


Genome-Wide Analysis of Oxidosqualene Cyclase Genes in Artemisia annua: Evolution, Expression, and Potential Roles in Triterpenoid Biosynthesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Genome Data Acquisition and OSC Gene Identification
2.2. Physicochemical Properties Analysis of AaOSCs
2.3. Gene Structure, Protein Feature Analysis, and Phylogenetic Analysis
2.4. Promoter Cis-Element and Functional Annotation
2.5. Expression Profiling
3. Results
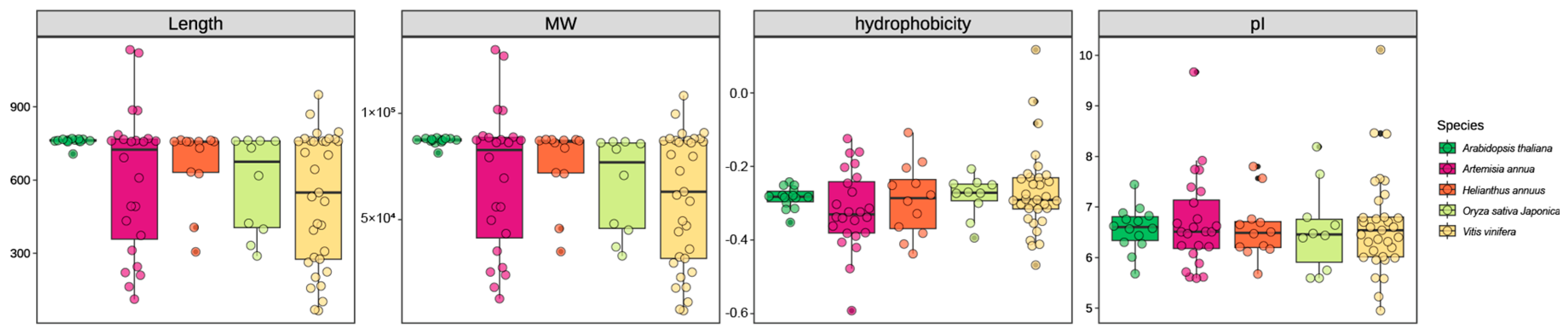
3.1. Genome-Wide Identification of AaOSCs and Physical and Chemical Characteristics
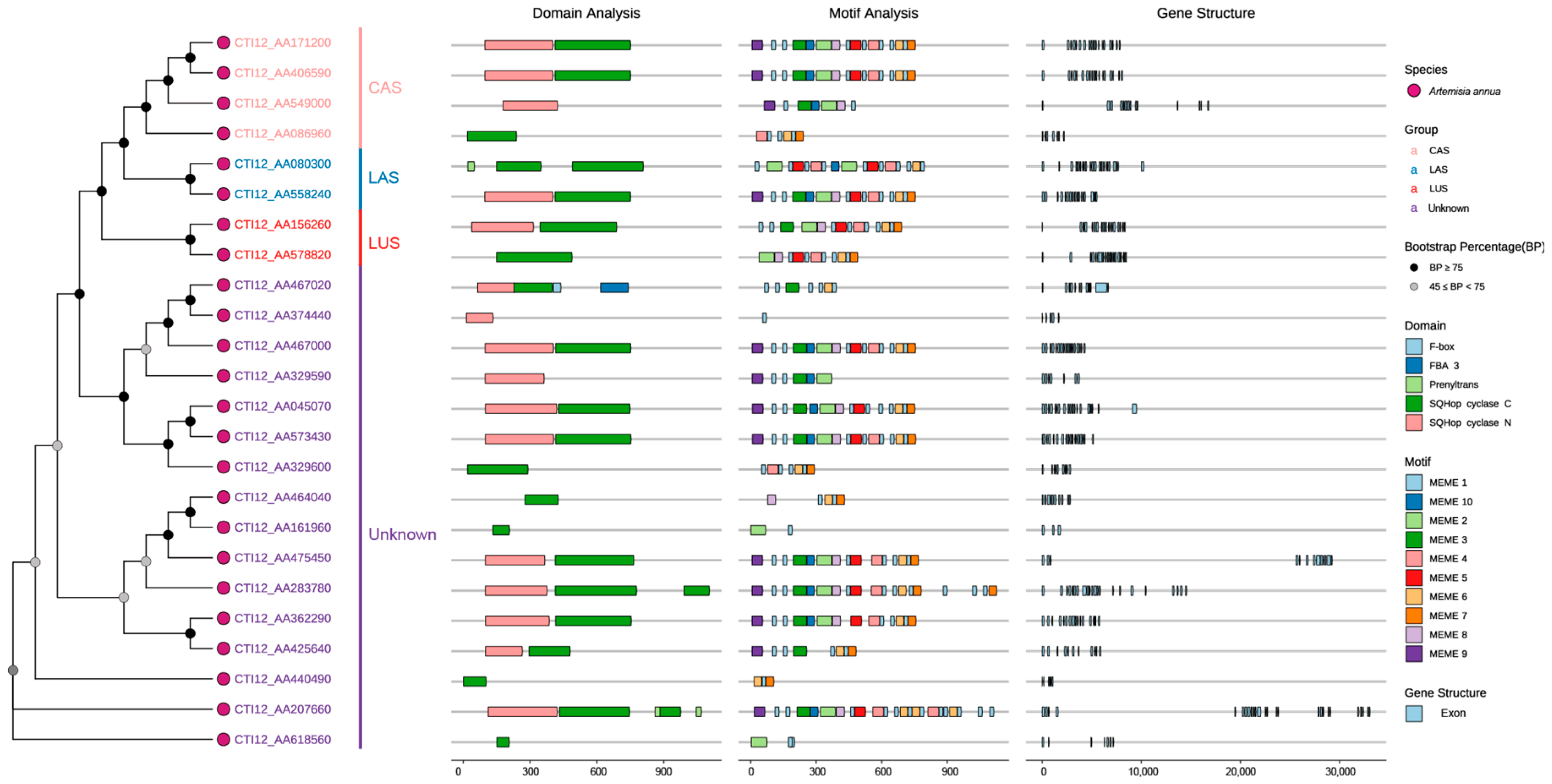
3.2. Gene Structure, Conserved Domain and Motif Analysis
3.3. Promoter Cis-Elements
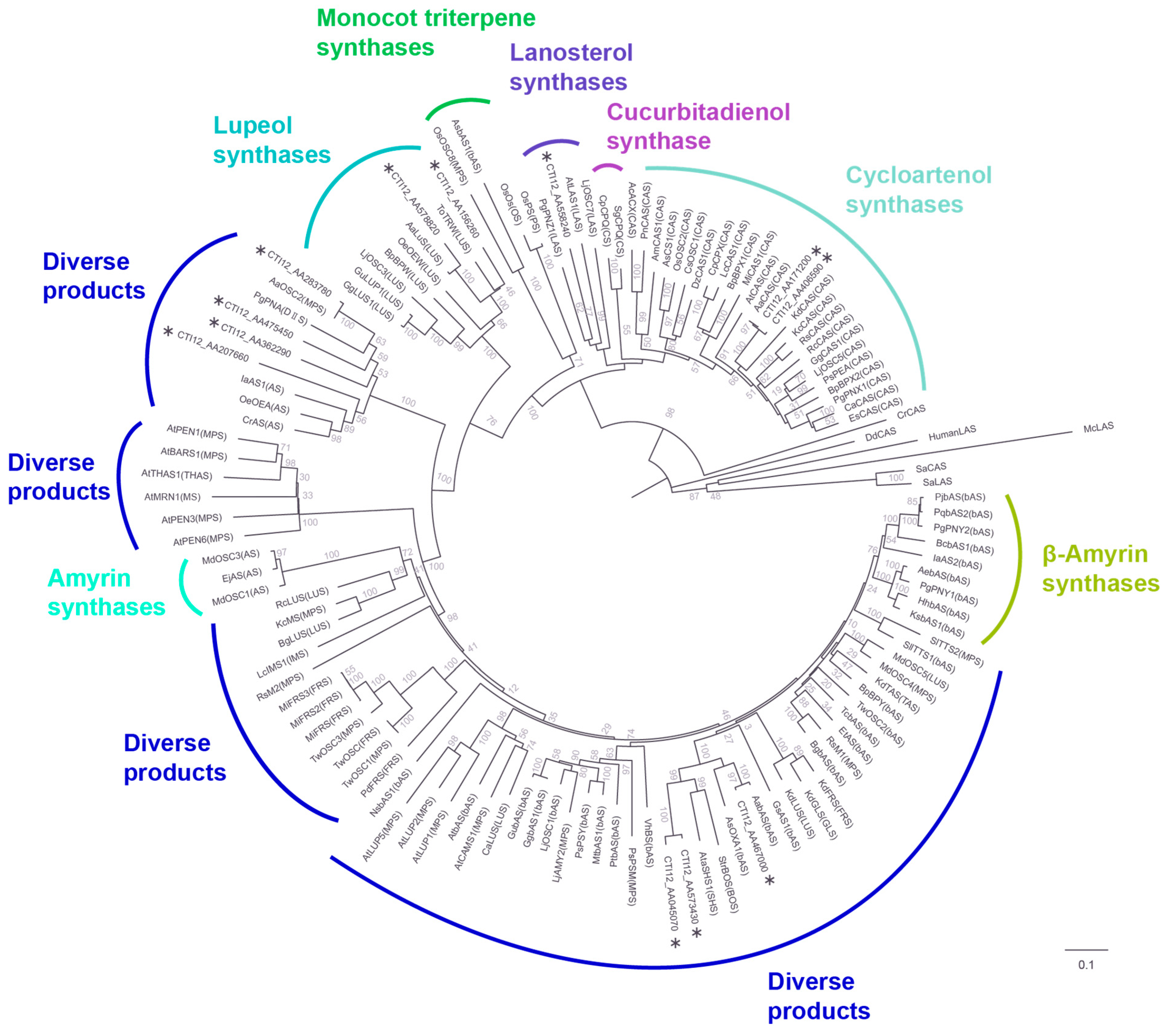
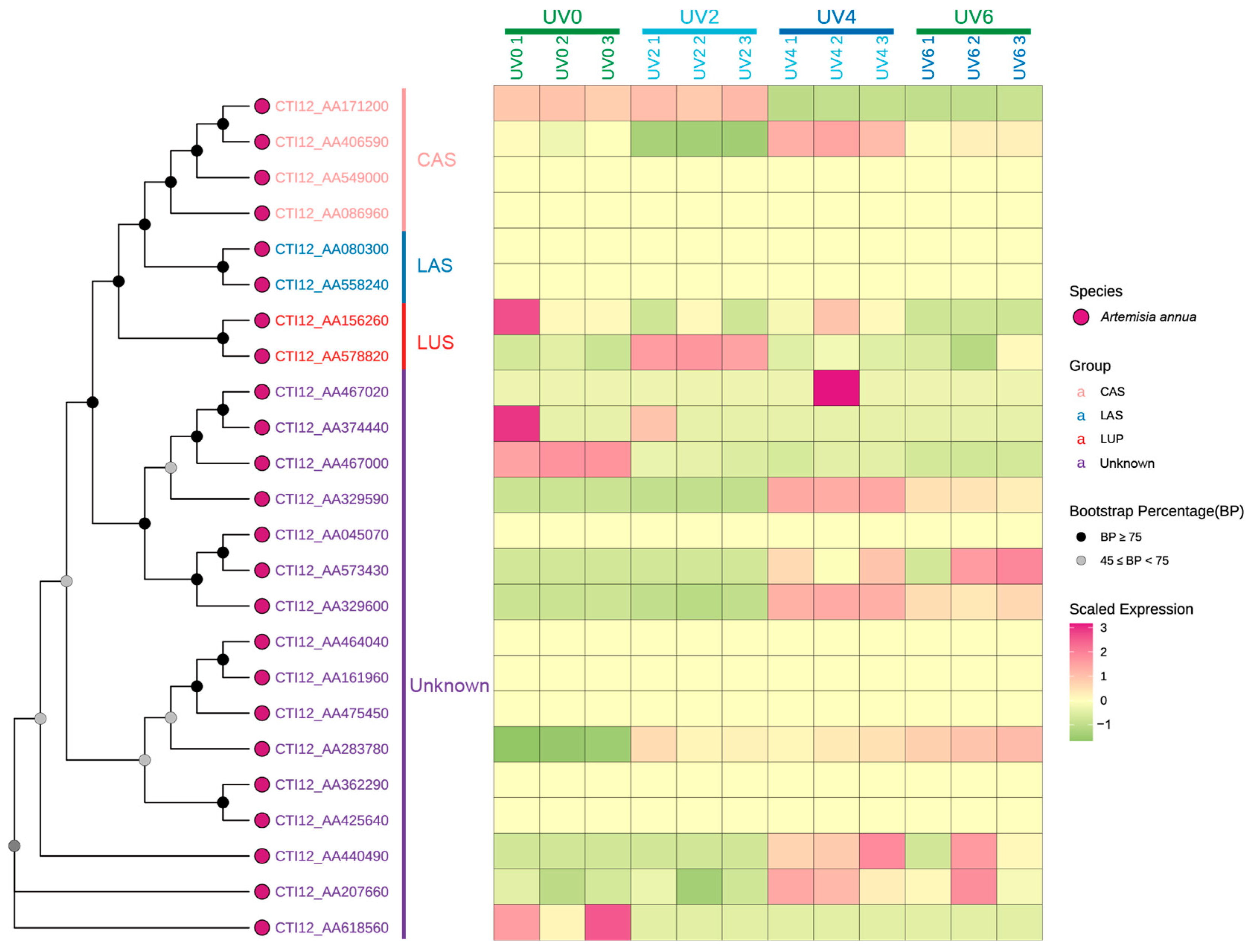
3.4. Phylogenetic Analysis
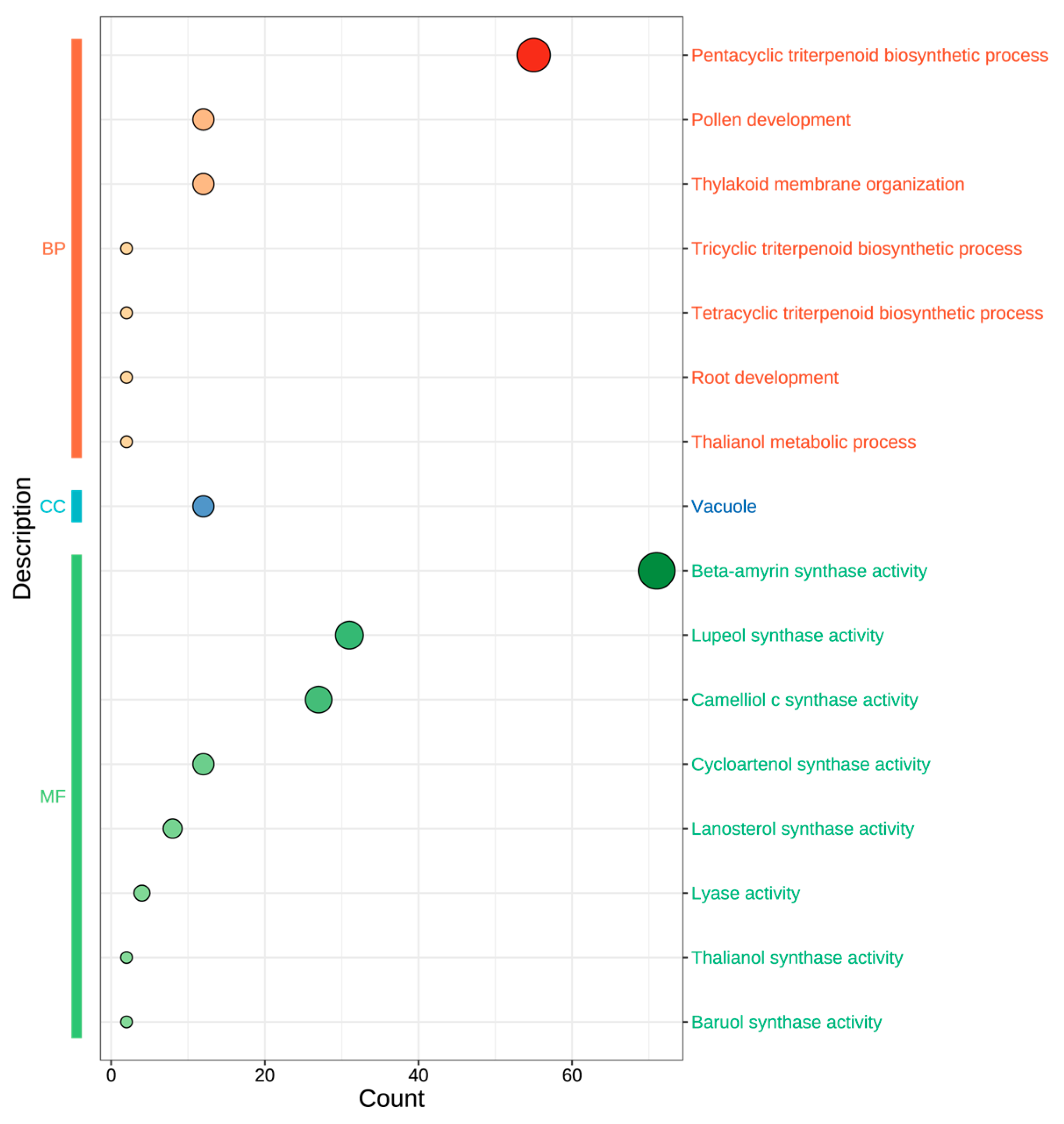
3.5. GO Annotation
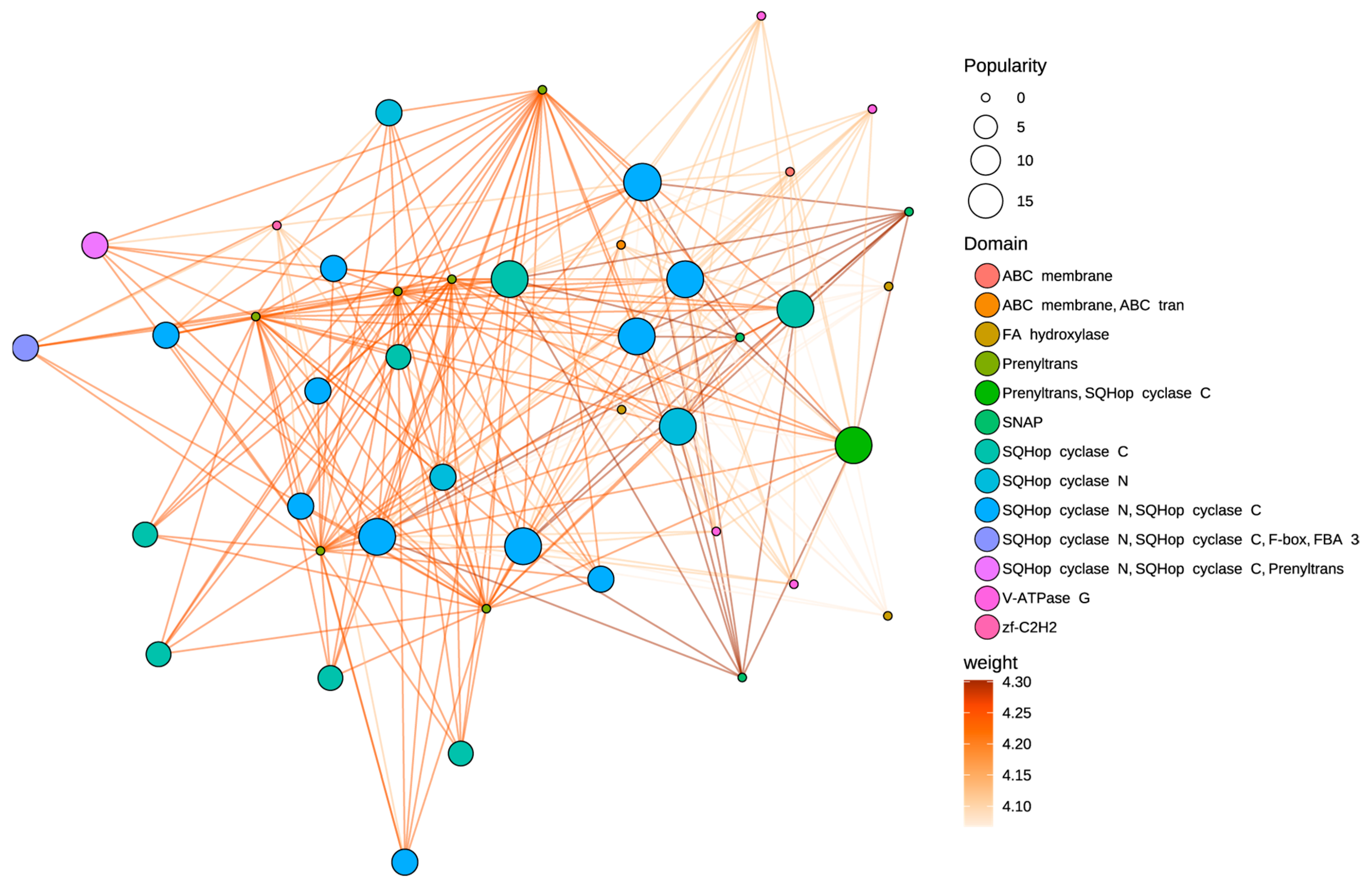
3.6. Protein–Protein Interaction
3.7. Expression Pattern in Tissues and Under UV Pressure
4. Discussion
4.1. Oxidosqualene Cyclase (OSC) Functional Diversification in A. annua
4.2. Evolutionary Mechanisms Enabling Metabolic Innovation
4.3. Transcriptional Regulation as an Adaptive Interface
4.4. Expression Dynamics Reflect Functional Prioritization
4.5. Bridging Knowledge Gaps
4.6. An Engineering Roadmap
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, Y.; Wang, J.; Li, L.; Song, W.; Li, M.; Hua, X.; Wang, Y.; Yuan, J.; Xue, Z. Natural Products of Pentacyclic Triterpenoids: From Discovery to Heterologous Biosynthesis. Nat. Prod. Rep. 2023, 40, 1303–1353. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, P.; Srivastava, V.; Bulone, V.; McKee, L.S. The Oxidosqualene Cyclase from the Oomycete Saprolegnia parasitica Synthesizes Lanosterol as a Single Product. Front. Microbiol. 2016, 7, 1802. [Google Scholar] [CrossRef] [PubMed]
- Thimmappa, R.; Geisler, K.; Louveau, T.; O’Maille, P.; Osbourn, A. Triterpene Biosynthesis in Plants. Annu. Rev. Plant Biol. 2014, 65, 225–257. [Google Scholar] [CrossRef] [PubMed]
- Thoma, R.; Schulz-Gasch, T.; D’Arcy, B.; Benz, J.; Aebi, J.; Dehmlow, H.; Hennig, M.; Stihle, M.; Ruf, A. Insight into Steroid Scaffold Formation from the Structure of Human Oxidosqualene Cyclase. Nature 2004, 432, 118–122. [Google Scholar] [CrossRef]
- Wang, J.; Xu, C.; Lun, Z.-R.; Meshnick, S.R. Unpacking ‘Artemisinin Resistance’. Trends Pharmacol. Sci. 2017, 38, 506–511. [Google Scholar] [CrossRef]
- Tu, Y. The Discovery of Artemisinin (qinghaosu) and Gifts from Chinese Medicine. Nat. Med. 2011, 17, 1217–1220. [Google Scholar] [CrossRef]
- White, N.J. Qinghaosu (Artemisinin): The Price of Success. Science 2008, 320, 330–334. [Google Scholar] [CrossRef]
- Li, Y.; Yang, Y.; Li, L.; Tang, K.; Hao, X.; Kai, G. Advanced Metabolic Engineering Strategies for Increasing Artemisinin Yield in Artemisia annua L. Hortic. Res. 2024, 11, uhad292. [Google Scholar]
- Zhang, F.; Lu, X.; Lv, Z.; Zhang, L.; Zhu, M.; Jiang, W.; Wang, G.; Sun, X.; Tang, K. Overexpression of the Artemisia Orthologue of ABA Receptor, AaPYL9, Enhances ABA Sensitivity and Improves Artemisinin Content in Artemisia annua L. PLoS ONE 2013, 8, e56697. PLoS ONE 2013, 8, e56697. [Google Scholar]
- Yu, Z.-X.; Li, J.-X.; Yang, C.-Q.; Hu, W.-L.; Wang, L.-J.; Chen, X.-Y. The Jasmonate-Responsive AP2/ERF Transcription Factors AaERF1 and AaERF2 Positively Regulate Artemisinin Biosynthesis in Artemisia annua L. Mol. Plant 2012, 5, 353–365. [Google Scholar]
- Osbourn, A. Saponins and Plant Defence-a Soap Story. Trends Plant Sci. 1996, 1, 4–9. [Google Scholar] [CrossRef]
- Mithöfer, A.; Boland, W. Plant Defense Against Herbivores: Chemical Aspects. Annu. Rev. Plant Biol. 2012, 63, 431–450. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Sawai, S. Triterpenoid Biosynthesis and Engineering in Plants. Front. Plant Sci. 2011, 2, 25. [Google Scholar]
- Szakiel, A.; Pączkowski, C.; Huttunen, S. Triterpenoid Content of Berries and Leaves of Bilberry Vaccinium myrtillus from Finland and Poland. J. Agric. Food. Chem. 2012, 60, 11839–11849. [Google Scholar] [CrossRef] [PubMed]
- Pichersky, E.; Lewinsohn, E. Convergent Evolution in Plant Specialized Metabolism. Annu. Rev. Plant Biol. 2011, 62, 549–566. [Google Scholar] [CrossRef] [PubMed]
- Osbourn, A. Secondary Metabolic Gene Clusters: Evolutionary Toolkits for Chemical Innovation. Trends Genet. 2010, 26, 449–457. [Google Scholar] [CrossRef]
- Seki, H.; Ohyama, K.; Sawai, S.; Mizutani, M.; Ohnishi, T.; Sudo, H.; Akashi, T.; Aoki, T.; Saito, K.; Muranaka, T. Licorice β-Amyrin 11-oxidase, a Cytochrome P450 with a Key Role in the Biosynthesis of the Triterpene Sweetener Glycyrrhizin. Proc. Natl. Acad. Sci. USA 2008, 105, 14204–14209. [Google Scholar] [CrossRef]
- Moses, T.; Pollier, J.; Shen, Q.; Soetaert, S.; Reed, J.; Erffelinck, M.-L.; Van Nieuwerburgh, F.C.W.; Vanden Bossche, R.; Osbourn, A.; Thevelein, J.M.; et al. OSC2 and CYP716A14v2 Catalyze the Biosynthesis of Triterpenoids for the Cuticle of Aerial Organs of Artemisia annua. Plant Cell 2015, 27, 286–301. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, M.; Ye, M.; Qiao, X. Site-directed Mutagenesis and Substrate Compatibility to Reveal the Structure-function Relationships of Plant Oxidosqualene Cyclases. Nat. Prod. Rep. 2021, 38, 2261–2275. [Google Scholar] [CrossRef]
- Abe, I.; Sankawa, U. Purification and Properties of Squalene-2, 3-epoxide Cyclases from Pea Seedlings. Chem. Pharm. Bull. 1992, 40, 1755–1760. [Google Scholar] [CrossRef]
- Corey, E.J.; Matsuda, S.P.; Bartel, B. Isolation of an Arabidopsis thaliana Gene Encoding Cycloartenol Synthase by Functional Expression in a Yeast Mutant Lacking Lanosterol Synthase by the Use of a Chromatographic Screen. Proc. Natl. Acad.Sci. USA 1993, 90, 11628–11632. [Google Scholar] [CrossRef] [PubMed]
- Morlacchi, P.; Wilson, W.K.; Xiong, Q.; Bhaduri, A.; Sttivend, D.; Kolesnikova, M.D.; Matsuda, S.P.T. Product Profile of PEN3: The Last Unexamined Oxidosqualene Cyclase in Arabidopsis thaliana. Org. Lett. 2009, 11, 2627–2630. [Google Scholar] [CrossRef] [PubMed]
- Segura, M.J.R.; Meyer, M.M.; Matsuda, S.P.T. Arabidopsis thaliana LUP1 Converts Oxidosqualene to Multiple Triterpene Alcohols and a Triterpene Diol. Org. Lett. 2000, 2, 2257–2259. [Google Scholar] [CrossRef]
- Zhang, H.; Hua, X.; Zheng, D.; Wu, H.; Li, C.; Rao, P.; Wen, M.; Choi, Y.-E.; Xue, Z.; Wang, Y.; et al. De Novo Biosynthesis of Oleanane-Type Ginsenosides in Saccharomyces cerevisiae Using Two Types of Glycosyltransferases from Panax ginseng. J. Agric. Food. Chem. 2022, 70, 2231–2240. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Meng, F.; Pan, X.; Qiu, X.; Li, C.; Lu, S. Chromosome-level Genome Assembly of Prunella vulgaris L. Provides Insights into Pentacyclic Triterpenoid Biosynthesis. Plant J. 2024, 118, 731–752. [Google Scholar] [CrossRef]
- Vernoud, V. β-Amyrin Synthase1 Controls the Accumulation of the Major Saponins Present in Pea (Pisum sativum). Plant Cell Physiol. 2021, 62, 784–797. [Google Scholar] [CrossRef]
- Xue, Z.; Duan, L.; Liu, D.; Guo, J.; Ge, S.; Dicks, J.; ÓMáille, P.; Osbourn, A.; Qi, X. Divergent Evolution of Oxidosqualene Cyclases in Plants. New Phytol. 2011, 193, 1022–1038. [Google Scholar] [CrossRef]
- Meesapyodsuk, D.; Balsevich, J.; Reed, D.W.; Covello, P.S. Saponin Biosynthesis in Saponaria vaccaria. cDNAs Encoding β-Amyrin Synthase and a Triterpene Carboxylic Acid Glucosyltransferase. Plant Physiol. 2007, 143, 959–969. [Google Scholar] [CrossRef]
- Sawai, S.; Akashi, T.; Sakurai, N.; Suzuki, H.; Shibata, D.; Ayabe, S.-i.; Aoki, T. Plant Lanosterol Synthase: Divergence of the Sterol and Triterpene Biosynthetic Pathways in Eukaryotes. Plant Cell Physiol. 2006, 47, 673–677. [Google Scholar] [CrossRef]
- Sun, J.; Xu, X.; Xue, Z.; Snyder, J.H.; Qi, X. Functional Analysis of a Rice Oxidosqualene Cyclase through Total Gene Synthesis. Mol. Plant 2013, 6, 1726–1729. [Google Scholar] [CrossRef]
- Haralampidis, K.; Bryan, G.; Qi, X.; Papadopoulou, K.; Bakht, S.; Melton, R.; Osbourn, A. A New Class of Oxidosqualene Cyclases Directs Synthesis of Antimicrobial Phytoprotectants in Monocots. Proc. Natl. Acad. Sci. USA 2001, 98, 13431–13436. [Google Scholar] [CrossRef]
- Shibuya, M.; Zhang, H.; Endo, A.; Shishikura, K.; Kushiro, T.; Ebizuka, Y. Two Branches of the Lupeol Synthase Gene in the Molecular Evolution of Plant Oxidosqualene Cyclases. Eur. J. Biochem. 1999, 266, 302–307. [Google Scholar] [CrossRef]
- Basyuni, M.; Oku, H.; Tsujimoto, E.; Kinjo, K.; Baba, S.; Takara, K. Triterpene Synthases from the Okinawan mangrove Tribe, Rhizophoraceae. FEBS J. 2007, 274, 5028–5042. [Google Scholar] [CrossRef]
- Hayashi, H.; Huang, P.; Takada, S.; Obinata, M.; Inoue, K.; Shibuya, M.; Ebizuka, Y. Differential Expression of Three Oxidosqualene Cyclase mRNAs in Glycyrrhiza glabra. Biol. Pharm. Bull. 2004, 27, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Guhling, O.; Hobl, B.; Yeats, T.; Jetter, R. Cloning and Characterization of a Lupeol Synthase Involved in the Synthesis of Epicuticular Wax Crystals on Stem and Hypocotyl Surfaces of Ricinus communis. Arch. Biochem. Biophys. 2006, 448, 60–72. [Google Scholar] [CrossRef]
- Herrera, J.B.R.; Bartel, B.; Wilson, W.K.; Matsuda, S.P.T. Cloning and Characterization of the Arabidopsis thaliana Lupeol Synthase Gene. Phytochemistry 1998, 49, 1905–1911. [Google Scholar] [CrossRef] [PubMed]
- Kirby, J.; Romanini, D.W.; Paradise, E.M.; Keasling, J.D. Engineering Triterpene Production in Saccharomyces cerevisiae-β-Amyrin Synthase from Artemisia annua. FEBS J. 2008, 275, 1852–1859. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J.; Tamura, K. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef] [PubMed]
- Moses, T.; Pollier, J.; Thevelein, J.M.; Goossens, A. Bioengineering of Plant (Tri)Terpenoids: From Metabolic Engineering of Plants to Synthetic Biology in Vivo and in Vitro. New Phytol. 2013, 200, 27–43. [Google Scholar] [CrossRef]
- Weng, J.-K.; Philippe, R.N.; Noel, J.P. The Rise of Chemodiversity in Plants. Science 2012, 336, 1667–1670. [Google Scholar] [CrossRef]
- Shen, Q.; Zhang, L.; Liao, Z.; Wang, S.; Yan, T.; Shi, P.; Liu, M.; Fu, X.; Pan, Q.; Wang, Y.; et al. The Genome of Artemisia annua Provides Insight into the Evolution of Asteraceae Family and Artemisinin Biosynthesis. Mol. Plant 2018, 11, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M. Plant Sterols and the Membrane Environment. Trends Plant Sci. 1998, 3, 170–175. [Google Scholar] [CrossRef]
- Richards, T.A.; Soanes, D.M.; Jones, M.D.M.; Vasieva, O.; Leonard, G.; Paszkiewicz, K.; Foster, P.G.; Hall, N.; Talbot, N.J. Horizontal Gene Transfer Facilitated the Evolution of Plant Parasitic Mechanisms in the Oomycetes. Proc. Natl. Acad. Sci. USA 2011, 108, 15258–15263. [Google Scholar] [CrossRef]
- Daum, G.; Lees, N.D.; Bard, M.; Dickson, R.J.Y. Biochemistry, Cell Biology and Molecular Biology of Lipids of Saccharomyces cerevisiae. Yeast 1998, 14, 1471–1510. [Google Scholar] [CrossRef]
- William Roy, S.; Gilbert, W. The Evolution of Spliceosomal Introns: Patterns, Puzzles and Progress. Nat. Rev. Genet. 2006, 7, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Filichkin, S.A.; Cumbie, J.S.; Dharmawardhana, P.; Jaiswal, P.; Chang, J.H.; Palusa, S.G.; Reddy, A.; Megraw, M.; Mockler, T. Environmental Stresses Modulate Abundance and Timing of Alternatively Spliced Circadian Transcripts in Arabidopsis. Mol. Plant 2015, 8, 207–227. [Google Scholar] [CrossRef]
- Hua, Z.; Vierstra, R.D. The Cullin-RING Ubiquitin-Protein Ligases. Annu. Rev. Plant Biol. 2011, 62, 299–334. [Google Scholar] [CrossRef]
- Tholl, D.; Lee, S. Terpene Specialized Metabolism in Arabidopsis thaliana. Arab. Book/Am. Soc. Plant Biol. 2011, 9, e0143. [Google Scholar]
- Santner, A.; Estelle, M. The Ubiquitin-Proteasome System Regulates Plant Hormone Signaling. Plant J. 2010, 61, 1029–1040. [Google Scholar] [CrossRef]
- Tokuriki, N.; Tawfik, D.S. Protein Dynamism and Evolvability. Science 2009, 324, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Fazio, G.C.; Matsuda, S.P.T. On the Origins of Triterpenoid Skeletal Diversity. Phytochemistry 2004, 65, 261–291. [Google Scholar] [CrossRef]
- Kushiro, T.; Shibuya, M.; Masuda, K.; Ebizuka, Y. Mutational Studies on Triterpene Synthases: Engineering Lupeol Synthase into β-Amyrin Synthase. J. Am. Chem. Soc. 2000, 122, 6816–6824. [Google Scholar] [CrossRef]
- Miettinen, K.; Pollier, J.; Buyst, D.; Arendt, P.; Csuk, R.; Sommerwerk, S.; Moses, T.; Mertens, J.; Sonawane, P.D.; Pauwels, L.; et al. The Ancient CYP716 Family is a Major Contributor to the Diversification of Eudicot Triterpenoid Biosynthesis. Nat. Commun. 2017, 8, 14153. [Google Scholar] [CrossRef] [PubMed]
- De Geyter, N.; Gholami, A.; Goormachtig, S.; Goossens, A. Transcriptional Machineries in Jasmonate-elicited Plant Secondary Metabolism. Trends Plant Sci. 2012, 17, 349–359. [Google Scholar] [CrossRef]
- Clark, S.E.; Running, M.P.; Meyerowitz, E.M. CLAVATA3 is a Specific Regulator of Shoot and Floral Meristem Development Affecting the Same Processes as CLAVATA1. Development 1995, 121, 2057–2067. [Google Scholar] [CrossRef]
- Jenkins, G.I. Photomorphogenic responses to ultraviolet-B light. Plant Cell Environ. 2017, 40, 2544–2557. [Google Scholar] [CrossRef]
- Carbonell-Bejerano, P.; Diago, M.-P.; Martínez-Abaigar, J.; Martínez-Zapater, J.M.; Tardáguila, J.; Núñez-Olivera, E. Solar Ultraviolet Radiation is Necessary to Enhance Grapevine Fruit Ripening Transcriptional and Phenolic Responses. BMC Plant Biol. 2014, 14, 1–16. [Google Scholar] [CrossRef]
- Babiychuk, E.; Bouvier-Navé, P.; Compagnon, V.; Suzuki, M.; Muranaka, T.; Van Montagu, M.; Kushnir, S.; Schaller, H. Allelic Mutant Series Reveal Distinct Functions for Arabidopsis Cycloartenol Synthase 1 in Cell Viability and Plastid Biogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 3163–3168. [Google Scholar] [CrossRef]
- Venkateshwaran, M.; Jayaraman, D.; Chabaud, M.; Genre, A.; Balloon, A.J.; Maeda, J.; Forshey, K.; den Os, D.; Kwiecien, N.W.; Coon, J.J.; et al. A Role for the Mevalonate Pathway in Early Plant Symbiotic Signaling. Proc. Natl. Acad. Sci. USA 2015, 112, 9781–9786. [Google Scholar] [CrossRef]
- Suzuki, M.; Kamide, Y.; Nagata, N.; Seki, H.; Ohyama, K.; Kato, H.; Masuda, K.; Sato, S.; Kato, T.; Tabata, S.J.T.P.J. Loss of Function of 3-hydroxy-3-methylglutaryl Coenzyme A Reductase 1 (HMG1) in Arabidopsis Leads to Dwarfing, Early Senescence and Male Sterility, and Reduced Sterol Levels. Plant J. 2004, 37, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Huang, P.; Inoue, K. Up-regulation of Soyasaponin Biosynthesis by Methyl Jasmonate in Cultured Cells of Glycyrrhiza glabra. Plant Cell Physiol. 2003, 44, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Kautsar, S.A.; Hong, Y.J.; Medema, M.H.; Bond, A.D.; Tantillo, D.J.; Osbourn, A. Unearthing a Sesterterpene Biosynthetic Repertoire in the Brassicaceae Through Genome Mining Reveals Convergent Evolution. Proc. Natl. Acad. Sci. USA 2017, 114, E6005–E6014. [Google Scholar] [CrossRef]
- Osbourn, A. Gene Clusters for Secondary Metabolic Pathways: An Emerging Theme in Plant Biology. Plant Physiol. 2010, 154, 531–535. [Google Scholar] [CrossRef]
- Graham, I.A.; Besser, K.; Blumer, S.; Branigan, C.A.; Czechowski, T.; Elias, L.; Guterman, I.; Harvey, D.; Isaac, P.G.; Khan, A.M.; et al. The Genetic Map of Artemisia annua L. Identifies Loci Affecting Yield of the Antimalarial Drug Artemisinin. Science 2010, 327, 328–331. [Google Scholar] [CrossRef] [PubMed]
- Ro, D.-K.; Paradise, E.M.; Ouellet, M.; Fisher, K.J.; Newman, K.L.; Ndungu, J.M.; Ho, K.A.; Eachus, R.A.; Ham, T.S.; Kirby, J.; et al. Production of the Antimalarial Drug Precursor Artemisinic Acid in Engineered Yeast. Nature 2006, 440, 940–943. [Google Scholar] [CrossRef]
- Kirby, J.; Keasling, J.D. Biosynthesis of Plant Isoprenoids: Perspectives for Microbial Engineering. Annu. Rev. Plant Biol. 2009, 60, 335–355. [Google Scholar] [CrossRef]
- Sainsbury, F.; Thuenemann, E.C.; Lomonossoff, G.P. pEAQ: Versatile Expression Vectors for Easy and Quick Transient Expression of Heterologous Proteins in Plants. Plant Biotechnol. J. 2009, 7, 682–693. [Google Scholar] [CrossRef]
- Reed, J.; Osbourn, A. Engineering Terpenoid Production Through Transient Expression in Nicotiana benthamiana. Plant Cell Rep. 2018, 37, 1431–1441. [Google Scholar] [CrossRef]
- Goodin, M.M.; Zaitlin, D.; Naidu, R.A.; Lommel, S.A. Nicotiana benthamiana: Its History and Future as a Model for Plant-pathogen Interactions. Mol. Plant-Microbe Interact. 2008, 21, 1015–1026. [Google Scholar] [CrossRef]
- Pavesi, G.; Mereghetti, P.; Mauri, G.; Pesole, G. Weeder Web: Discovery of Transcription Factor Binding Sites in a Set of Sequences from Co-regulated Genes. Nucleic Acids Res. 2004, 32, W199–W203. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.S.; Mortazavi, A.; Myers, R.M.; Wold, B. Genome-Wide Mapping of in Vivo Protein-DNA Interactions. Science 2007, 316, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Bohlmann, J.; Meyer-Gauen, G.; Croteau, R. Plant Terpenoid Synthases: Molecular Biology and Phylogenetic Analysis. Proc. Natl. Acad. Sci. USA 1998, 95, 4126–4133. [Google Scholar] [CrossRef]
- Ober, D. Gene Duplications and the Time Thereafter—Examples from Plant Secondary Metabolism. Plant Biol. 2010, 12, 570–577. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Karunanithi, P.S.; Zerbe, P. Terpene Synthases as Metabolic Gatekeepers in the Evolution of Plant Terpenoid Chemical Diversity. Front. Plant Sci. 2019, 10, 1166. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, C.; Xu, S.; Guo, X. Genome-Wide Analysis of Oxidosqualene Cyclase Genes in Artemisia annua: Evolution, Expression, and Potential Roles in Triterpenoid Biosynthesis. Curr. Issues Mol. Biol. 2025, 47, 545. https://doi.org/10.3390/cimb47070545
Guo C, Xu S, Guo X. Genome-Wide Analysis of Oxidosqualene Cyclase Genes in Artemisia annua: Evolution, Expression, and Potential Roles in Triterpenoid Biosynthesis. Current Issues in Molecular Biology. 2025; 47(7):545. https://doi.org/10.3390/cimb47070545
Chicago/Turabian StyleGuo, Changfeng, Si Xu, and Xiaoyun Guo. 2025. "Genome-Wide Analysis of Oxidosqualene Cyclase Genes in Artemisia annua: Evolution, Expression, and Potential Roles in Triterpenoid Biosynthesis" Current Issues in Molecular Biology 47, no. 7: 545. https://doi.org/10.3390/cimb47070545
APA StyleGuo, C., Xu, S., & Guo, X. (2025). Genome-Wide Analysis of Oxidosqualene Cyclase Genes in Artemisia annua: Evolution, Expression, and Potential Roles in Triterpenoid Biosynthesis. Current Issues in Molecular Biology, 47(7), 545. https://doi.org/10.3390/cimb47070545