
Drug Design, Synthesis and Biological Evaluation of Heterocyclic Molecules as Anti-Inflammatory Agents

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Docking Studies
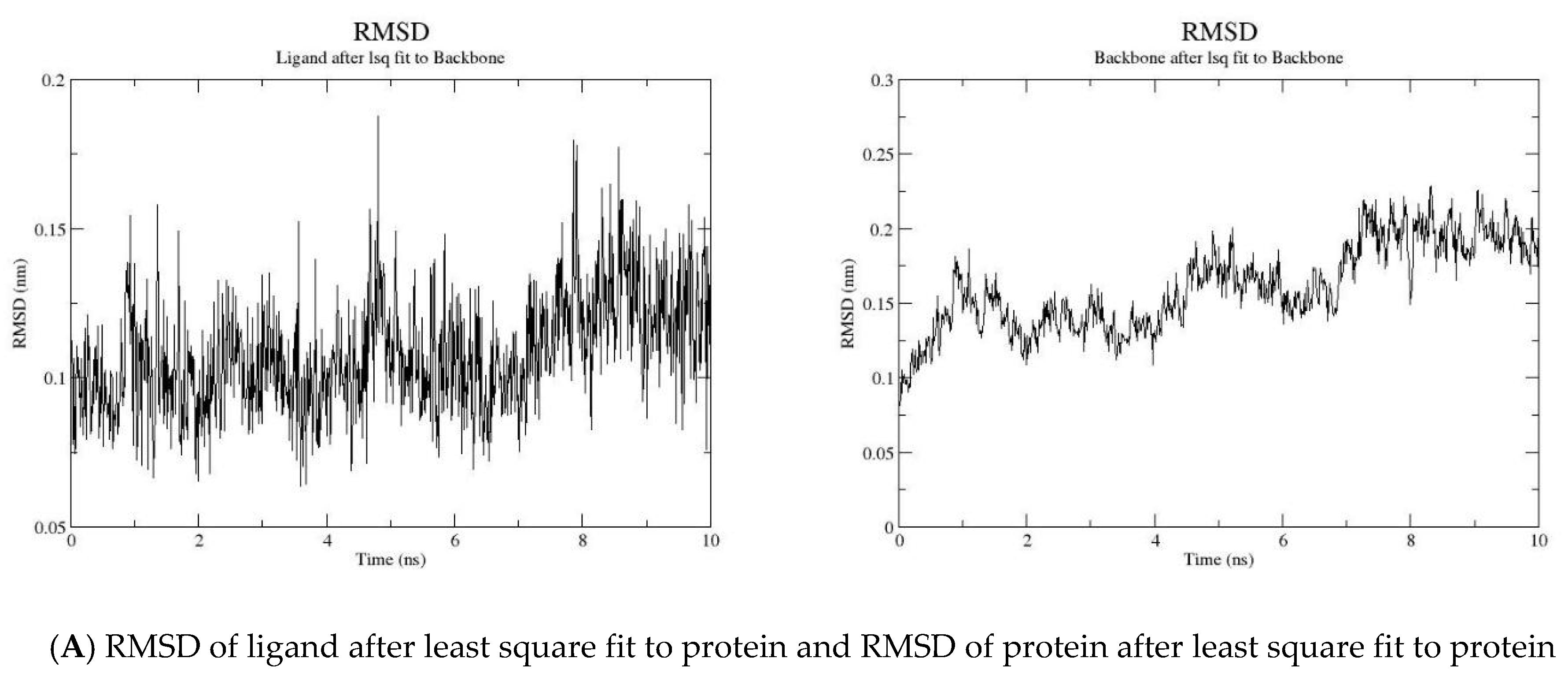
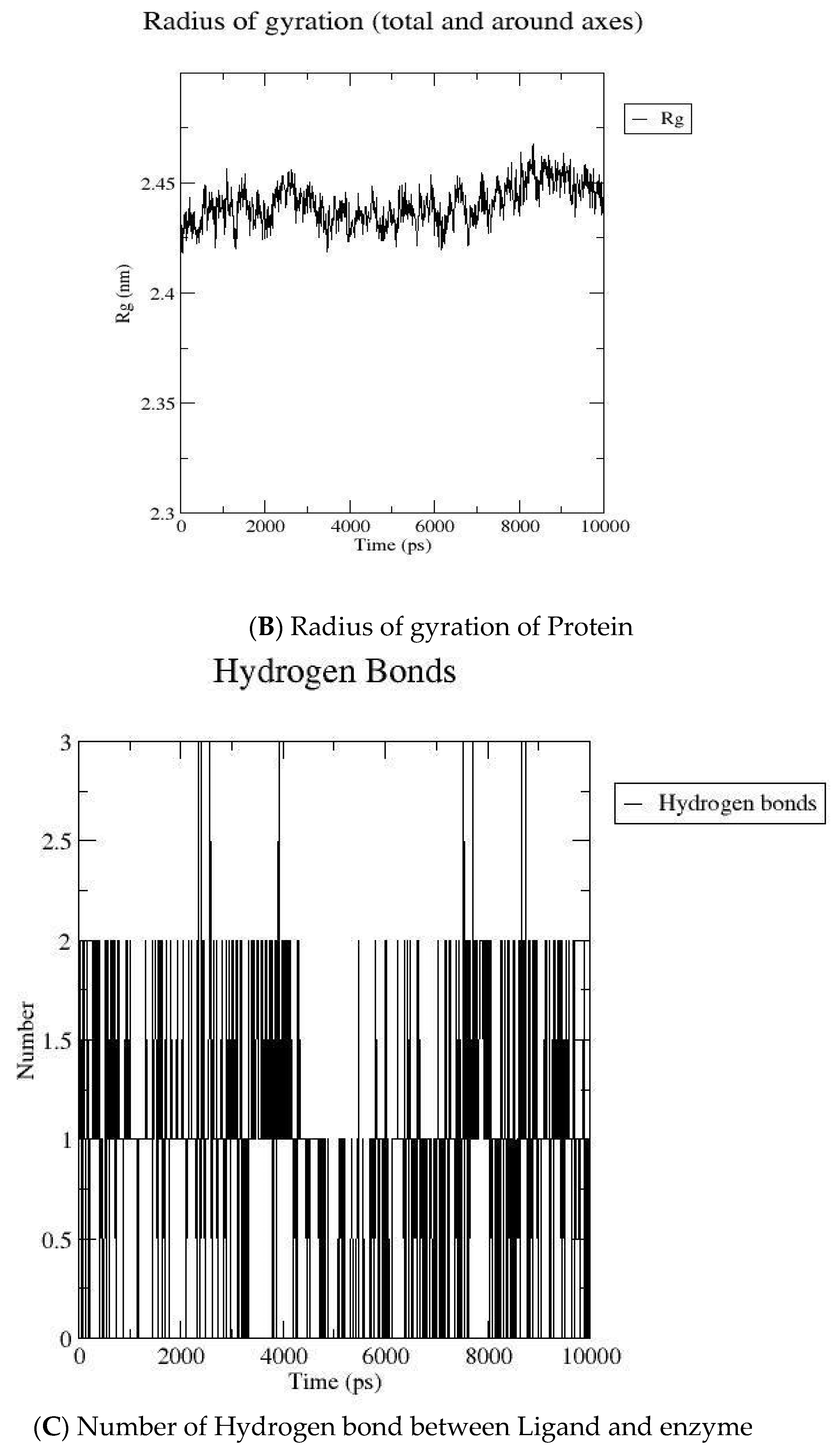
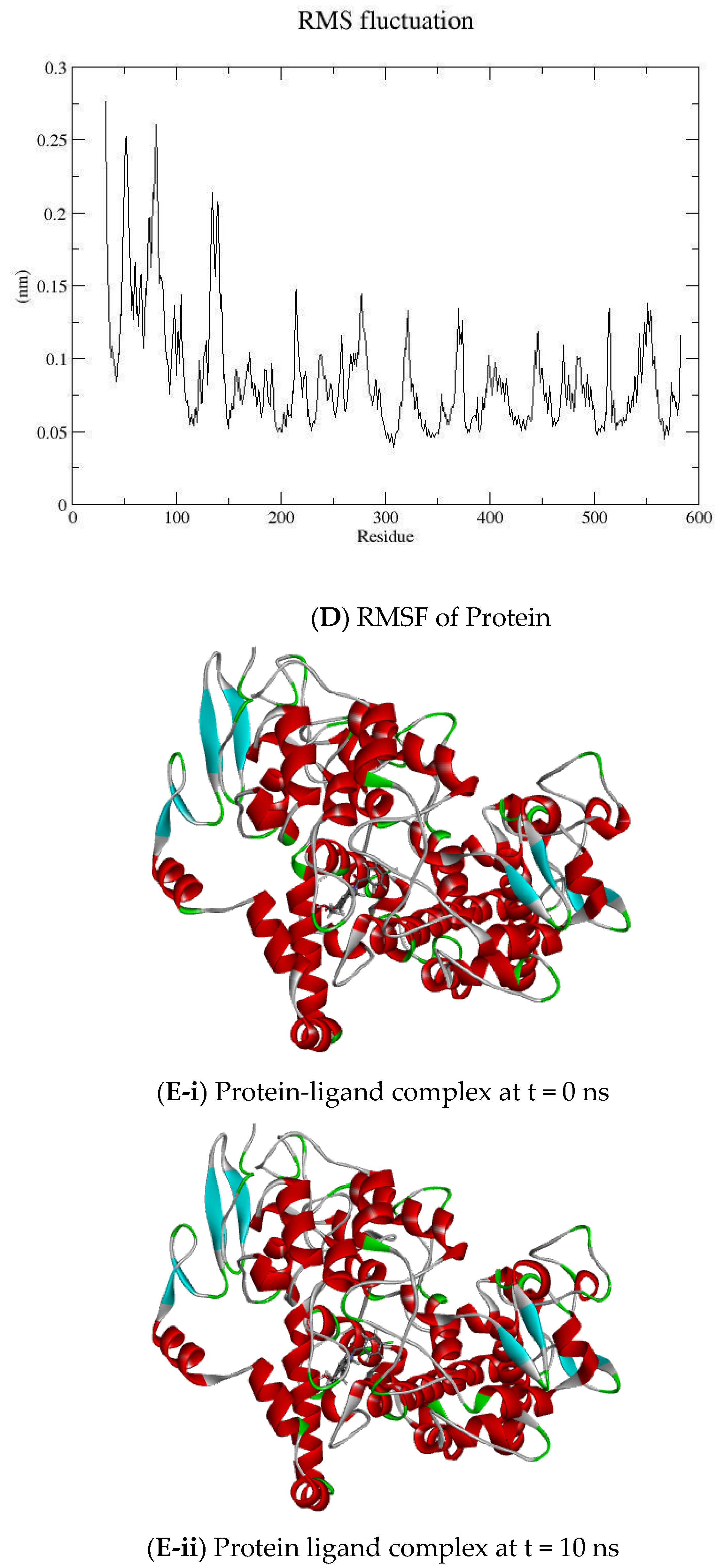
2.2. MD Simulation
2.3. Acute Toxicity Prediction Using PASS Online Software
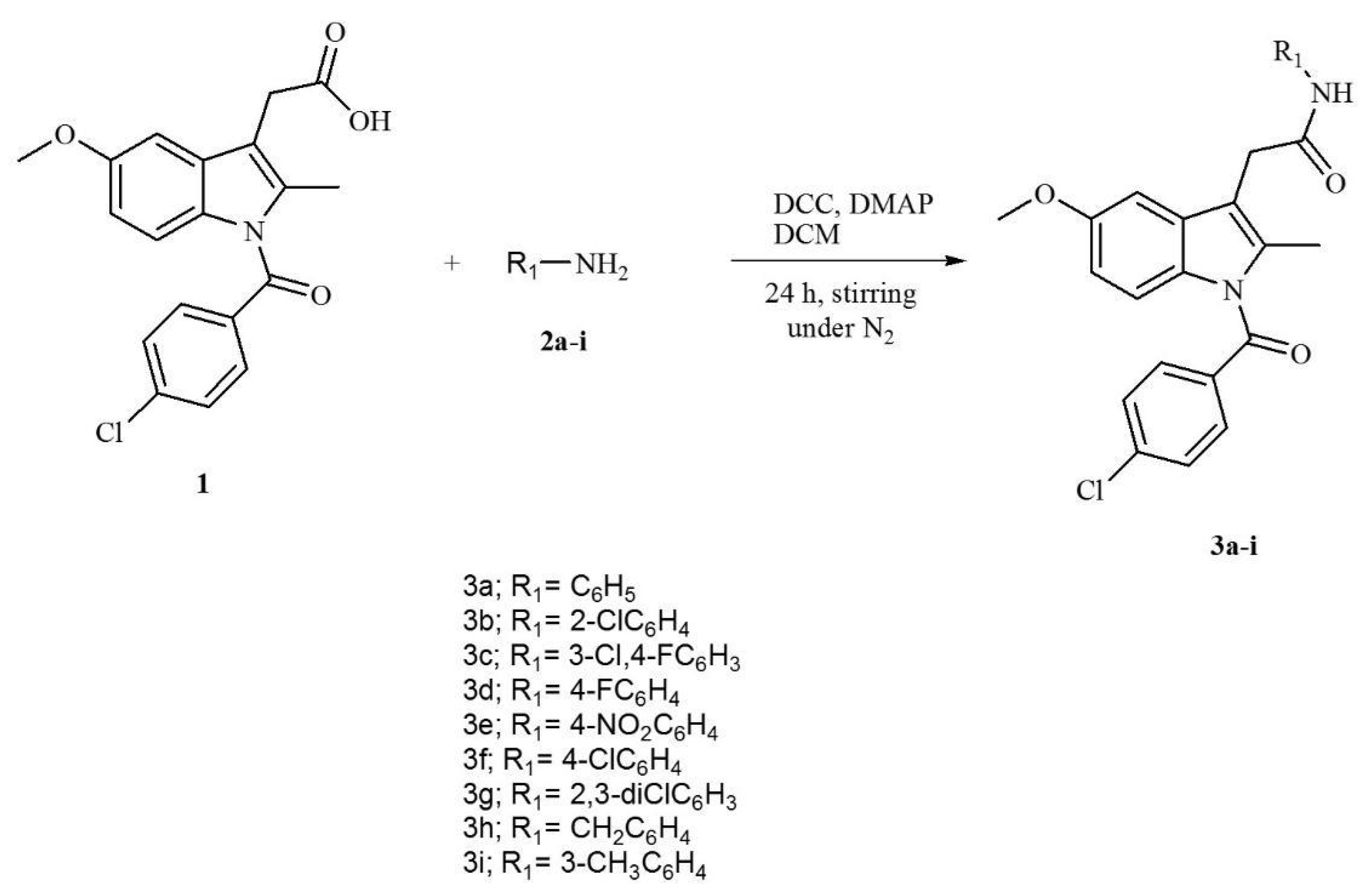
2.4. Synthesis of Indomethacin Derivatives (3a–3i)
2.4.1. Synthesis of 2-(1(4-chlorobenzoyl),5-methoxy-2-methyl-1H-indol-3-yl)-N-phenylacetamide 3a
2.4.2. Synthesis of 2-(1-(4-chlorobenzoyl),5-methoxy-2-methyl-1H-indol-3-yl)-N-(2-chlorophenyl)acetamide 3b
2.4.3. Synthesis of N-(3-chloro-4-fluorophenyl)-2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)acetamide 3c
2.4.4. Synthesis of 2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)-N-(4-fluorophenyl)acetamide 3d
2.4.5. Synthesis of 2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)-N-(4-nitrophenyl)acetamide 3e
2.4.6. Synthesis of 2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)-N-(4-chlorophenyl)acetamide 3f
2.4.7. Synthesis of 2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)-N-(2,6-dichlorophenyl)acetamide 3g
2.4.8. Synthesis of N-benzyl-2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)acetamide 3h
2.4.9. Synthesis of 2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)-N-m-tolylacetamide 3i
2.5. Pharmacological Screening
2.5.1. In Vitro COX-1 and COX-2 Enzymatic Assay
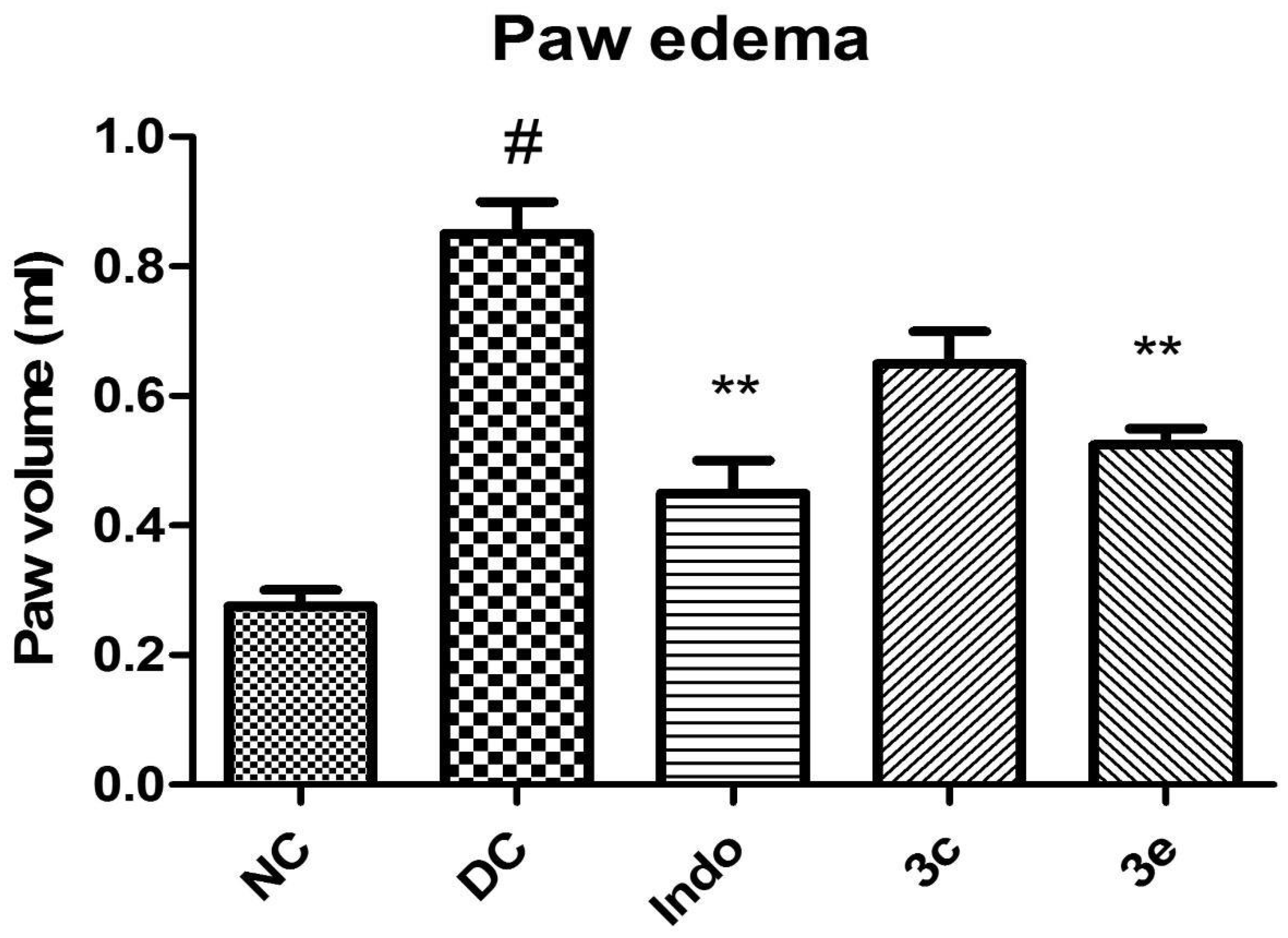
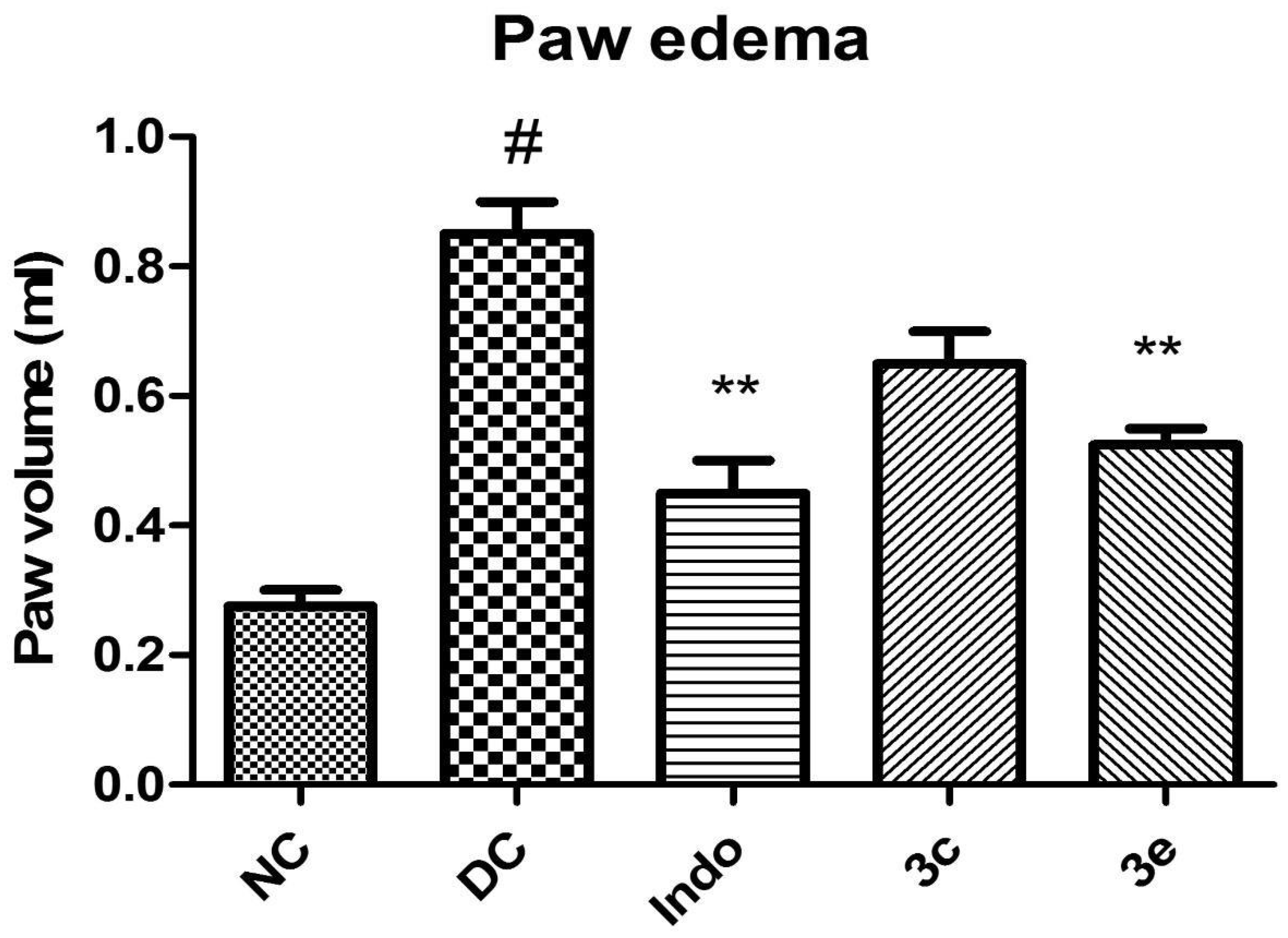
2.5.2. In Vivo Carrageenan-Induced Rat Paw Oedema Study
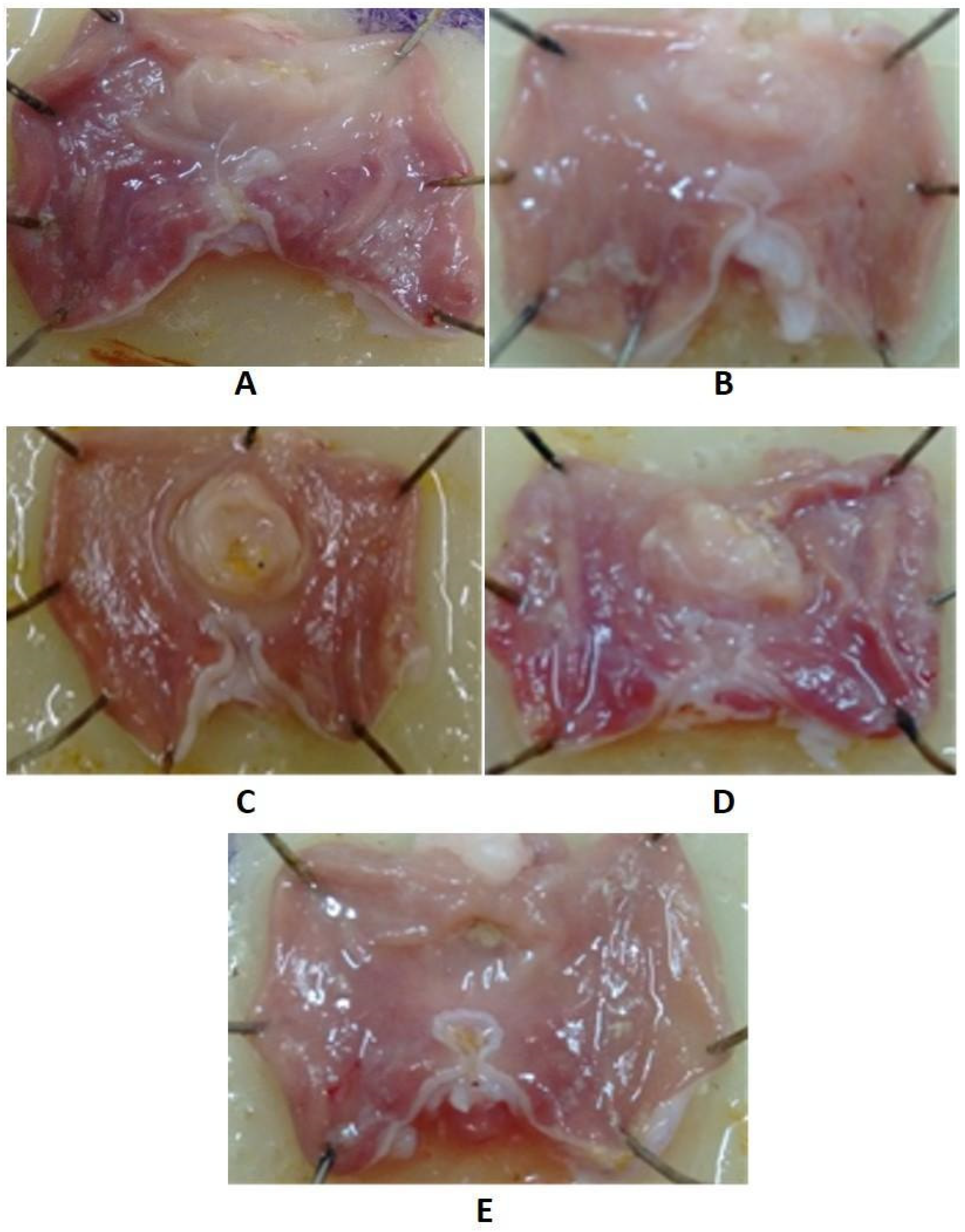
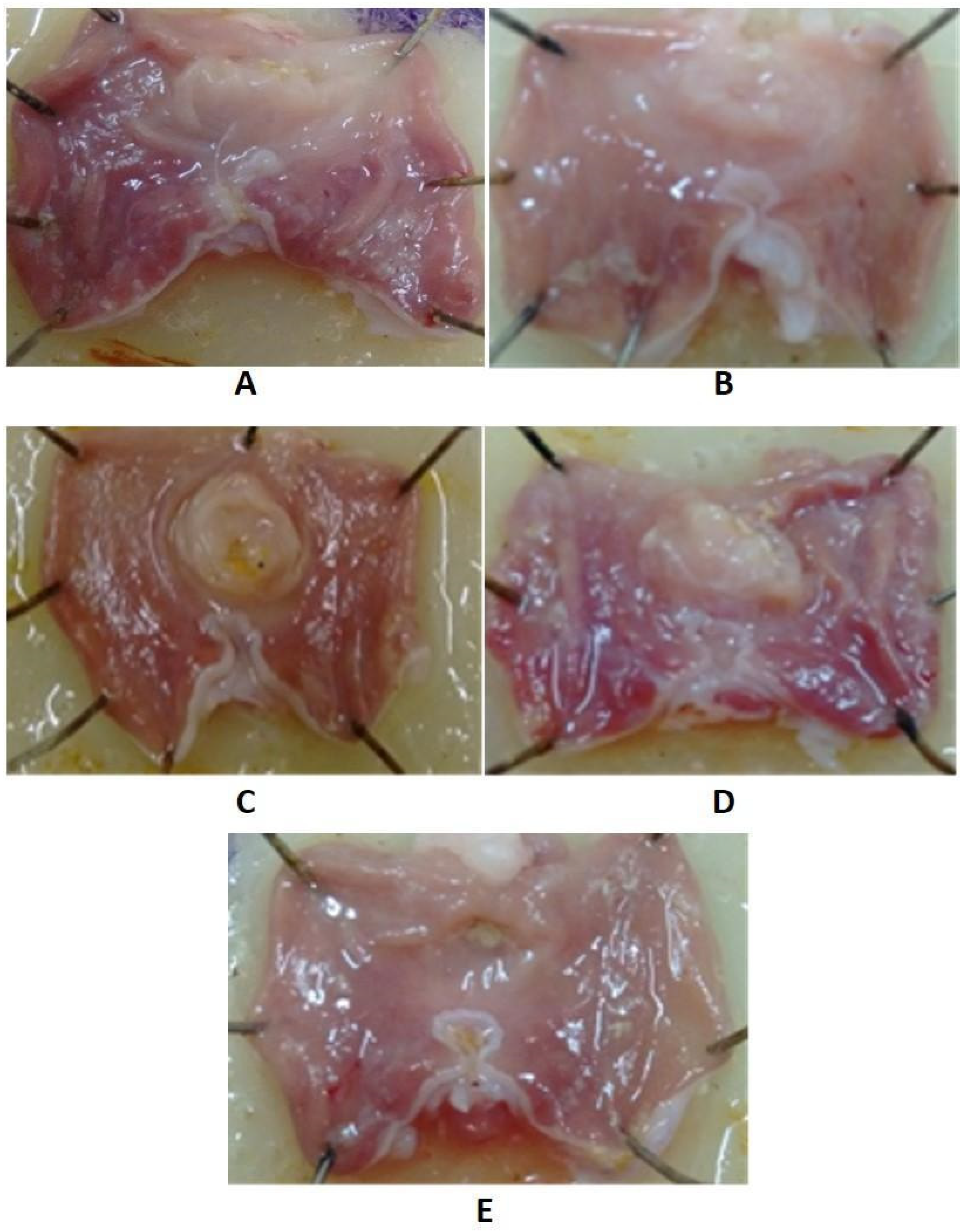
2.5.3. The Effect of Indomethacin Derivatives on the Gastric Mucosa
3. Results
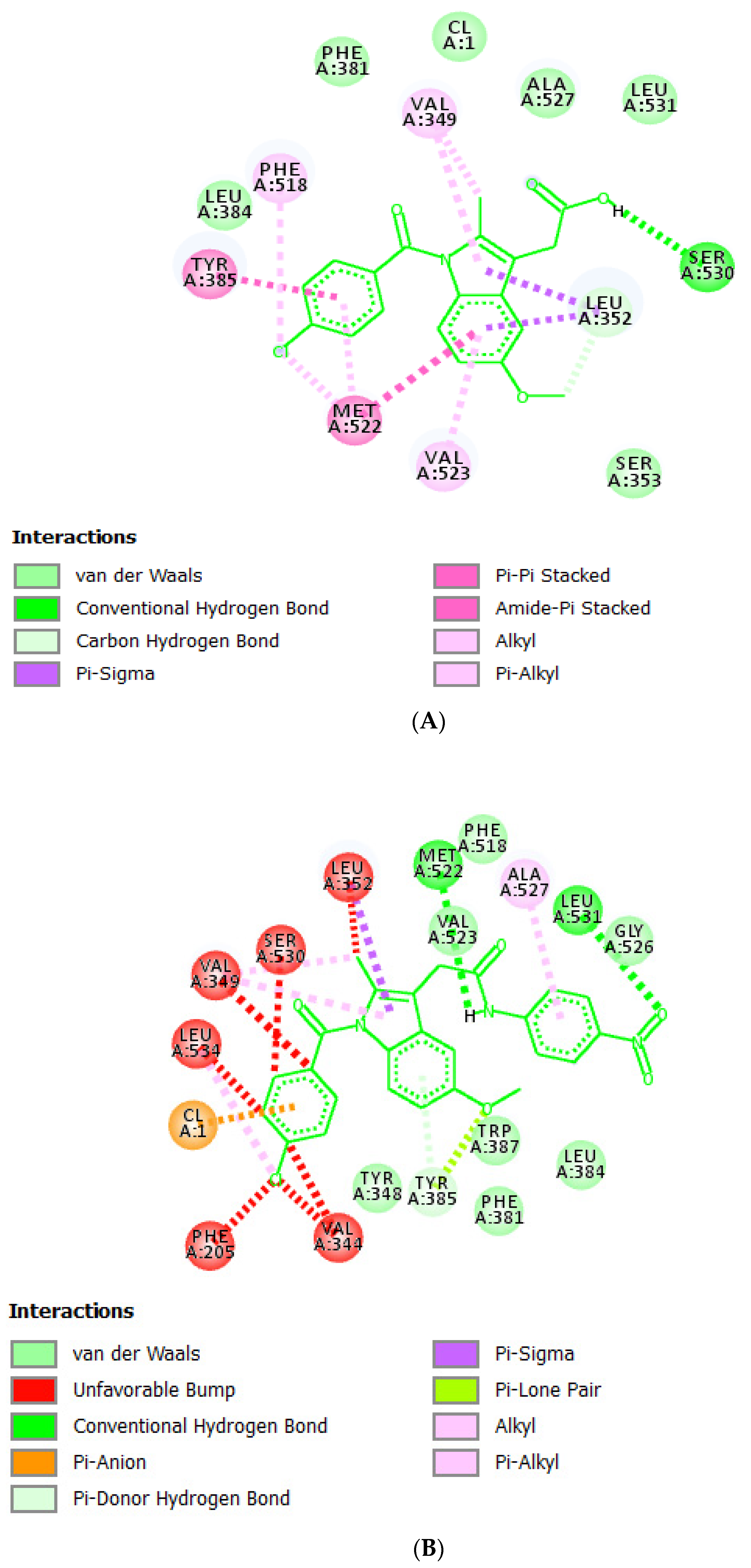
3.1. Molecular Docking Studies
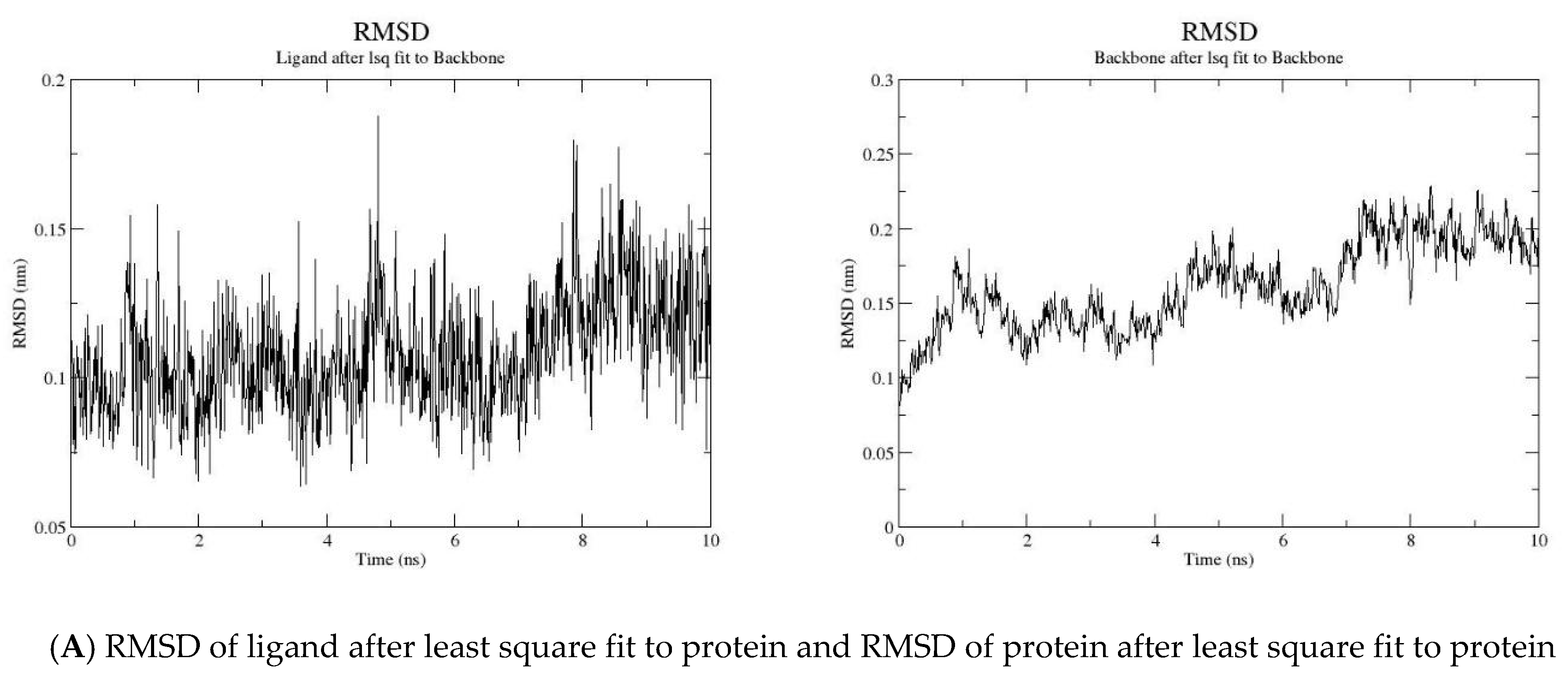
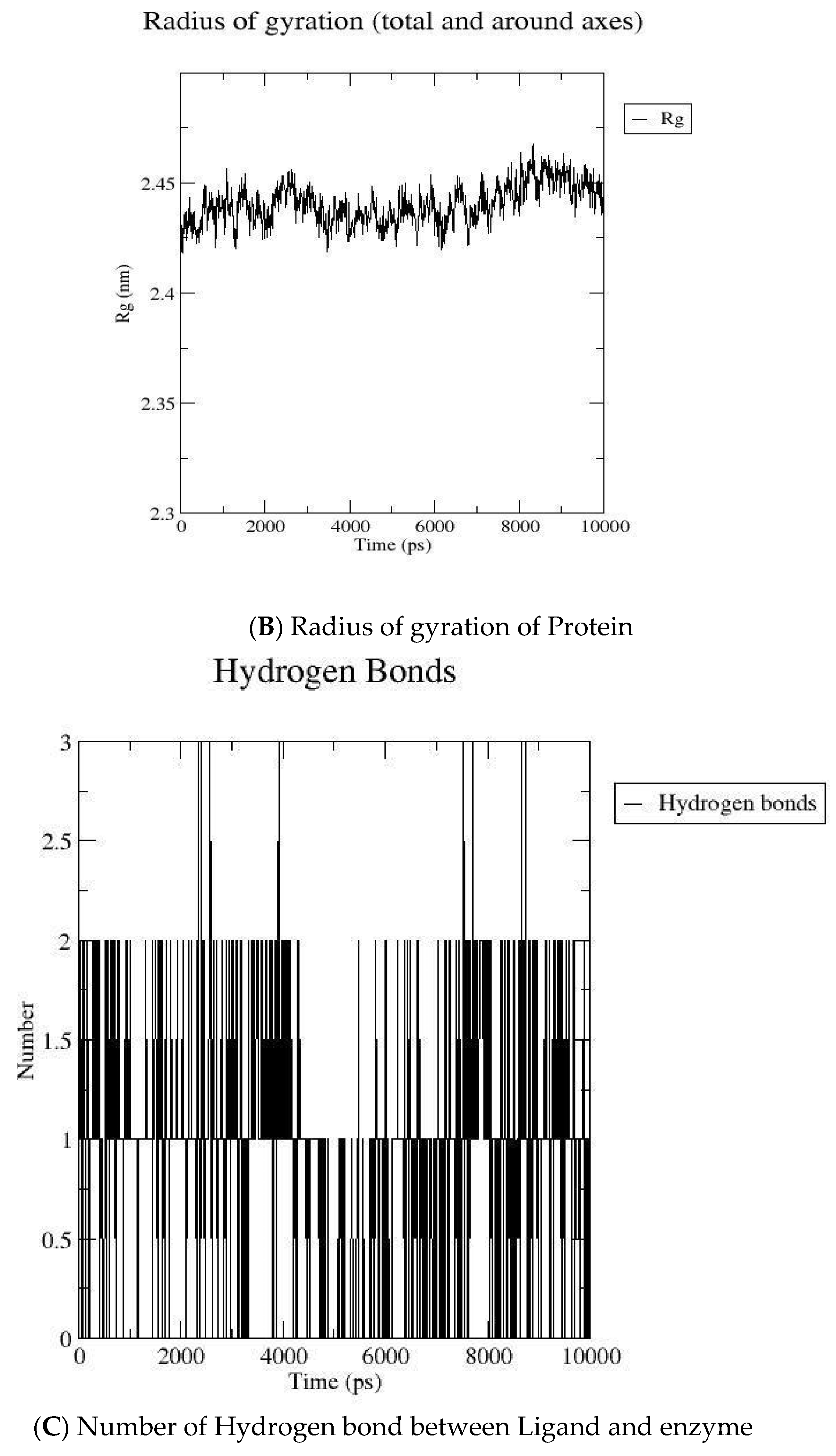
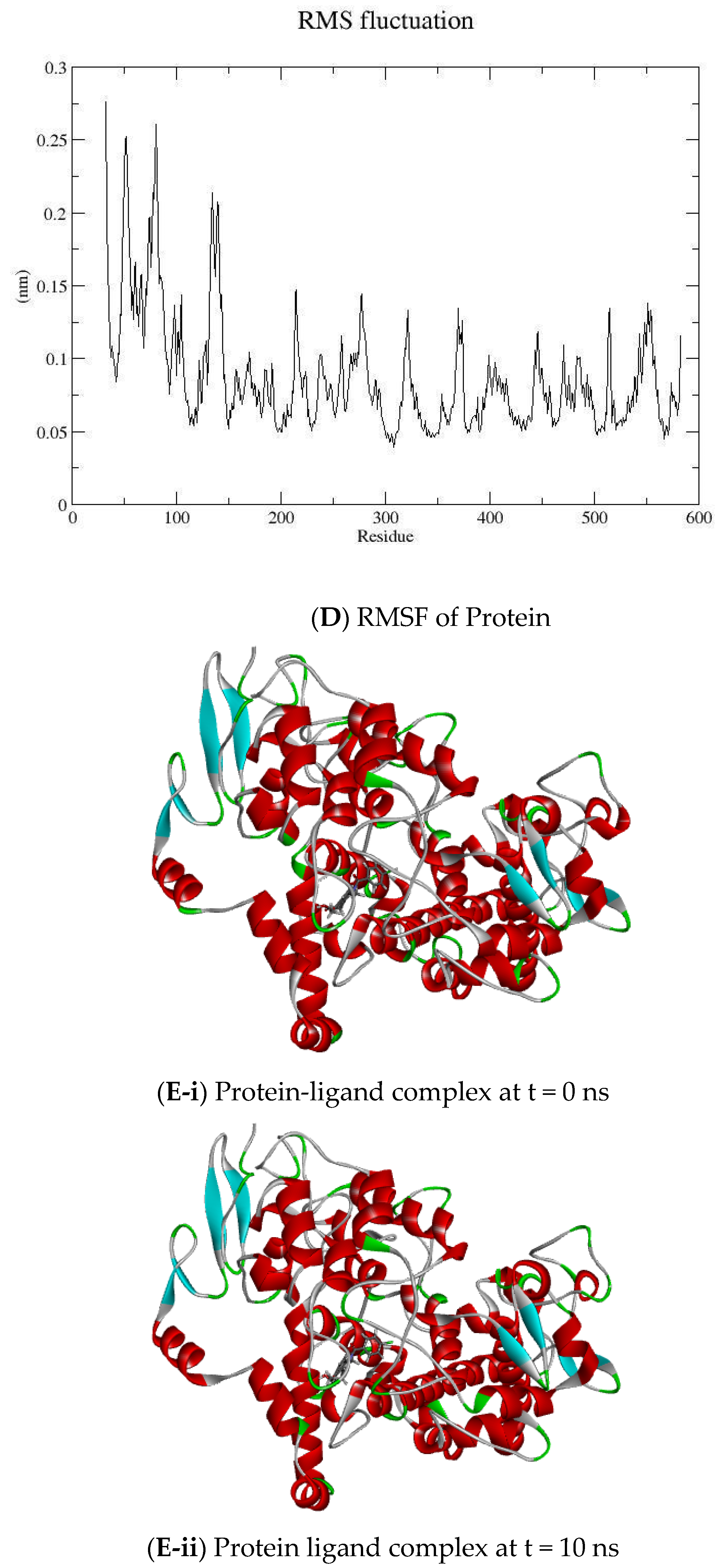
3.2. Molecular Dynamic Simulation
3.3. LD50 Predictions Using GUSAR
3.4. Physical Characterization of Indomethacin Derivatives
3.4.1. Structural Characterization of 2-(1(4-chlorobenzoyl),5-methoxy-2-methyl-1H-indol-3-yl)-N-phenylacetamide 3a
3.4.2. Structural Characterization of 2-(1-(4-chlorobenzoyl),5-methoxy-2-methyl-1H-indol-3-yl)-N-(2-chlorophenyl)acetamide 3b
3.4.3. Structural Characterization of N-(3-chloro-4-fluorophenyl)-2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)acetamide 3c
3.4.4. Structural Characterization of 2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)-N-(4-fluorophenyl)acetamide 3d
3.4.5. Structural Characterization of 2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)-N-(4-nitrophenyl)acetamide 3e
3.4.6. Structural Characterization of 2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)-N-(4-chlorophenyl)acetamide 3f
3.4.7. Structural Characterization of 2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)-N-(2,6-dichlorophenyl)acetamide 3g
3.5. Biological Screening
3.5.1. In Vitro Biological Screening of Indomethacin Analogues
3.5.2. In Vivo Pharmacological Screening
3.5.3. Determination of Ulcerogenic Effect
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kalgutkar, A.S.; Rowlinson, S.W.; Crews, B.C.; Marnett, L.J. Amide Derivatives of Meclofenamic Acid as Selective Cyclooxygenase-2 Inhibitors. Bioorg. Med. Chem. Lett. 2002, 12, 521–524. [Google Scholar] [CrossRef]
- Marnett, L.J.; Rowlinson, S.W.; Goodwin, D.C.; Kalgutkar, A.S.; Lanzo, C.A. Arachidonic Acid Oxygenation by COX-1 and COX-2. Mechanisms of Catalysis and Inhibition. J. Biol. Chem. 1999, 274, 22903–22906. [Google Scholar] [CrossRef] [Green Version]
- Dogne, J.-M.; Leval, X.; Hanson, J.; Frederich, M.; Lambermont, B.; Ghuysen, A.; Casini, A.; Masereel, B.; Ruan, K.-H.; Pirotte, B.; et al. New Developments on Thromboxane and Prostacyclin Modulators Part I: Thromboxane Modulators. Curr. Med. Chem. 2004, 11, 1223–1241. [Google Scholar] [CrossRef]
- Aljadhey, H.; Tu, W.; Hansen, R.A.; Blalock, S.; Brater, D.C.; Murray, M.D. Risk of Hyperkalemia Associated with Selective COX-2 Inhibitors. Pharmacoepidemiol. Drug Saf. 2010, 19, 1194–1198. [Google Scholar] [CrossRef] [Green Version]
- Bansal, S.; Bala, M.; Suthar, S.K.; Choudhary, S.; Bhattacharya, S.; Bhardwaj, V.; Singla, S.; Joseph, A. Design and Synthesis of Novel 2-Phenyl-5-(1,3-Diphenyl-1H-Pyrazol-4-Yl)-1,3,4-Oxadiazoles as Selective COX-2 Inhibitors with Potent Anti-Inflammatory Activity. Eur. J. Med. Chem. 2014, 80, 167–174. [Google Scholar] [CrossRef]
- Patrono, C.; Baigent, C. Nonsteroidal Anti-Inflammatory Drugs and the Heart. Circulation 2014, 129, 907–916. [Google Scholar] [CrossRef] [Green Version]
- Kalgutkar, A.S.; Marnett, A.B.; Crews, B.C.; Remmel, R.P.; Marnett, L.J. Ester and Amide Derivatives of the Nonsteroidal Antiinflammatory Drug, Indomethacin, as Selective Cyclooxygenase-2 Inhibitors. J. Med. Chem. 2000, 43, 2860–2870. [Google Scholar] [CrossRef]
- Savjani, J.K.; Mulamkattil, S.; Variya, B.; Patel, S. Molecular Docking, Synthesis and Biological Screening of Mefenamic Acid Derivatives as Anti-in Fl Ammatory Agents. Eur. J. Pharmacol. 2017, 801, 28–34. [Google Scholar] [CrossRef]
- Talley, J.J.; Brown, D.L.; Carter, J.S.; Graneto, M.J.; Koboldt, C.M.; Masferrer, J.L.; Perkins, W.E.; Rogers, R.S.; Shaffer, A.F.; Zhang, Y.Y.; et al. 4-[5-Methyl-3-Phenylisoxazol-4-Yl]-Benzenesulfonamide, Valdecoxib: A Potent and Selective Inhibitor of COX-2. J. Med. Chem. 2000, 43, 775–777. [Google Scholar] [CrossRef]
- Woods, K.W.; McCroskey, R.W.; Michaelides, M.R.; Wada, C.K.; Hulkower, K.I.; Bell, R.L. Thiazole Analogues of the NSAID Indomethacin as Selective COX-2 Inhibitors. Bioorg. Med. Chem. Lett. 2001, 11, 1325–1328. [Google Scholar] [CrossRef]
- Ganga Reddy, V.; Srinivasa Reddy, T.; Lakshma Nayak, V.; Prasad, B.; Reddy, A.P.; Ravikumar, A.; Taj, S.; Kamal, A. Design, Synthesis and Biological Evaluation of N-((1-Benzyl-1H-1,2,3-Triazol-4-Yl)Methyl)-1,3-Diphenyl-1H-Pyrazole-4-Carboxamides as CDK1/Cdc2 Inhibitors. Eur. J. Med. Chem. 2016, 122, 164–177. [Google Scholar] [CrossRef]
- Korobko, D. Synthesis of the Row of New Functional Derivatives of 7-Arylalkyl-8-Hydrazine Theophyllines. ScienceRise 2016, 3, 39. [Google Scholar] [CrossRef]
- Available online: http://www.pharmaexpert.ru/passonline/ (accessed on 22 May 2020).
- Jang, M. Cancer Chemopreventive Activity of Resveratrol, a Natural Product Derived from Grapes. Science 1997, 275, 218–220. [Google Scholar] [CrossRef] [Green Version]
- Esser, R.; Berry, C.; Du, Z.; Dawson, J.; Fox, A.; Fujimoto, R.A.; Haston, W.; Kimble, E.F.; Koehler, J.; Peppard, J.; et al. Preclinical Pharmacology of Lumiracoxib: A Novel Selective Inhibitor of Cyclooxygenase-2. Br. J. Pharmacol. 2005, 144, 538–550. [Google Scholar] [CrossRef] [Green Version]
- Chigayo, K.; Mojapelo, P.; Bessong, P.; Gumbo, J. The Preliminary Assessment of Anti-Microbial Activity of Hplc Separated Components of Kirkia wilmsii. Afr. J. Tradit. Complement. Altern. Med. 2014, 11, 275. [Google Scholar]
- Bali, A.; Ohri, R.; Deb, P.K. Synthesis, Evaluation and Docking Studies on 3-Alkoxy-4-Methanesulfonamido Acetophenone Derivatives as Non Ulcerogenic Anti-Inflammatory Agents. Eur. J. Med. Chem. 2012, 49, 397–405. [Google Scholar] [CrossRef]
- Vasantha, K.; Basavarajaswamy, G.; Vaishali Rai, M.; Boja, P.; Pai, V.R.; Shruthi, N.; Bhat, M. Rapid ‘One-Pot’ Synthesis of a Novel Benzimidazole-5-Carboxylate and Its Hydrazone Derivatives as Potential Anti-Inflammatory and Antimicrobial Agents. Bioorg. Med. Chem. Lett. 2015, 25, 1420–1426. [Google Scholar] [CrossRef]
- FitzGerald, G.A. COX-2 and beyond: Approaches to Prostaglandin Inhibition in Human Disease. Nat. Rev. Drug Discov. 2003, 2, 879–890. [Google Scholar] [CrossRef]
- Dogne, J.-M.; Hanson, J.; Supuran, C.; Pratico, D. Coxibs and Cardiovascular Side-Effects: From Light to Shadow. Curr. Pharm. Des. 2006, 12, 971–975. [Google Scholar] [CrossRef]
- Li, M.; Yin, L.; Cai, M.; Zhang, W.; Huang, Y.; Wang, X.; Zhu, X.; Shen, J. Design, Synthesis, and Anti-Inflammatory Evaluation of a Series of Novel Amino Acid-Binding 1,5-Diarylpyrazole Derivatives. Acta Pharmacol. Sin. 2005, 26, 865–872. [Google Scholar] [CrossRef] [Green Version]
- Smale, S.; Bjarnason, I. Determining Small Bowel Integrity Following Drug Treatment. Br. J. Clin. Pharmacol. 2003, 56, 284–291. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Code | GOLD Score | Interaction |
|---|---|---|
| Indomethacin | 45.07 | Val523, Ser530, Tyr385 |
| 3a | 50.08 | Ser530, Tyr385 |
| 3b | 51.09 | Tyr385, Trp387 |
| 3c | 49.66 | Val 523, Ser 530 |
| 3d | 46.55 | Ser530, Tyr385 |
| 3e | 48.11 | Trp 387, Ser 530 |
| 3f | 48.03 | Val 523, Ser 530 |
| 3g | 47.34 | Leu 384, Val 523 |
| 3h | 45.06 | Ser530, Tyr385 |
| 3i | 57.88 | Val 523, Ser 530, Tyr 385 Trp 387, Leu 384 |
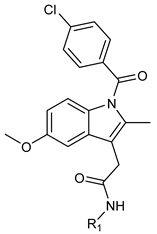
| Compound Code | R1 | LD50 (mg/kg) | |||
|---|---|---|---|---|---|
| IP | IV | Oral | SC | ||
| Indomethacin | - | 62.3 b * | 60.9 a # | 80.8 b * | 50.8 b * |
| 3a | C6H5 | 347.7 a # | 82.85 a # | 452.7 a # | 449.8 b # |
| 3b | 2-ClC6H4 | 312.4 a # | 36.64 a * | 391.6 a # | 318.7 b # |
| 3c | 3-Cl,4-FC6H3 | 271 a # | 60.33 a # | 458.6 a # | 2870 a $ |
| 3d | 4-FC6H4 | 309.2 a # | 43.26 a # | 344.6 a # | 2245 a & |
| 3e | 4-NO2C6H4 | 384.7 a # | 44.16 a # | 417.5 a # | 227.3 b # |
| 3f | 4-ClC6H4 | 322.6 a # | 45..7 a # | 546.2 a # | 343.6 b # |
| 3g | 2,3-dichloroC6H3 | 260.3 a # | 44.23 a # | 403.7 a # | 347.8 b # |
| 3h | CH2C6H5 | 414.5 a # | 71.13 a # | 337.8 a # | 374.7 b # |
| 3i | 3-CH3C6H4 | 334.4 a # | 45.68 a # | 480.6 a # | 1494 a & |
| Compound Code | IUPAC Name | IC50 COX-2 (µM) | IC50 COX-1 (µM) | Selectivity |
|---|---|---|---|---|
| Indomethacin | 2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1-H-indol-3-yl)acetic acid | 1.06 ± 0.31 | 3.50 ± 0.05 | 3.29 |
| 3a | 2-(1(4-chlorobenzoyl),5-methoxy-2-methyl-1H-indol-3-yl)-N-phenylacetamide | 2.97 ± 0.11 | 6.04 ± 0.14 | 2.04 |
| 3b | 2-(1-(4-chlorobenzoyl),5-methoxy-2-methyl-1H-indol-3-yl)-N-(2-chlorophenyl)acetamide | 3.28 ± 0.16 | 4.61 ± 0.31 | 1.41 |
| 3c | N-(3-chloro-4-fluorophenyl)-2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)acetamide | 1.39 ± 0.32 | 1.06 ± 0.10 | 0.76 |
| 3d | 2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)-N-(4-fluorophenyl)acetamide | 2.45 ± 0.03 | 5.0 ± 0.47 | 2.04 |
| 3e | 2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)-N-(4-nitrophenyl)acetamide | 0.34 ± 0.31 | 1.15 ± 0.30 | 3.32 |
| 3f | 2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)-N-(4-chlorophenyl)acetamide | 4.71 ± 0.4 | 3.55 ± 0.2 | 0.75 |
| 3g | 2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)-N-(2,6-dichlorophenyl)acetamide | 2.91 ± 0.38 | 4.14 ± 0.25 | 1.42 |
| 3h | N-benzyl-2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)acetamide | 4.73 ± 0.15 | 4.99 ± 0.11 | 1.06 |
| 3i | 2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)-N-m-tolylacetamide | 4.22 ± 0.23 | 4.71 ± 0.22 | 1.12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savjani, J.; Variya, B.; Patel, S.; Mulamkattil, S.; Amin, H.; Butani, S.; Allam, A.; Ajarem, J.; Shah, H. Drug Design, Synthesis and Biological Evaluation of Heterocyclic Molecules as Anti-Inflammatory Agents. Molecules 2022, 27, 1262. https://doi.org/10.3390/molecules27041262
Savjani J, Variya B, Patel S, Mulamkattil S, Amin H, Butani S, Allam A, Ajarem J, Shah H. Drug Design, Synthesis and Biological Evaluation of Heterocyclic Molecules as Anti-Inflammatory Agents. Molecules. 2022; 27(4):1262. https://doi.org/10.3390/molecules27041262
Chicago/Turabian StyleSavjani, Jignasa, Bhavesh Variya, Snehal Patel, Suja Mulamkattil, Harsh Amin, Shital Butani, Ahmed Allam, Jamaan Ajarem, and Harsh Shah. 2022. "Drug Design, Synthesis and Biological Evaluation of Heterocyclic Molecules as Anti-Inflammatory Agents" Molecules 27, no. 4: 1262. https://doi.org/10.3390/molecules27041262
APA StyleSavjani, J., Variya, B., Patel, S., Mulamkattil, S., Amin, H., Butani, S., Allam, A., Ajarem, J., & Shah, H. (2022). Drug Design, Synthesis and Biological Evaluation of Heterocyclic Molecules as Anti-Inflammatory Agents. Molecules, 27(4), 1262. https://doi.org/10.3390/molecules27041262