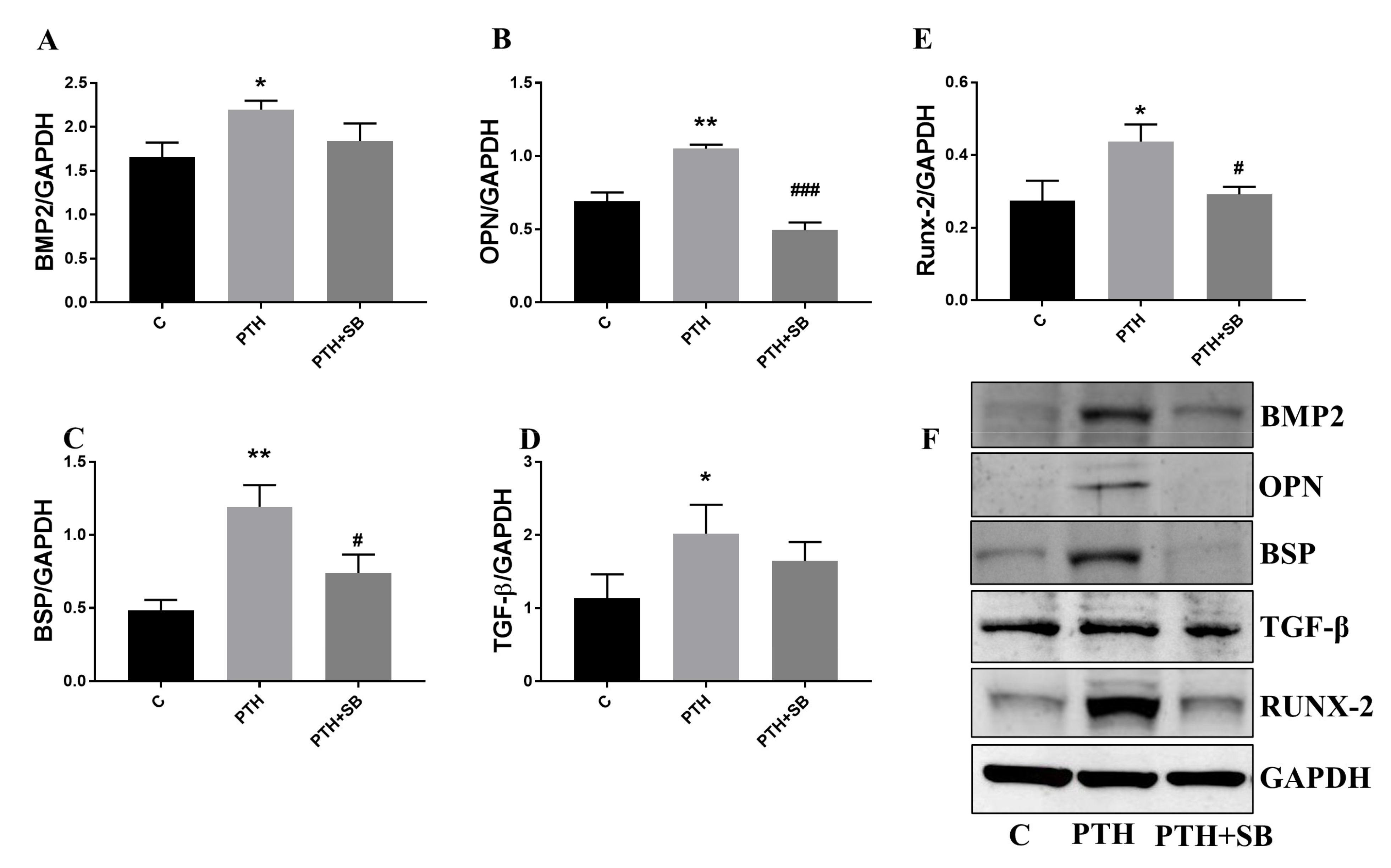
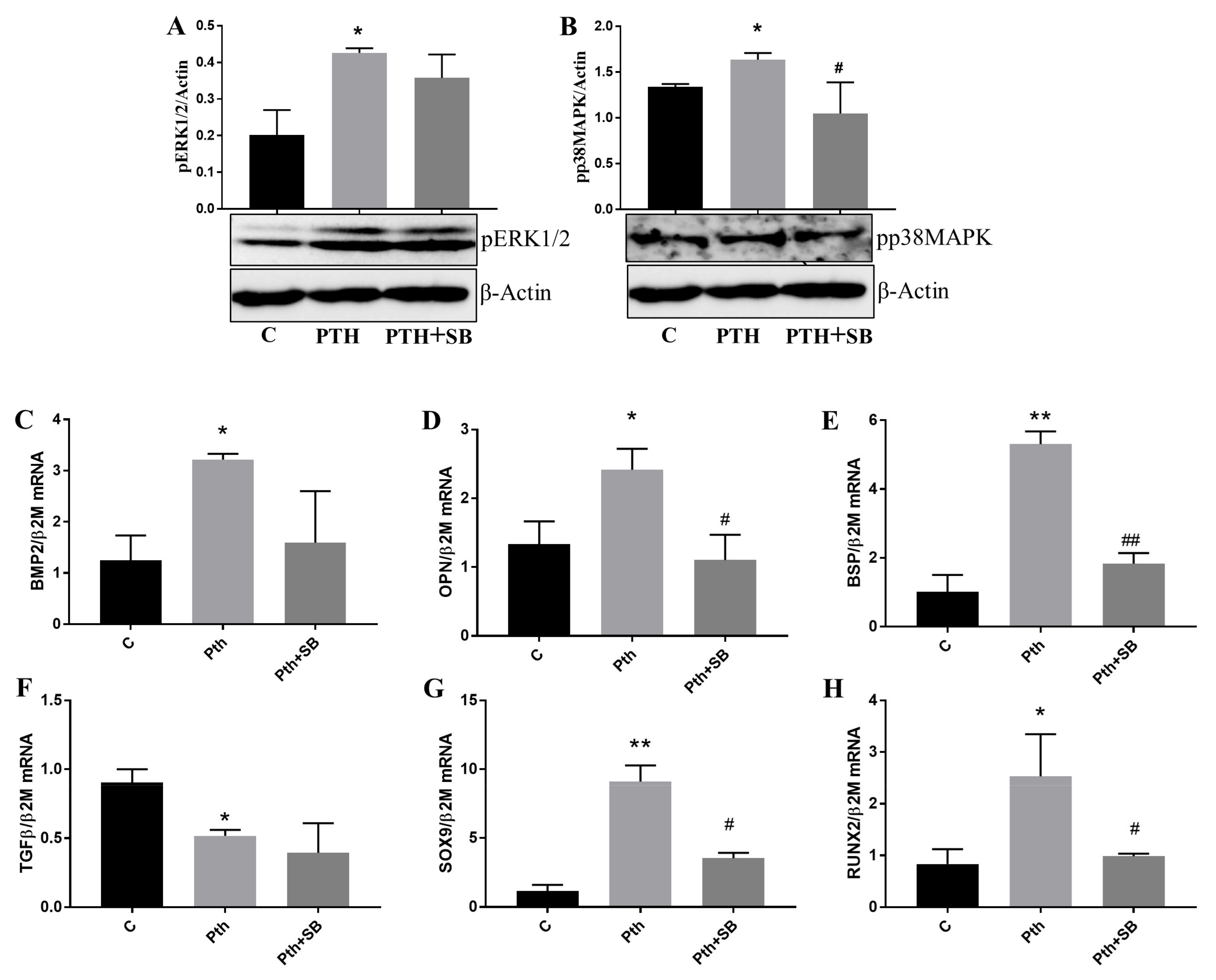
2.5. PTH Activates p38MAPK and ERK1/2 Kinases and Upregulates Molecules Associated with Valve Calcification in VEC
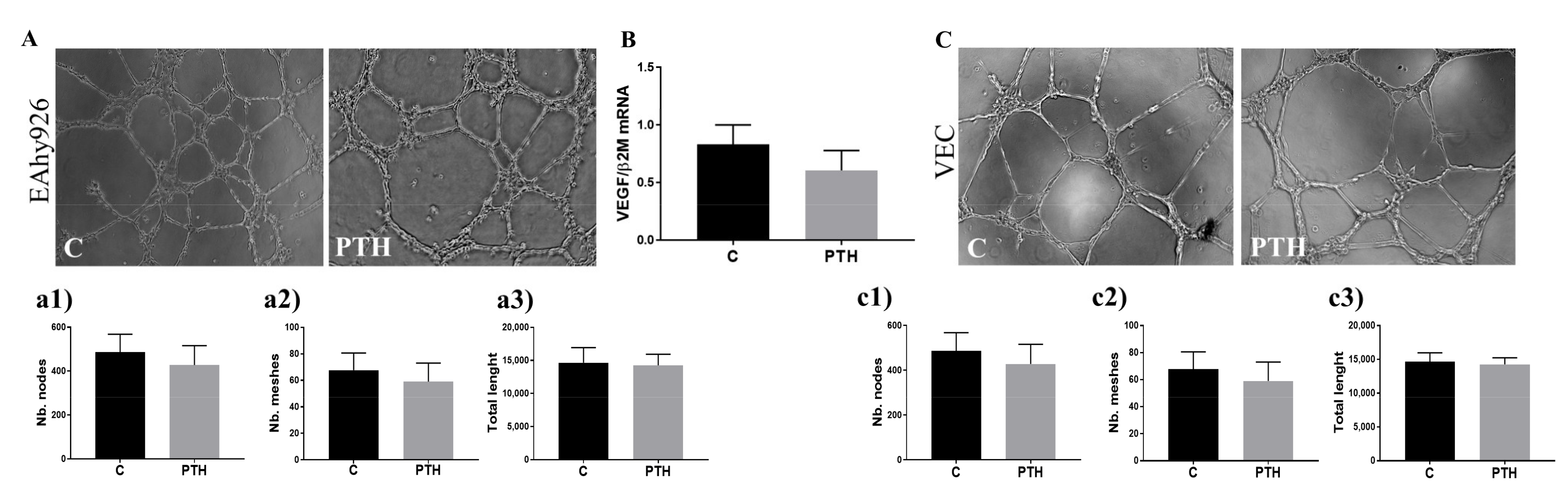
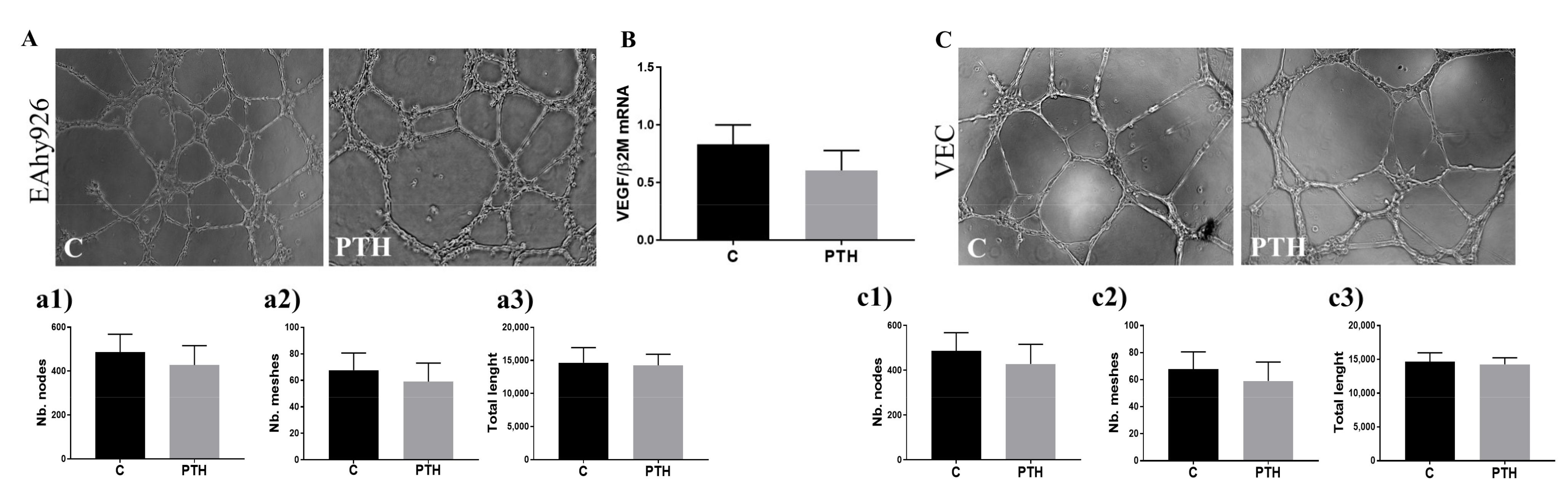
2.6. PTH Does Not Have an Angiogenic Effect on Endothelial Cells
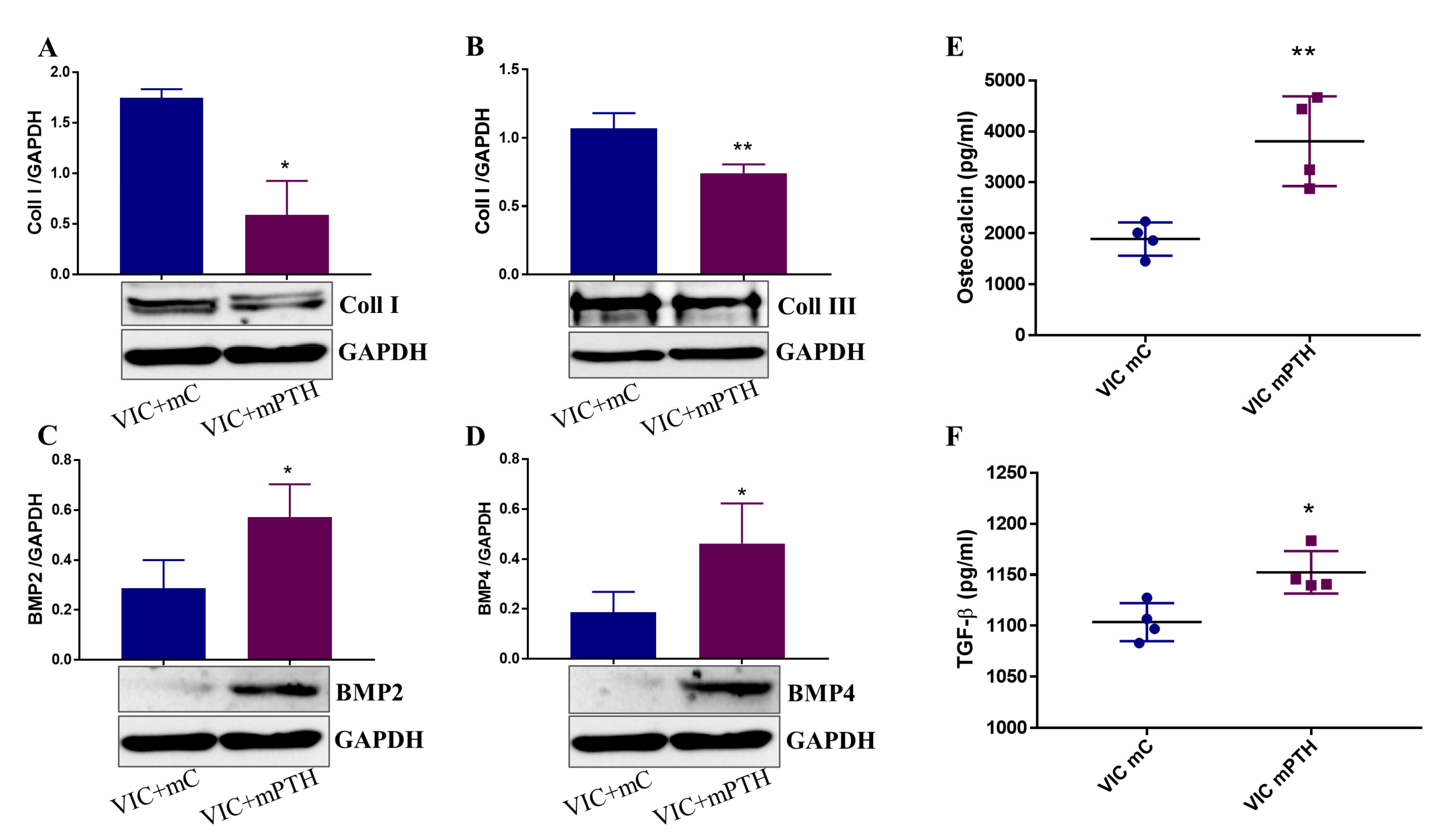
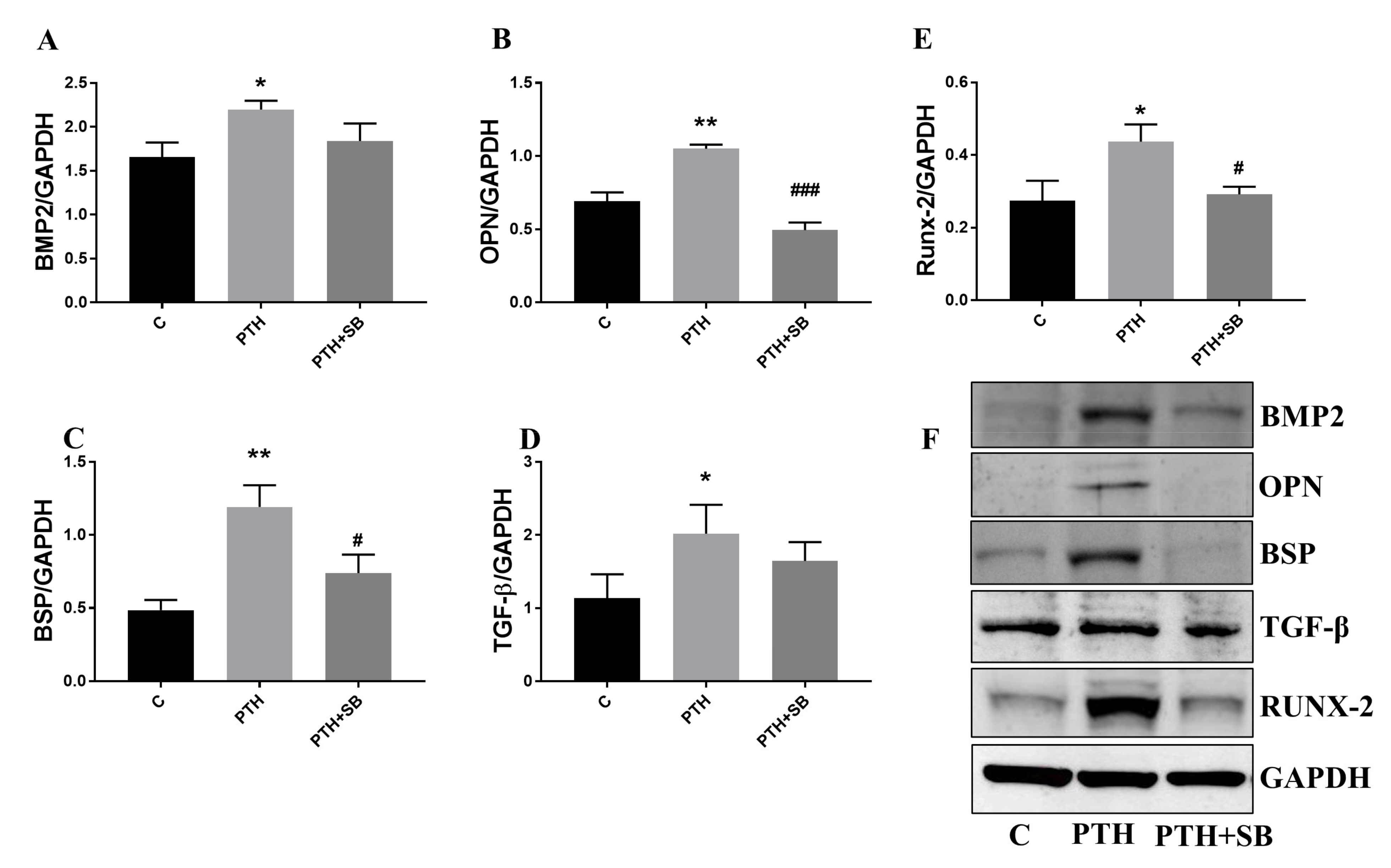
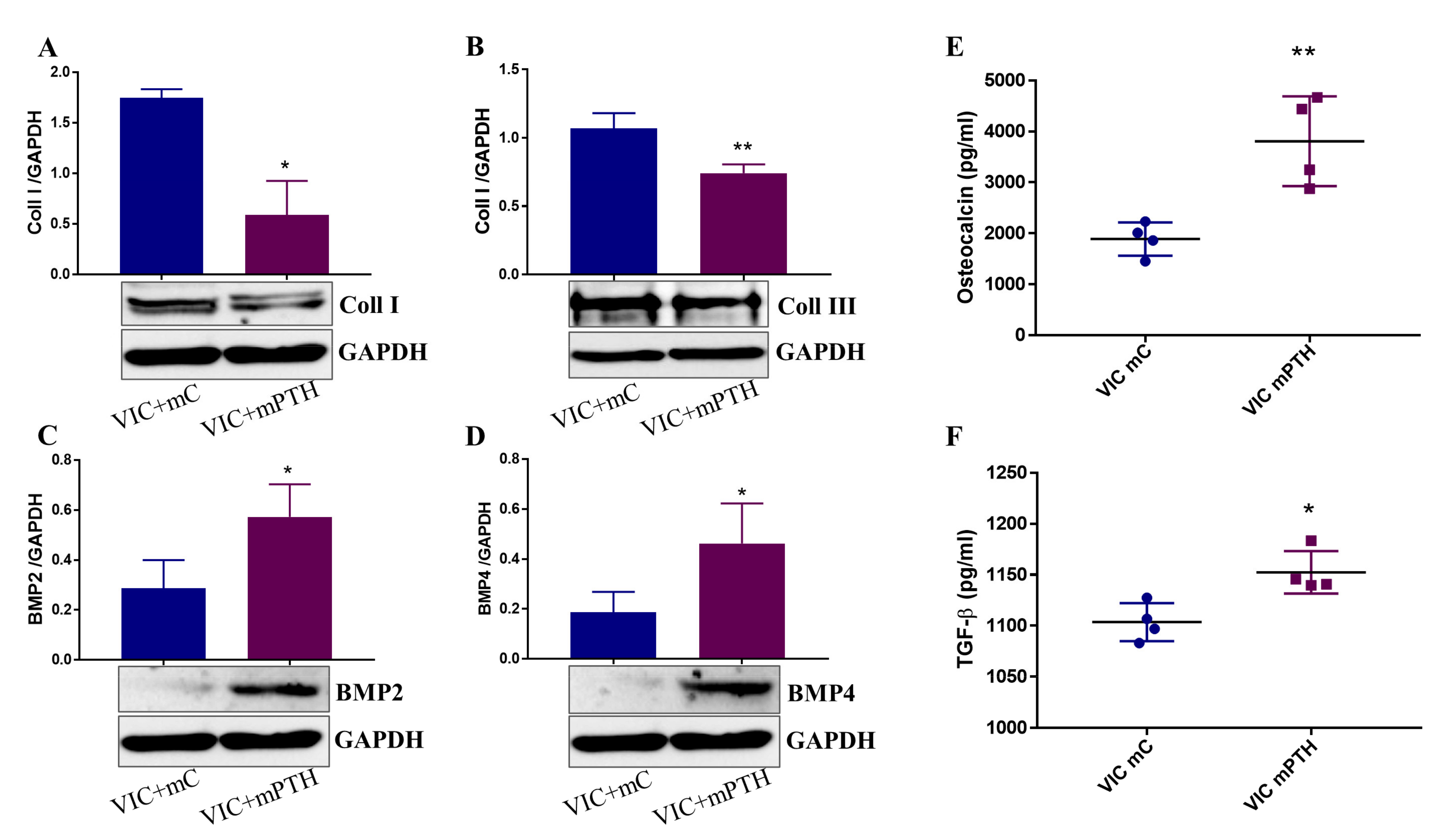
2.7. Mediators Secreted by Endothelial Cells Exposed to PTH Induce the Switch of VIC to an Osteogenic Phenotype
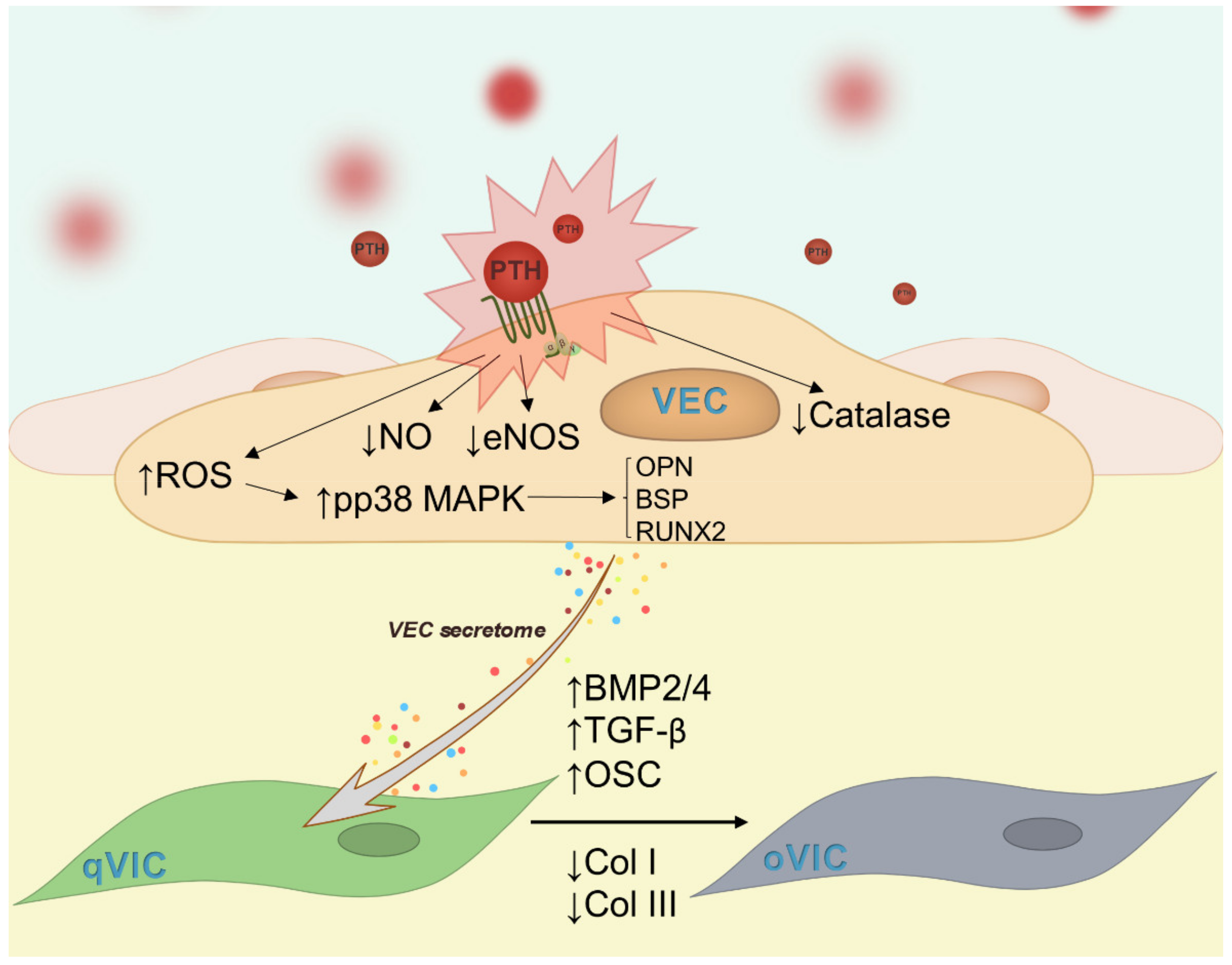
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Experimental design
4.3. Immunofluorescence of Valve Sections and VEC
4.4. Nitic Oxide (NO) Assays
4.5. Evaluation of ROS Production
4.6. Cell Energy Assay
4.7. Quantitative RT-PCR
4.8. Western Blot
4.9. Matrigel Assay
4.10. Enzyme-Linked Immunosorbent Assay (ELISA)
4.11. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neves, K.-R.; Graciolli, F.-G.; Dos Reis, L.; Neves, C.-L.; Magalhães, A.-O.; Custódio, M.-R.; Batista, D.-G.; Jorgetti, V.; Moyses, R. Vascular calcification: Contribution of parathyroid hormone in renal failure. Kidney Int. 2007, 71, 1262–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraser, W.D. Hyperparathyroidism. Lancet 2009, 374, 145–158. [Google Scholar] [CrossRef]
- Fukagawa, M.; Nakanishi, S.; Fujii, H.; Hamada, Y.; Abe, T. Regulation of parathyroid function in chronic kidney disease (CKD). Clin. Exp. Nephrol. 2006, 10, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Van Ballegooijen, A.J.; Reinders, I.; Visser, M.; Brouwer, I.A. Parathyroid hormone and cardiovascular disease events: A systematic review and meta-analysis of prospective studies. Am. Heart J. 2013, 165, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Isales, C.M.; Sumpio, B.; Bollag, R.J.; Zhong, Q.; Ding, K.-H.; Du, W.; Rodriguez-Commes, J.; Lopez, R.; Rosales, O.R.; Gasalla-Herraiz, J.; et al. Functional parathyroid hormone receptors are present in an umbilical vein endothelial cell line. Am. J. Physiol. Metab. 2000, 279, E654–E662. [Google Scholar] [CrossRef] [PubMed]
- Clemens, T.L.; Cormier, S.; Eichinger, A.; Endlich, K.; Fiaschi-Taesch, N.; Fischer, E.; Friedman, P.A.; Karaplis, A.C.; Massfelder, T.; Rossert, J.; et al. Parathyroid hormone-related protein and its receptors: Nuclear functions and roles in the renal and cardiovascular systems, the placental trophoblasts and the pancreatic islets. Br. J. Pharmacol. 2001, 134, 1113–1136. [Google Scholar] [CrossRef]
- Fujii, H. Association between Parathyroid Hormone and Cardiovascular Disease. Ther. Apher. Dial. 2018, 22, 236–241. [Google Scholar] [CrossRef] [Green Version]
- Lishmanov, A.; Dorairajan, S.; Pak, Y.; Chaudhary, K.; Chockalingam, A. Elevated serum parathyroid hormone is a cardiovascular risk factor in moderate chronic kidney disease. Int. Urol. Nephrol. 2012, 44, 541–547. [Google Scholar] [CrossRef]
- Iwata, S.; Walker, M.; Di Tullio, M.R.; Hyodo, E.; Jin, Z.; Liu, R.; Sacco, R.L.; Homma, S.; Silverberg, S.J. Aortic Valve Calcification in Mild Primary Hyperparathyroidism. J. Clin. Endocrinol. Metab. 2012, 97, 132–137. [Google Scholar] [CrossRef] [Green Version]
- Iwata, S.; Hyodo, E.; Yanagi, S.; Hayashi, Y.; Nishiyama, H.; Kamimori, K.; Ota, T.; Matsumura, Y.; Homma, S.; Yoshiyama, M. Parathyroid hormone and systolic blood pressure accelerate the progression of aortic valve stenosis in chronic hemodialysis patients. Int. J. Cardiol. 2013, 163, 256–259. [Google Scholar] [CrossRef]
- Mazzone, A.; Venneri, L.; Berti, S. Aortic valve stenosis and coronary artery disease: Pathophysiological and clinical links. J. Cardiovasc. Med. 2007, 8, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Demer, L.L.; Tintut, Y. Vascular calcification: Pathobiology of a multifaceted disease. Circulation 2008, 117, 2938–2948. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, E.; Nahrendorf, M.; Sosnovik, D.; Lok, V.M.; Jaffer, F.A.; Aikawa, M.; Weissleder, R. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation 2007, 115, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Rutkovskiy, A.; Malashicheva, A.; Sullivan, G.; Bogdanova, M.; Kostareva, A.; Stensløkken, K.; Fiane, A.; Vaage, J. Valve Interstitial Cells: The Key to Understanding the Pathophysiology of Heart Valve Calcification. J. Am. Heart Assoc. 2017, 6, e006339. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Zhao, D.; Yuan, P.; Li, J.; Yun, Y.; Cui, Y.; Zhang, T.; Ma, J.; Sun, L.; Ma, H.; et al. Endothelial-to-Mesenchymal Transition in Calcific Aortic Valve Disease. Acta Cardiol. Sin. 2020, 36, 183–194. [Google Scholar] [PubMed]
- Yuan, C.; Ni, L.; Zhang, C.; Hu, X.; Wu, X. Vascular calcification: New insights into endothelial cells. Microvasc. Res. 2020, 134, 104105. [Google Scholar] [CrossRef] [PubMed]
- Rattazzi, M.; Pauletto, P. Valvular endothelial cells: Guardians or destroyers of aortic valve integrity? Atherosclerosis 2015, 242, 396–398. [Google Scholar] [CrossRef]
- Cheng, Z.Y.; Ye, T.; Ling, Q.Y.; Wu, T.; Wu, G.Y.; Zong, G.J. Parathyroid hormone promotes osteoblastic differentiation of endothelial cells via the extracellular signal-regulated protein kinase 1/2 and nuclear factor-κB signaling pathways. Exp. Ther. Med. 2018, 15, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Butcher, J.T.; Nerem, R.M. Valvular endothelial cells and the mechanoregulation of valvular pathology. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2007, 29, 1445–1457. [Google Scholar] [CrossRef] [Green Version]
- Gould, S.T.; Matherly, E.E.; Smith, J.N.; Heistad, D.D.; Anseth, K.S. The role of valvular endothelial cell paracrine signaling and matrix elasticity on valvular interstitial cell activation. Biomaterials 2014, 35, 3596–3606. [Google Scholar] [CrossRef] [Green Version]
- Richards, J.; El-Hamamsy, I.; Chen, S.; Sarang, Z.; Sarathchandra, P.; Yacoub, M.H.; Chester, A.H.; Butcher, J.T. Side-specific endothelial-dependent regulation of aortic valve calcification: Interplay of hemodynamics and nitric oxide signaling. Am. J. Pathol. 2013, 182, 1922–1931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heistad, D.D. Oxidative stress and vascular disease: 2005 Duff lecture. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 689–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Carballo, E.; Gámez, B.; Ventura, F. p38 MAPK Signaling in Osteoblast Differentiation. Front. Cell Dev. Biol. 2016, 4, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rey, A.; Manen, D.; Rizzoli, R.; Ferrari, S.; Caverzasio, J. Evidences for a role of p38 MAP kinase in the stimulation of alkaline phosphatase and matrix mineralization induced by parathyroid hormone in osteoblastic cells. Bone 2007, 41, 59–67. [Google Scholar] [CrossRef]
- Ding, Q.; Sun, P.; Zhou, H.; Wan, B.; Yin, J.; Huang, Y.; Li, Q.; Yin, G.; Fan, J. Lack of endogenous parathyroid hormone delays fracture healing by inhibiting vascular endothelial growth factor-mediated angiogenesis. Int. J. Mol. Med. 2018, 42, 171–181. [Google Scholar] [CrossRef]
- Rajamannan, N.M.; Bonow, R.O.; Rahimtoola, S.H. Calcific aortic stenosis: An update. Nat. Clin. Pract. Cardiovasc. Med. 2007, 4, 254–262. [Google Scholar] [CrossRef]
- Rodriguez, K.J.; Piechura, L.M.; Porras, A.M.; Masters, K.S. Manipulation of valve composition to elucidate the role of collagen in aortic valve calcification. BMC Cardiovasc. Disord. 2014, 14, 29. [Google Scholar] [CrossRef] [Green Version]
- Vadana, M.; Cecoltan, S.; Ciortan, L.; Macarie, R.D.; Tucureanu, M.M.; Mihaila, A.C.; Droc, I.; Butoi, E.; Manduteanu, I. Molecular mechanisms involved in high glucose-induced valve calcification in a 3D valve model with human valvular cells. J. Cell. Mol. Med. 2020, 24, 6350–6361. [Google Scholar] [CrossRef]
- Menon, V.; Lincoln, J. The Genetic Regulation of Aortic Valve Development and Calcific Disease. Front. Cardiovasc. Med. 2018, 5, 162. [Google Scholar] [CrossRef]
- Yi, B.; Zeng, W.; Lv, L.; Hua, P. Changing epidemiology of calcific aortic valve disease: 30-year trends of incidence, prevalence, and deaths across 204 countries and territories. Aging 2021, 13, 12710–12732. [Google Scholar] [CrossRef]
- Wu, M.; Zhang, J.-D.; Tang, R.-N.; Crowley, S.D.; Liu, H.; Lv, L.-L.; Ma, K.-L.; Liu, B.-C. Elevated PTH induces endothelial-to-chondrogenic transition in aortic endothelial cells. Am. J. Physiol. Physiol. 2017, 312, F436–F444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambardella, J.; De Rosa, M.; Sorriento, D.; Prevete, N.; Fiordelisi, A.; Ciccarelli, M.; Trimarco, B.; De Luca, N.; Iaccarino, G. Parathyroid Hormone Causes Endothelial Dysfunction by Inducing Mitochondrial ROS and Specific Oxidative Signal Transduction Modifications. Oxidative Med. Cell. Longev. 2018, 2018, 9582319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.Y.; Ye, Z.X.; Wang, X.F.; Chang, J.; Yang, M.W.; Zhong, H.H.; Hong, F.F.; Yang, S.L. Nitric oxide bioavailability dysfunction involves in atherosclerosis. Biomed. Pharmacother. 2018, 97, 423–428. [Google Scholar] [CrossRef]
- Miller, J.D.; Chu, Y.; Brooks, R.M.; Richenbacher, W.E.; Peña-Silva, R.; Heistad, D.D. Dysregulation of Antioxidant Mechanisms Contributes to Increased Oxidative Stress in Calcific Aortic Valvular Stenosis in Humans. J. Am. Coll. Cardiol. 2008, 52, 843–850. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Huang, R.T.; Hamanaka, R.B.; Krause, M.; Oh, M.J.; Kuo, C.H.; Nigdelioglu, R.; Meliton, A.Y.; Witt, L.; Dai, G.; et al. HIF-1α is required for disturbed flow-induced metabolic reprogramming in human and porcine vascular endothelium. eLife 2017, 30, e25217. [Google Scholar] [CrossRef] [PubMed]
- Quintero, M.; Colombo, S.L.; Godfrey, A.; Moncada, S. Mitochondria as signaling organelles in the vascular endothelium. Proc. Natl. Acad. Sci. USA 2006, 103, 5379–5384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumas, S.J.; García-Caballero, M.; Carmeliet, P. Metabolic Signatures of Distinct Endothelial Phenotypes. Trends. Endocrinol. Metab. 2020, 31, 580–595. [Google Scholar] [CrossRef]
- Wik, J.A.; Phung, D.; Kolan, S.; Haraldsen, G.; Skålhegg, B.S.; Hol, F.J. Inflammatory activation of endothelial cells increases glycolysis and oxygen consumption despite inhibiting cell proliferation. FEBS Open Bio 2021, 11, 1719–1730. [Google Scholar] [CrossRef]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell. 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Wang, L.; Tang, R.; Zhang, Y.; Chen, S.; Guo, Y.; Wang, X.; Liu, Z.; Liu, H.; Zhang, X.; Liu, B.C. PTH-induced EndMT via miR-29a-5p/GSAP/Notch1 pathway contributed to valvular calcification in rats with CKD. Cell Prolif. 2021, 54, e13018. [Google Scholar] [CrossRef]
- Mazrouei, S.; Sharifpanah, F.; Caldwell, R.W.; Franz, M.; Shatanawi, A.; Muessig, J.; Fritzenwanger, M.; Schulze, P.C.; Jung, C. Regula-tion of MAP kinase-mediated endothelial dysfunction in hyperglycemia via arginase I and eNOS dysregulation. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1398–1411. [Google Scholar] [CrossRef]
- Reustle, A.; Torzewski, M. Role of p38 MAPK in Atherosclerosis and Aortic Valve Sclerosis. Int. J. Mol. Sci. 2018, 19, 3761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, G.; Jiang, D.; Gopalakrishnan, R.; Franceschi, R.T. Fibroblast Growth Factor 2 Induction of the Osteocalcin Gene Requires MAPK Activity and Phosphorylation of the Osteoblast Transcription Factor, Cbfa1/Runx2. J. Biol. Chem. 2002, 277, 36181–36187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thouverey, C.; Caverzasio, J. Suppression of p38α MAPK Signaling in Osteoblast Lineage Cells Impairs Bone Anabolic Action of Parathyroid Hormone. J. Bone Miner. Res. 2016, 31, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Syväranta, S.; Helske, S.; Laine, M.; Lappalainen, J.; Kupari, M.; Mäyränpää, M.I.; Lindstedt, K.A.; Kovanen, P.T. Vascular endothelial growth factor-secreting mast cells and myofibroblasts: A novel self-perpetuating angiogenic pathway in aortic valve stenosis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1220–1227. [Google Scholar] [CrossRef] [Green Version]
- James, A.W.; Lachaud, G.; Shen, J.; Asatrian, G.; Nguyen, V.; Zhang, X.; Ting, K.; Soo, C. A Review of the Clinical Side Effects of Bone Morphogenetic Protein-2. Tissue Eng. Part B Rev. 2016, 22, 284–297. [Google Scholar] [CrossRef]
- Kennedy, J.A.; Hua, X.; Mishra, K.; Murphy, G.A.; Rosenkranz, A.C.; Horowitz, J.D. Inhibition of calcifying nodule formation in cultured porcine aortic valve cells by nitric oxide donors. Eur. J. Pharmacol. 2009, 602, 28–35. [Google Scholar] [CrossRef]
- World Medical Association declaration of Helsinki. Recommendations guiding physicians in biomedical research involving human subjects. JAMA 1997, 277, 925–926. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | GenBank® Accession Number | Sequences of Oligonucleotide Primers | Predicted Size (bp) |
|---|---|---|---|
| TGF-β 1 | NM_000660.7 | Fw: 5′-ccacctgcaagaccatcgac-3′ Rv: 3′-ctggcgagccttagtttggac-5′ | 91 |
| Osteopontin | NM_001040058.2 | Fw: 5′-gaagtttcgcagacctgacat-3′ Rv: 5′-gtatgcaccattcaactcctcg-3′ | 91 |
| BMP-2 | NM_001200.4 | Fw: 5′-actaccagaaacgagtgggaa-3′ Rv: 5′-gcatctgttctcggaaaacct-3′ | 113 |
| BSP | NM_004967.4 | Fw:5′gaacctcgtggggacaattac-3′ Rv:3′ catcatagccatcgtagccttg-5′ | 79 |
| VEGF | NM_001025366.3 | Fw:5′-agggcagaatcatcacgaagt-3′ Rv: 3′agggtctcgattggatggca-5′ | 75 |
| Sox9 | NM_000346.4 | Fw:5′agcgaacgcacatcaagac-3′ Rv:3′ ctgtaggcgatctgttgggg-5′ | 85 |
| RUNX2 | NM_001024630.4 | Fw: 5′-ccgcctcagtgatttagggc-3′ Rv: 5′-gggtctgtaatctgactctgtcc-3′ | 132 |
| β2-microglobulin | NM_001101.4 | Fw: 5′-catgtacgttgctatccaggc-3′ Rv: 5′-ctccttaatgtcacgcacgat-3′ | 250 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vadana, M.; Cecoltan, S.; Ciortan, L.; Macarie, R.D.; Mihaila, A.C.; Tucureanu, M.M.; Gan, A.-M.; Simionescu, M.; Manduteanu, I.; Droc, I.; et al. Parathyroid Hormone Induces Human Valvular Endothelial Cells Dysfunction That Impacts the Osteogenic Phenotype of Valvular Interstitial Cells. Int. J. Mol. Sci. 2022, 23, 3776. https://doi.org/10.3390/ijms23073776
Vadana M, Cecoltan S, Ciortan L, Macarie RD, Mihaila AC, Tucureanu MM, Gan A-M, Simionescu M, Manduteanu I, Droc I, et al. Parathyroid Hormone Induces Human Valvular Endothelial Cells Dysfunction That Impacts the Osteogenic Phenotype of Valvular Interstitial Cells. International Journal of Molecular Sciences. 2022; 23(7):3776. https://doi.org/10.3390/ijms23073776
Chicago/Turabian StyleVadana, Mihaela, Sergiu Cecoltan, Letitia Ciortan, Razvan D. Macarie, Andreea C. Mihaila, Monica M. Tucureanu, Ana-Maria Gan, Maya Simionescu, Ileana Manduteanu, Ionel Droc, and et al. 2022. "Parathyroid Hormone Induces Human Valvular Endothelial Cells Dysfunction That Impacts the Osteogenic Phenotype of Valvular Interstitial Cells" International Journal of Molecular Sciences 23, no. 7: 3776. https://doi.org/10.3390/ijms23073776
APA StyleVadana, M., Cecoltan, S., Ciortan, L., Macarie, R. D., Mihaila, A. C., Tucureanu, M. M., Gan, A.-M., Simionescu, M., Manduteanu, I., Droc, I., & Butoi, E. (2022). Parathyroid Hormone Induces Human Valvular Endothelial Cells Dysfunction That Impacts the Osteogenic Phenotype of Valvular Interstitial Cells. International Journal of Molecular Sciences, 23(7), 3776. https://doi.org/10.3390/ijms23073776