
Molecular Docking and Drug-Likeness of Salicornia-Derived Phytochemicals Against HER Receptors

Abstract
1. Introduction
2. Materials and Methods
2.1. Retrieval and Preparation of Proteins
2.2. Ligand Selection and Preparation
2.3. Molecular Docking Analysis and Visualization
2.4. In Silico ADME Analysis
2.5. Toxicity Analysis of Selected Compounds
3. Results and Discussion
3.1. Molecular Docking Analysis
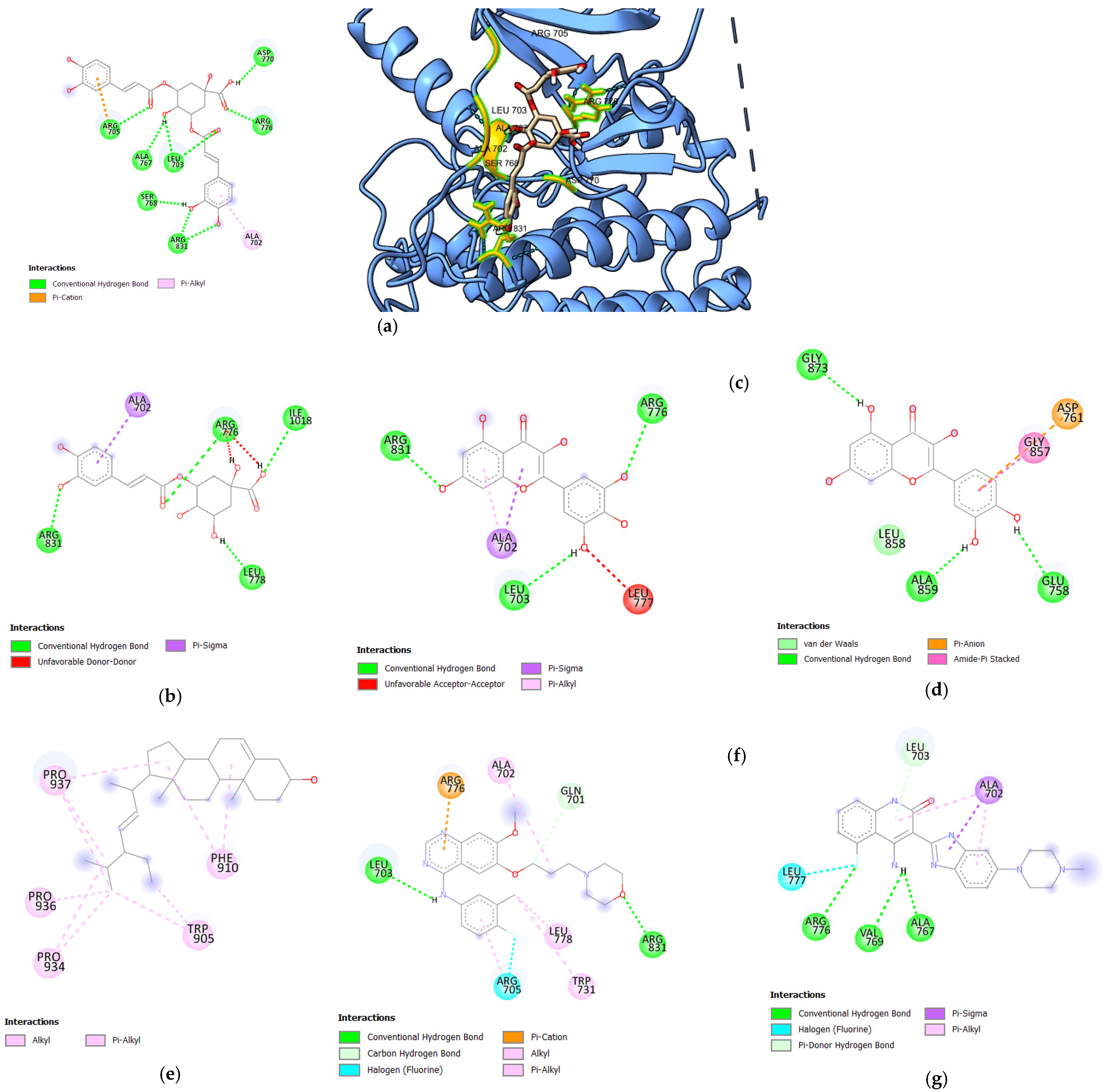
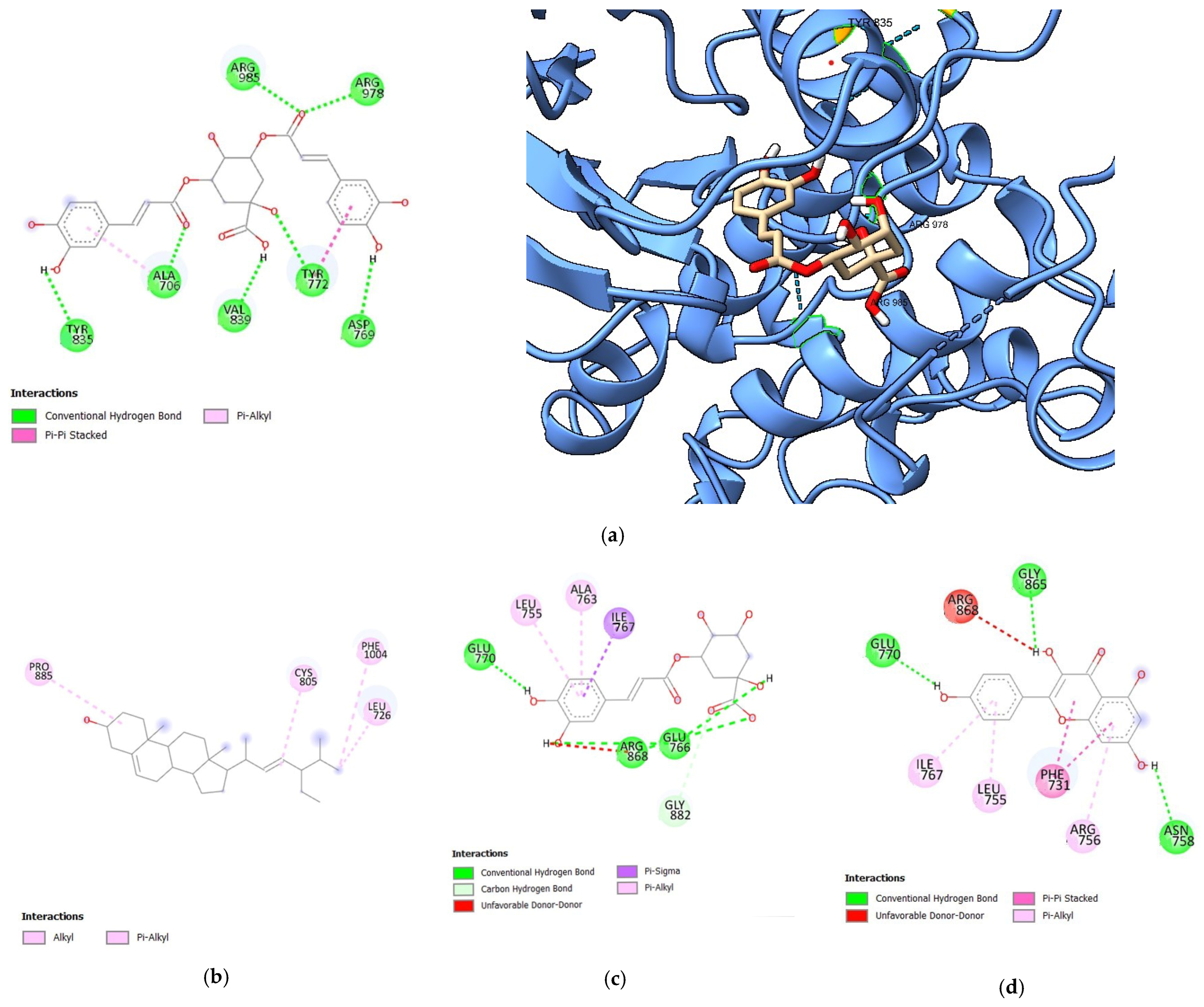
3.1.1. Molecular Docking Analysis of Bioactive Compounds from S. herbacea and S. brachiata with HER1 Receptor
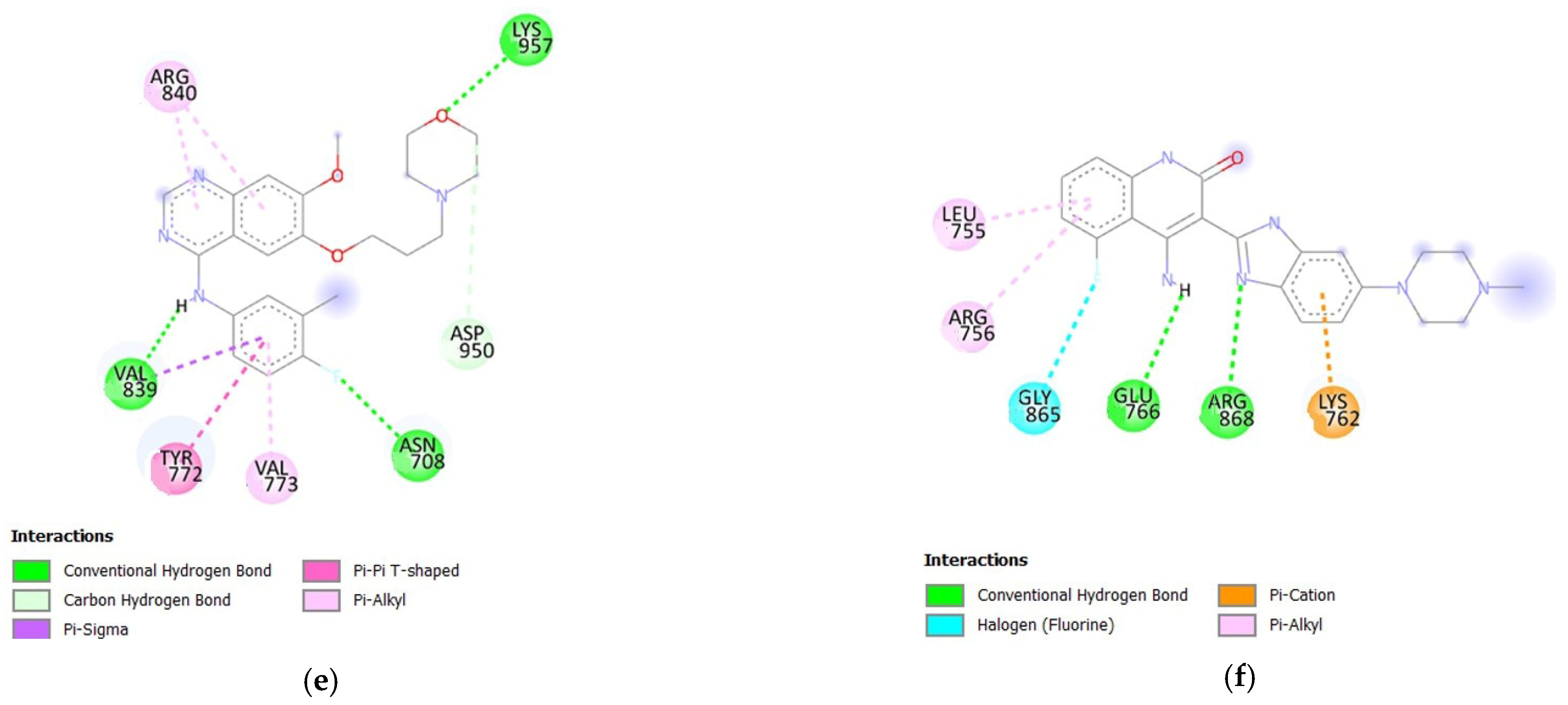
3.1.2. Molecular Docking Analysis of Bioactive Compounds from S. herbacea and S. brachiata with the HER2 Receptor
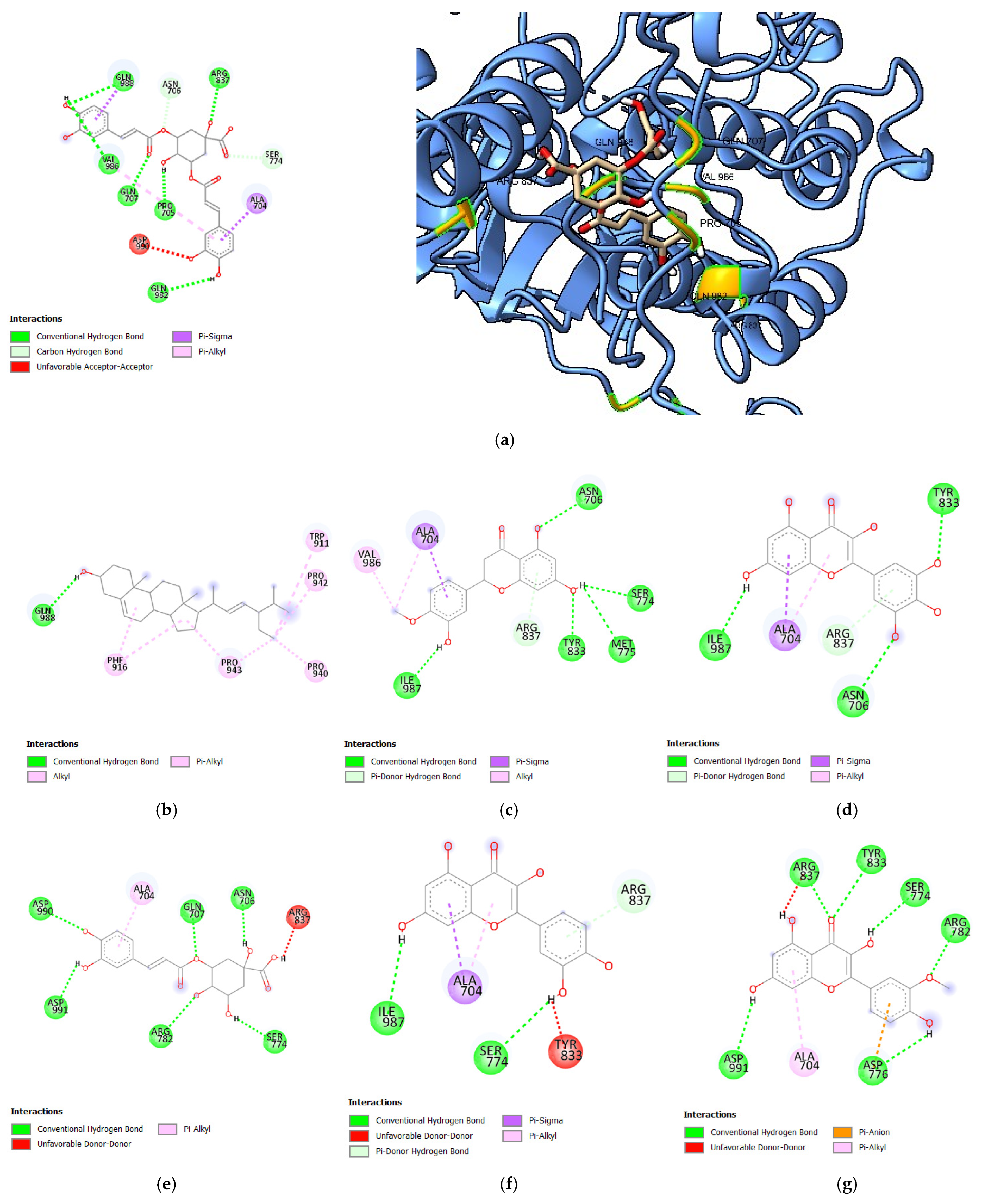
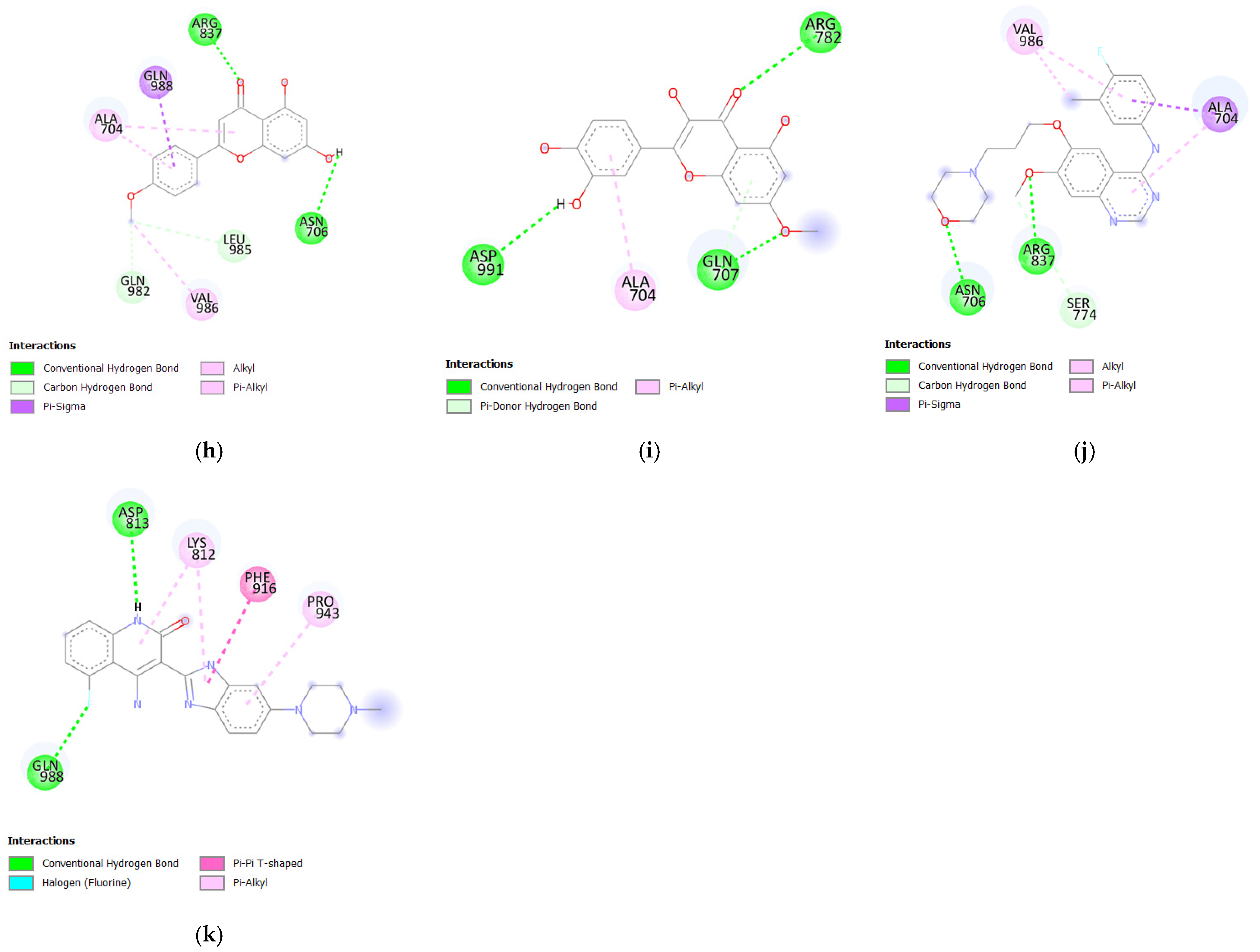
3.1.3. Molecular Docking Analysis of Bioactive Compounds from S. herbacea and S. brachiata with the HER4 Receptor
3.2. Drug-Likeness Properties of the Selected Phytochemicals According to Lipinski’s Rule of Five
3.3. Pharmacokinetic Properties of Selected Phytochemicals According to SwissADME Analysis
3.4. Toxicity Analysis
3.5. Prediction of Organ-Specific Toxicity of the Selected Compounds in S. herbacea
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer Statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Arthur, R.S.; Kabat, G.C.; Kim, M.Y.; Wild, R.A.; Shadyab, A.H.; Wactawski-Wende, J.; Ho, G.Y.F.; Reeves, K.W.; Kuller, L.H.; Luo, J.; et al. Metabolic Syndrome and Risk of Endometrial Cancer in Postmenopausal Women: A Prospective Study. Cancer Causes Control 2019, 30, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Bauso, L.V.; La Fauci, V.; Munaò, S.; Bonfiglio, D.; Armeli, A.; Maimone, N.; Longo, C.; Calabrese, G. Biological Activity of Natural and Synthetic Peptides as Anticancer Agents. Int. J. Mol. Sci. 2024, 25, 7264. [Google Scholar] [CrossRef]
- Singh, A. Global Burden of Five Major Types of Gastrointestinal Cancer. Gastroenterol. Rev. 2024, 19, 236–254. [Google Scholar] [CrossRef]
- Zhou, J.; Sun, H.; Wang, Z.; Cong, W.; Zeng, M.; Zhou, W.; Bie, P.; Liu, L.; Wen, T.; Kuang, M.; et al. Guidelines for the Diagnosis and Treatment of Primary Liver Cancer (2022 Edition). Liver Cancer 2023, 12, 405–444. [Google Scholar] [CrossRef]
- Boța, M.; Vlaia, L.; Jîjie, A.-R.; Marcovici, I.; Crişan, F.; Oancea, C.; Dehelean, C.A.; Mateescu, T.; Moacă, E.-A. Exploring Synergistic Interactions between Natural Compounds and Conventional Chemotherapeutic Drugs in Preclinical Models of Lung Cancer. Pharmaceuticals 2024, 17, 598. [Google Scholar] [CrossRef]
- Newman, D.J. Non-Insulin-Based Drug Entities Used to Treat Diabetes Type 2 Disease (T2DM), Based on Natural Products from All Sources. J. Nat. Prod. 2024, 87, 629–637. [Google Scholar] [CrossRef]
- Chantrill, L.A.; Nagrial, A.M.; Watson, C.; Johns, A.L.; Martyn-Smith, M.; Simpson, S.; Mead, S.; Jones, M.D.; Samra, J.S.; Gill, A.J.; et al. Precision Medicine for Advanced Pancreas Cancer: The Individualized Molecular Pancreatic Cancer Therapy (IMPaCT) Trial. Clin. Cancer Res. 2015, 21, 2029–2037. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Siridewa, K.; De Silva, W.; Ratnayake, R.M.C.S.; Wijesundara, S.; Perera, D.; Attanayake, R.N. Species Identification and Pollination Biology of an Economically Important True Halophyte, Salicornia brachiata Roxb. Aquat. Bot. 2025, 196, 103827. [Google Scholar] [CrossRef]
- Ekanayake, S.; Egodawatta, C.; Attanayake, R.N.; Perera, D. From Salt Pan to Saucepan: Salicornia, a Halophytic Vegetable with an Array of Potential Health Benefits. Food Front. 2023, 4, 641–676. [Google Scholar] [CrossRef]
- Kang, S.; Kim, M.-R.; Chiang, M.; Hong, J. Evaluation and Comparison of Functional Properties of Freshwater-Cultivated Glasswort (Salicornia herbacea L.) with Naturally-Grown Glasswort. Food Sci. Biotechnol. 2015, 24, 2245–2250. [Google Scholar] [CrossRef]
- Kim, S.; Lee, E.-Y.; Hillman, P.F.; Ko, J.; Yang, I.; Nam, S.-J. Chemical Structure and Biological Activities of Secondary Metabolites from Salicornia europaea L. Molecules 2021, 26, 2252. Molecules 2021, 26, 2252. [Google Scholar] [CrossRef]
- Sánchez-Gavilán, I.; Ramírez, E.; de la Fuente, V. Bioactive Compounds in Salicornia patula Duval-Jouve: A Mediterranean Edible Euhalophyte. Foods 2021, 10, 410. [Google Scholar] [CrossRef]
- Lee, W.-J.; Shin, Y.-W.; Kim, D.-E.; Kweon, M.-H.; Kim, M. Effect of Desalted Salicornia europaea L. Ethanol Extract (PM-EE) on the Subjects Complaining Memory Dysfunction without Dementia: A 12-Week, Randomized, Double-Blind, Placebo-Controlled Clinical Trial. Sci. Rep. 2020, 10, 19914. [Google Scholar] [CrossRef]
- Nájar, A.M.; López Azcárate, C.; Domínguez Ruiz, C.; Núñez-Jurado, D.; de Torres, R.; López, R.; Camino-Moya, M.; Magni, E.; Montero-Ramirez, E.; Bocero, A.; et al. Evaluating the Clinical Impact of a Polyphenol-Rich Extract from Salicornia ramosissima on Patients with Transient Ischemic Attack and Minor Stroke. Nutrients 2024, 16, 4307. [Google Scholar] [CrossRef]
- On, J.-Y.; Kim, S.-H.; Kim, J.-M.; Park, S.; Kim, K.-H.; Lee, C.-H.; Kim, S.-K. Effects of Fermented Artemisia annua L. and Salicornia herbacea L. on Inhibition of Obesity In Vitro and In Mice. Nutrients 2023, 15, 2022. [Google Scholar] [CrossRef]
- Lim, B.; Lin, Y.; Navin, N. Advancing Cancer Research and Medicine with Single-Cell Genomics. Cancer Cell 2020, 37, 456–470. [Google Scholar] [CrossRef]
- Ko, Y.-C.; Choi, H.S.; Kim, J.-H.; Kim, S.-L.; Yun, B.-S.; Lee, D.-S. Coriolic Acid (13-(S)-Hydroxy-9Z, 11E-Octadecadienoic Acid) from Glasswort (Salicornia herbacea L.) Suppresses Breast Cancer Stem Cell through the Regulation of c-Myc. Molecules 2020, 25, 4950. [Google Scholar] [CrossRef]
- Doi, N.; Togari, H.; Minagi, K.; Nakaoji, K.; Hamada, K.; Tatsuka, M. Protective Effects of Salicornia europaea on UVB-Induced Misoriented Cell Divisions in Skin Epithelium. Cosmetics 2020, 7, 44. [Google Scholar] [CrossRef]
- Karadeniz, F.; Kim, J.A.; Ahn, B.N.; Kwon, M.S.; Kong, C.S. Effect of Salicornia herbacea on Osteoblastogenesis and Adipogenesis in Vitro. Mar. Drugs 2014, 12, 5132–5147. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, W.; Ansari, M.A.; Alsayari, A.; Almaghaslah, D.; Wahab, S.; Alomary, M.N.; Jamal, Q.M.S.; Khan, F.A.; Ali, A.; Alam, P.; et al. In Vitro, Molecular Docking and In Silico ADME/Tox Studies of Emodin and Chrysophanol against Human Colorectal and Cervical Carcinoma. Pharmaceuticals 2022, 15, 1348. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.A.; Hossain, M.S.; Hasnat, S.; Shuvo, M.H.; Akter, S.; Maria, M.A.; Tahcin, A.; Hossain, M.A.; Hoque, M.N. In-Silico Study Unveils Potential Phytocompounds in Andrographis Paniculata against E6 Protein of the High-Risk HPV-16 Subtype for Cervical Cancer Therapy. Sci. Rep. 2024, 14, 17182. [Google Scholar] [CrossRef]
- Chebbac, K.; Abchir, O.; Chalkha, M.; El Moussaoui, A.; El Barnossi, A.; Lafraxo, S.; Chtita, S.; Salamatullah, A.M.; Bourhia, M.; Dauelbait, M.; et al. Phytochemical Analysis, Antimicrobial and Antioxidant Activities of Essential Oils of the Species Artemisia Mesatlantica Maire: In Vitro and in Silico Approaches. CyTA-J. Food 2024, 22, 2388269. [Google Scholar] [CrossRef]
- Amaya-Rodriguez, C.A.; Carvajal-Zamorano, K.; Bustos, D.; Alegría-Arcos, M.; Castillo, K. A Journey from Molecule to Physiology and in Silico Tools for Drug Discovery Targeting the Transient Receptor Potential Vanilloid Type 1 (TRPV1) Channel. Front. Pharmacol. 2024, 14, 1251061. [Google Scholar] [CrossRef]
- Brogi, S.; Ramalho, T.C.; Kuca, K.; Medina-Franco, J.L.; Valko, M. Editorial: In Silico Methods for Drug Design and Discovery. Front. Chem. 2020, 8, 612. [Google Scholar] [CrossRef]
- Yun, C.-H.; Boggon, T.J.; Li, Y.; Woo, S.; Greulich, H.; Meyerson, M.; Eck, M.J. Crystal Structure of EGFR Kinase Domain in Complex with AFN941. Worldw. Protein Data Bank 2007, 3, 217–227. [Google Scholar]
- Du, Y.; Shi, W.W.; He, Y.X.; Yang, Y.H.; Zhou, C.Z.; Chen, Y. Structures of the Substrate-Binding Protein Provide Insights into the Multiple Compatible Solutes Binding Specificities of Bacillus Subtilis ABC Transporter OpuC. Worldw. Protein Data Bank 2011, 436, 283–289. [Google Scholar] [CrossRef]
- Wood, E.R.; Shewchuk, L.M.; Ellis, B.; Brignola, P.; Brashear, R.L.; Caferro, T.R.; Dickerson, S.H.; Dickson, H.D.; Donaldson, K.H.; Gaul, M.; et al. 6-Ethynylthieno[3,2-d]- and 6-Ethynylthieno[2,3-d] Pyrimidin-4-Anilines as Tunable Covalent Modifiers of ErbB Kinases. Proc. Natl. Acad. Sci. USA 2008, 105, 2773–2778. [Google Scholar] [CrossRef]
- Sathish, S.; Devaraju, P.; Julius, A.; Sohn, H.; Madhavan, T. Identification of Selective Inhibitors for Janus Kinase 1: An Integrated Drug Repurposing Strategy for Breast Cancer. Chem. Pap. 2024, 78, 245–262. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.C.; Goddard, T.D.; Pettersen, E.F.; Couch, G.S.; Pearson, Z.J.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 2023, 32, e4792. [Google Scholar] [CrossRef] [PubMed]
- BIOVIA, Dassault Systèmes, Discovery Studio, Release 2024. San. Diego: Dassault Systèmes. Available online: https://www.3ds.com/products/biovia/discovery-studio (accessed on 15 December 2024).
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Jayaram, B.; Singh, T.; Mukherjee, G.; Mathur, A.; Shekhar, S.; Shekhar, V. Sanjeevini: A Freely Accessible Web-Server for Target-Directed Lead Molecule Discovery. BMC Bioinform. 2012, 13, S7. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A Webserver for the Prediction of Toxicity of Chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef]
- Başar, Y.; Yenigün, S.; Gül, F.; Ozen, T.; Demirtas, İ.; Alma, M.H.; Temel, S. Phytochemical Profiling, Molecular Docking and ADMET Prediction of Crude Extract of Atriplex nitens schkuhr for the Screening of Antioxidant and Urease Inhibitory. Int. J. Chem. Technol. 2024, 8, 60–68. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug. Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Degfie, T.; Endale, M.; Begna, T.; Jung, C.; Dekebo, A. GC-MS Profiling and in Silico Pharmacokinetic Properties of Essential Oils Hydrodistilled from Leaves of Capparis Tomentosa and Cadaba Rotundifolia. Bull. Chem. Soc. Ethiop. 2024, 39, 351–366. [Google Scholar] [CrossRef]
- Amin, M.L. P-Glycoprotein Inhibition for Optimal Drug Delivery. Drug Target Insights 2013, 7, DTI.S12519. [Google Scholar] [CrossRef] [PubMed]
- Antonovic, L.; Martinez, M. Role of the Cytochrome P450 Enzyme System in Veterinary Pharmacokinetics: Where Are We Now? Where Are We Going? Future Med. Chem. 2011, 3, 855–879. [Google Scholar] [CrossRef]
- Lei, Z.; Tian, Q.; Teng, Q.; Wurpel, J.N.D.; Zeng, L.; Pan, Y.; Chen, Z. Understanding and Targeting Resistance Mechanisms in Cancer. MedComm 2023, 4, e265. [Google Scholar] [CrossRef] [PubMed]
- Drwal, M.N.; Banerjee, P.; Dunkel, M.; Wettig, M.R.; Preissner, R. ProTox: A Web Server for the in-Silico Prediction of Rodent Oral Toxicity. Nucleic Acids Res. 2014, 42, W53–W58. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Vayer, P.; Tanwar, S.; Poyet, J.-L.; Tsaioun, K.; Villoutreix, B.O. Drug Discovery and Development: Introduction to the General Public and Patient Groups. Front. Drug Discov. 2023, 3, 1201419. [Google Scholar] [CrossRef]
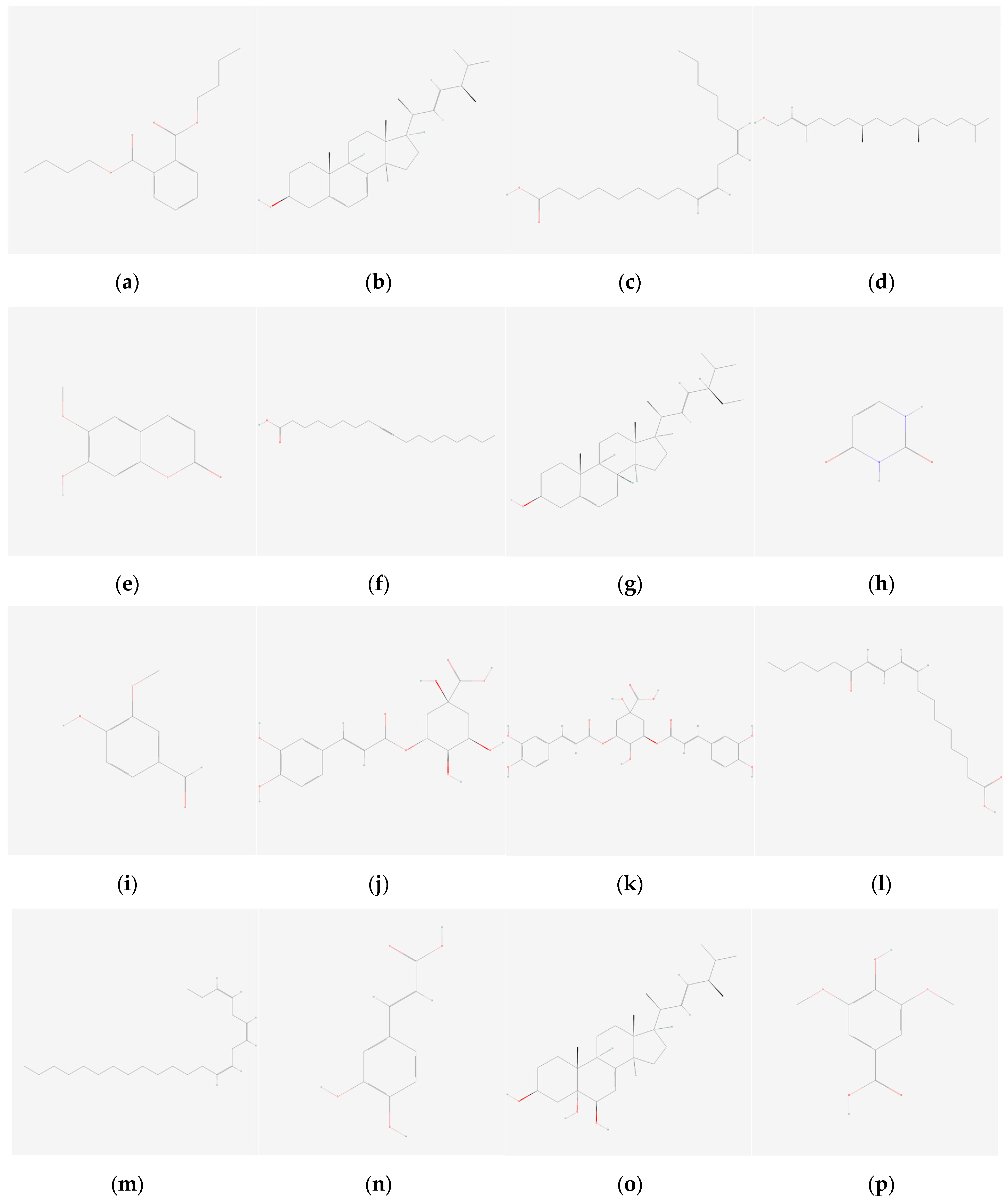
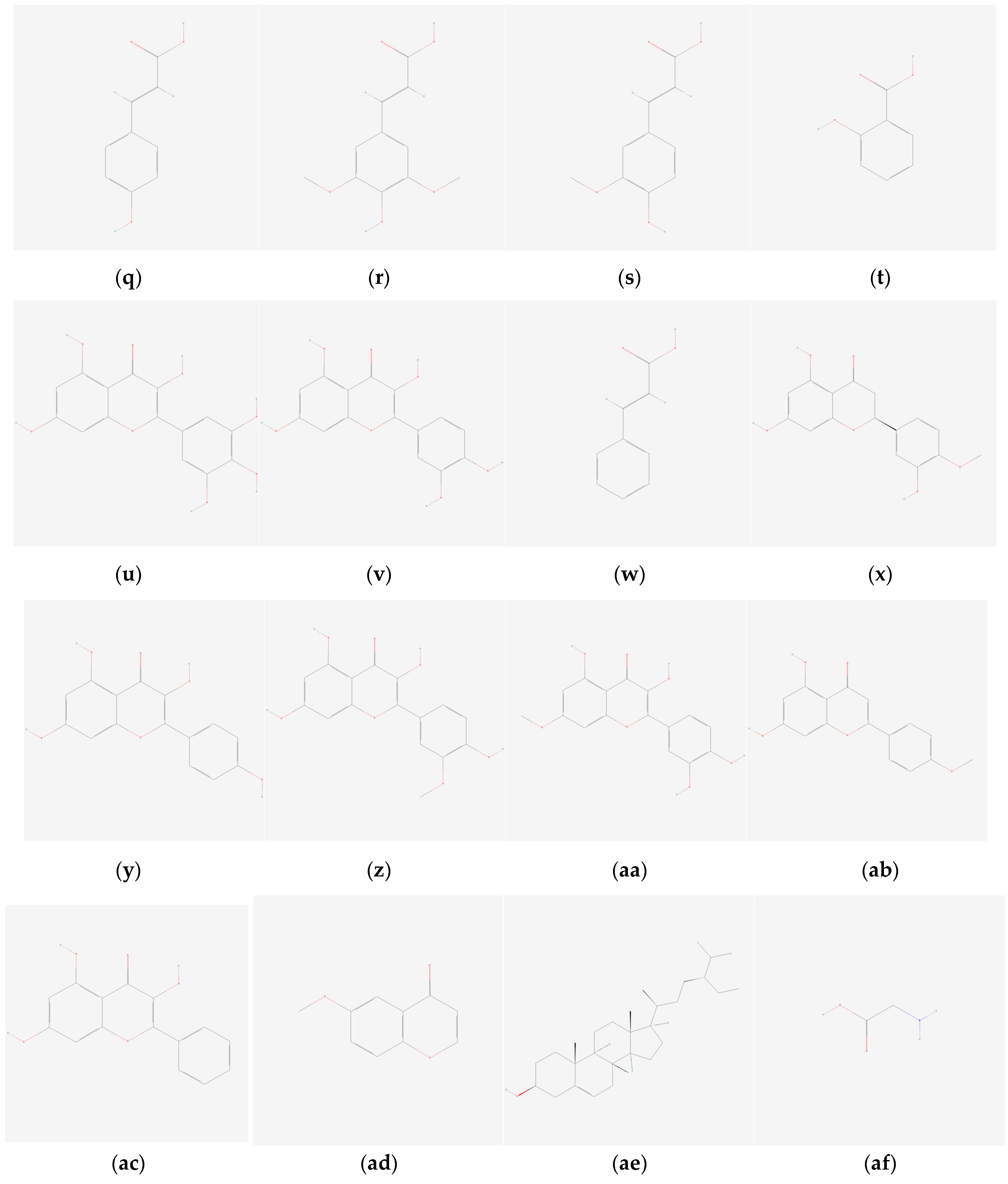
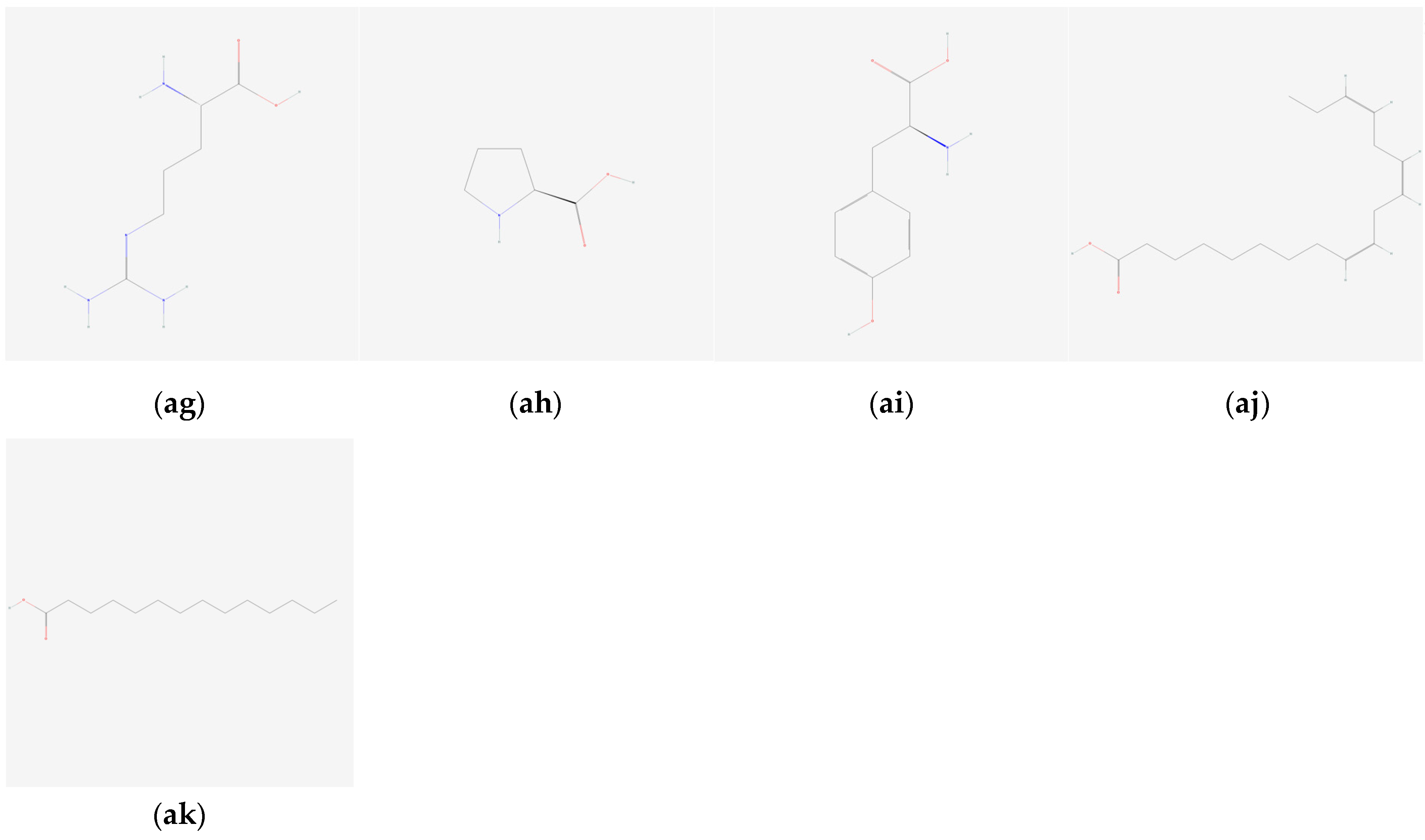

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phytochemical | Binding Energy (ΔG) (kcal/mol) | Number of H Bonds | Amino Acids Involved in Hydrogen Bonding |
|---|---|---|---|
| 3,5-di-O-caffeoylquinic acid | −8.7 | 7 | ASP770, ARG767, LEU703, SER768, ARG831, ALA767, ALA766, ARG705 |
| 3-O-caffeoylquinic acid | −7.7 | 4 | ARG831, ARG776, ILE1018, LEU778 |
| Myricetin | −7.6 | 3 | ARG831, ARG776, LEU703 |
| Quercetin | −7.5 | 3 | GLY873, ALA859, GLU758 |
| Stigmasterol | −7.5 | 0 | - |
| Gefitinib | −7.4 | 2 | LEU703, ARG831 |
| Dovitinib | −8.1 | 3 | ARG776, VAL769, ALA767 |
| Phytochemical | Binding Energy (ΔG) (kcal/mol) | Number of H Bonds | Amino Acids Involved in Hydrogen Bonding |
|---|---|---|---|
| 3,5-di-O-caffeoylquinic acid | −8.5 | 7 | ARG98, ASP769, ARG978, TYR835, ALA706, VAL839, TYR772 |
| Stigmasterol | −8.1 | 0 | - |
| 3-O-caffeoylquinic acid | −8.0 | 3 | GLU770, ARG868, GLU766 |
| Kaempferol | −7.8 | 3 | GLU770, GLY865, ASN758 |
| Gefitinib | −7.8 | 3 | LYS 957, VAL839, ASN708 |
| Dovitinib | −9.0 | 2 | GLU766, ARG868 |
| Phytochemical | Binding Energy (ΔG) (kcal/mol) | Number of H Bonds | Amino Acids Involved in Hydrogen Bonding |
|---|---|---|---|
| 3,5-di-O-caffeoylquinic acid | −8.3 | 5 | GLN988, ARG837, VAL986, GLN707, PRO705, GLN 982 |
| Stigmasterol | −7.9 | 1 | GLN988 |
| Hesperitin | −7.7 | 5 | ASN706, ILE987, TYR833, MET775, SER774 |
| Myricetin | −7.6 | 3 | ILE987, ASN706, TYR833 |
| 3-O-caffeoylquinic acid | −7.5 | 6 | ASP990, ASP991, GLN707, ARG782, ASN706, SER774 |
| Quercetin | −7.5 | 2 | ILE987, SER774 |
| Isorhamnetin | −7.4 | 6 | ASP991, ARG837, TYR833, SER774, ARG782, ASP776 |
| Acacetin | −7.3 | 2 | ARG837, ASN706 |
| Rhamnetin | −7.3 | 3 | ARG782, ASP991, GLN707 |
| Gefitinib | −7.2 | 2 | ASN706, ARG837 |
| Dovitinib | −8.5 | 2 | ASP813, GLN988 |
| Phytochemical | Mass (g/mol) | Hydrogen Bond Donor | Hydrogen Bond Acceptor | Log P | Molar Refractivity |
|---|---|---|---|---|---|
| 3,5-di-O-caffeoylqunic acid | 516.45 | 7 | 12 | 0.81 | 126.90 |
| Myricetin | 318.24 | 6 | 8 | 1.69 | 80.06 |
| Quercetin | 302.24 | 5 | 7 | 1.99 | 78.03 |
| Hesperitin | 302.28 | 3 | 6 | 2.19 | 78.06 |
| Isorhamnetin | 478.40 | 7 | 12 | −0.24 | 114.63 |
| Stigmasterol | 412.69 | 1 | 1 | 7.80 | 132.75 |
| Rhamnetin | 82.50 | 4 | 7 | 2.29 | 82.50 |
| Kaempferol | 286.24 | 4 | 6 | 1.90 | 76.01 |
| Acacetin | 284.26 | 2 | 5 | 3.35 | 78.46 |
| Gefitinib | 446.90 | 1 | 7 | 4.32 | 121.66 |
| Dovitinib | 392.43 | 3 | 4 | 2.21 | 120.28 |
| 3-O-caffeoylquinic acid | 354.31 | 6 | 9 | −0.75 | 83.50 |
| Phytochemical | ESOL (Log S) | GIA | BBB Permeant | P-gp Substrate | CYP3A4 Inhibitor | CYPIA2 Inhibitors | Bioavailability Score |
|---|---|---|---|---|---|---|---|
| 3,5-di-O-caffeoylquinic acid | −3.65 | Low | No | Yes | No | No | 0.11 |
| 3-O-caffeoylquinic acid | −1.62 | Low | No | No | No | No | 0.11 |
| Myricetin | −3.01 | Low | No | No | Yes | Yes | 0.55 |
| Quercetin | −3.16 | High | No | No | Yes | Yes | 0.55 |
| Hesperitin | −3.62 | High | No | Yes | Yes | Yes | 0.55 |
| Isorhamnetin | −3.26 | Low | No | Yes | No | No | 0.17 |
| Stigmasterol | −7.46 | Low | No | No | No | No | 0.55 |
| Rhamnetin | −3.36 | High | No | No | Yes | Yes | 0.55 |
| Gefitinib | −5.05 | High | Yes | No | Yes | No | 0.56 |
| Dovitinib | −3.66 | High | No | Yes | No | Yes | 0.55 |
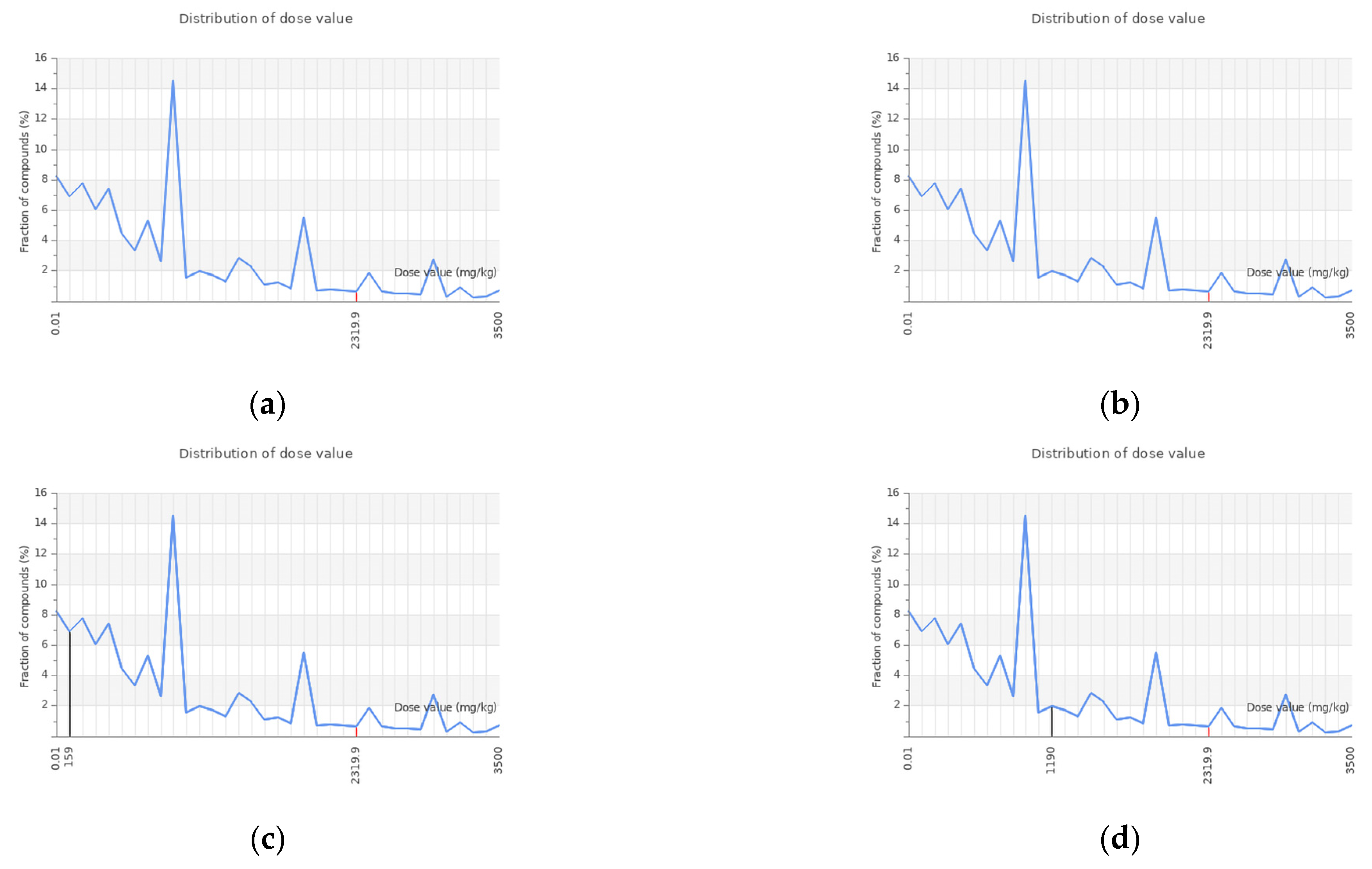
| Compounds Name | Oral LD50 Value (mg/Kg) | Predicted Toxicity Class | Prediction Accuracy (%) | Average Similarity (%) |
|---|---|---|---|---|
| 3,5-di-O-caffeoylquinic acid | 5000 | 5 | 71.63 | 69.26 |
| 3-O-caffeoylquinic acid | 5000 | 5 | 71.21 | 69.26 |
| Myricetin | 159 | 3 | 100 | 100 |
| Quercetin | 159 | 3 | 100 | 100 |
| Stigmasterol | 890 | 4 | 89.38 | 70.97 |
| Kaempferol | 3919 | 5 | 82.46 | 70.97 |
| Isorhamnetin | 5000 | 5 | 87.48 | 70.97 |
| Rhamnetin | 5000 | 5 | 86.79 | 70.97 |
| Hesperitin | 2000 | 4 | 77.77 | 69.26 |
| Acacetin | 4000 | 5 | 83.54 | 70.97 |
| Gefitinib | 2935 | 5 | 51.88 | 67.38 |
| Dovitinib | 1072 | 4 | 53.59 | 67.38 |
| Compounds Name | Hepatotoxicity | Neurotoxicity | Nephrotoxicity | Respirotoxicity | Cardiotoxicity |
|---|---|---|---|---|---|
| 3,5-di-O-caffeoylquinic acid | 0.70 I | 0.87 I | 0.52 A | 0.52 A | 0.88 I |
| 3-O-caffeoylquinic acid | 0.72 I | 0.89 I | 0.56 A | 0.57 A | 0.99 I |
| Myricetin | 0.69 I | 0.89 I | 0.62 A | 0.83 A | 0.99 I |
| Quercetin | 0.69 I | 0.89 I | 0.62 A | 0.83 A | 0.99 I |
| Stigmasterol | 0.87 I | 0.54 A | 0.89 I | 0.82 A | 0.85 I |
| Kaempferol | 0.68 I | 0.89 I | 0.62 A | 0.83 A | 0.91 I |
| Isorhamnetin | 0.72 I | 0.88 I | 0.64 A | 0.85 A | 0.82 A |
| Rhamnetin | 0.73 I | 0.86 I | 0.59 A | 0.82 A | 0.71 I |
| Hesperitin | 0.70 I | 0.87 I | 0.67 A | 0.86 A | 0.99 A |
| Acacetin | 0.72 I | 0.84 I | 0.63 A | 0.78 A | 0.64 A |
| Gefitinib | 0.73 A | 0.83 A | 0.51 I | 0.98 A | 0.81 I |
| Dovitinib | 0.54 A | 0.96 A | 0.75 I | 0.98 A | 0.87 I |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Withana, T.N.; Perera, D.; Fernando, T.D. Molecular Docking and Drug-Likeness of Salicornia-Derived Phytochemicals Against HER Receptors. Curr. Issues Mol. Biol. 2025, 47, 495. https://doi.org/10.3390/cimb47070495
Withana TN, Perera D, Fernando TD. Molecular Docking and Drug-Likeness of Salicornia-Derived Phytochemicals Against HER Receptors. Current Issues in Molecular Biology. 2025; 47(7):495. https://doi.org/10.3390/cimb47070495
Chicago/Turabian StyleWithana, Thiwanga N., Dinum Perera, and Tharani D. Fernando. 2025. "Molecular Docking and Drug-Likeness of Salicornia-Derived Phytochemicals Against HER Receptors" Current Issues in Molecular Biology 47, no. 7: 495. https://doi.org/10.3390/cimb47070495
APA StyleWithana, T. N., Perera, D., & Fernando, T. D. (2025). Molecular Docking and Drug-Likeness of Salicornia-Derived Phytochemicals Against HER Receptors. Current Issues in Molecular Biology, 47(7), 495. https://doi.org/10.3390/cimb47070495