
Reactive Oxygen Species as Key Molecules in the Pathogenesis of Alcoholic Fatty Liver Disease and Nonalcoholic Fatty Liver Disease: Future Perspectives


Abstract
1. Introduction
2. ROS Pathogenesis in ALD and NAFLD
2.1. ROS Production in NAFLD (Figure 1)
- (1)
- Metabolic dysregulation: Approximately 15% of hepatic fatty acids are derived directly from dietary intake. Chronic overnutrition leads to excessive lipid accumulation in hepatocytes, forming lipid droplets. This stored fat can subsequently be broken down into free fatty acids (FFAs), further contributing to hepatic stress and inflammation [11].
- (2)
- Insulin resistance: The accumulation of lipid droplets and FFAs from metabolic dysregulation can impair insulin signaling. This weakened insulin response reduces the suppression of adipocyte lipolysis, leading to an excessive release of FFAs into the bloodstream. These FFAs are then transported to the liver, compounding the cycle of fat accumulation and liver dysfunction [12,13].
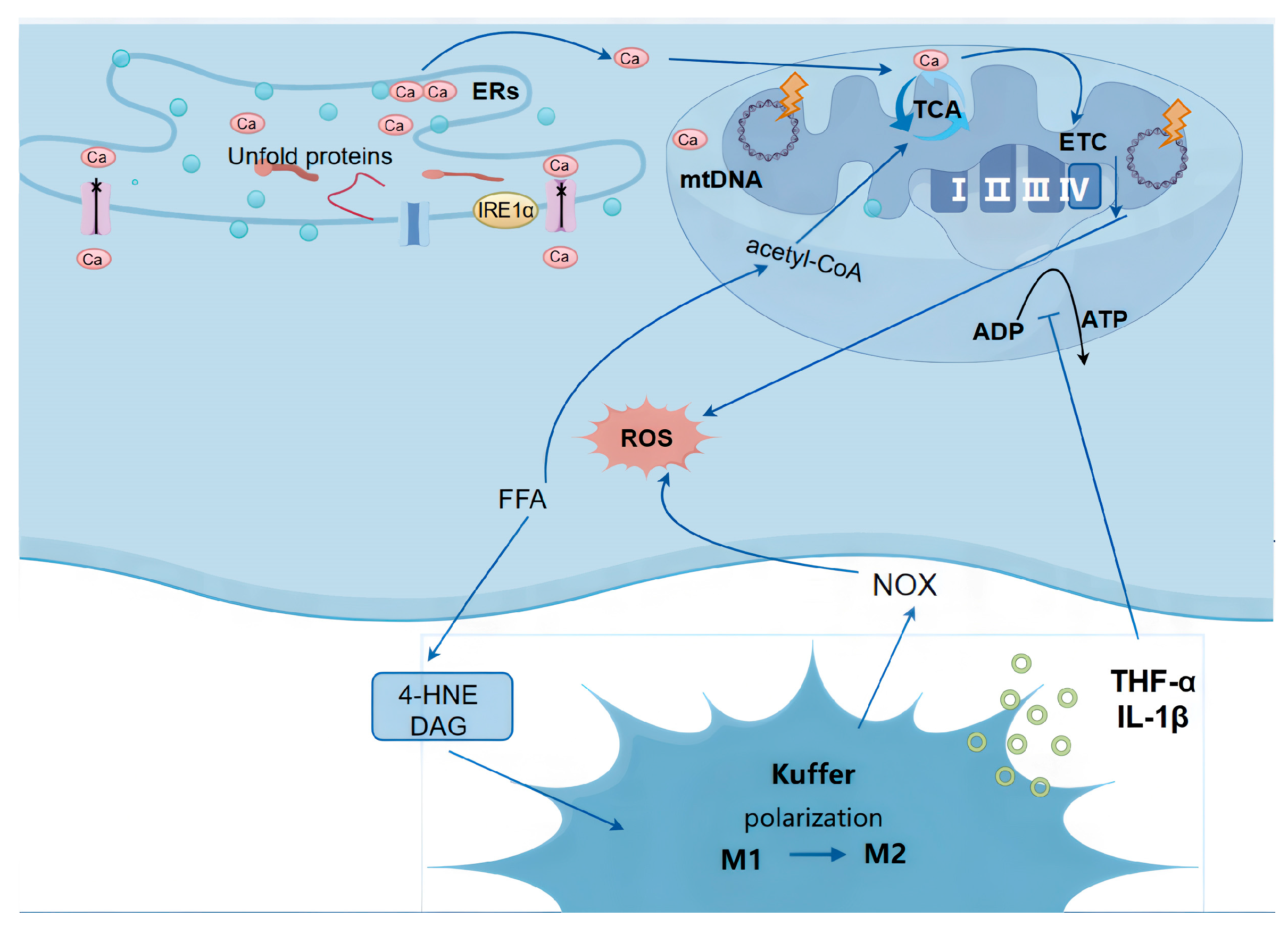
2.1.1. Mitochondrial Dysfunction
2.1.2. Endoplasmic Reticulum Stress (ER Stress)
2.1.3. Lipotoxicity
2.2. ROS Production in ALD (Figure 2)

2.2.1. Alcohol Dehydrogenase (ADH) and Aldehyde Dehydrogenase 2 (ALDH2) Pathway
2.2.2. Microsomal Ethanol Oxidizing System (MEOS)
2.2.3. Catalase Pathway
2.3. Similarities and Differences in ROS Production Between ALD and NAFLD
2.3.1. Common Sources of ROS in ALD and NAFLD
2.3.2. Key Differences in ROS Generation Between ALD and NAFLD
- (1)
- Distinct Initial Triggers of ROS Production
- (2)
- Different Dominant Pathways of ROS Generation
3. Mechanisms of ROS Action in ALD and NAFLD
3.1. Shared Mechanisms of ROS in ALD and NAFLD Pathogenesis (Figure 3)
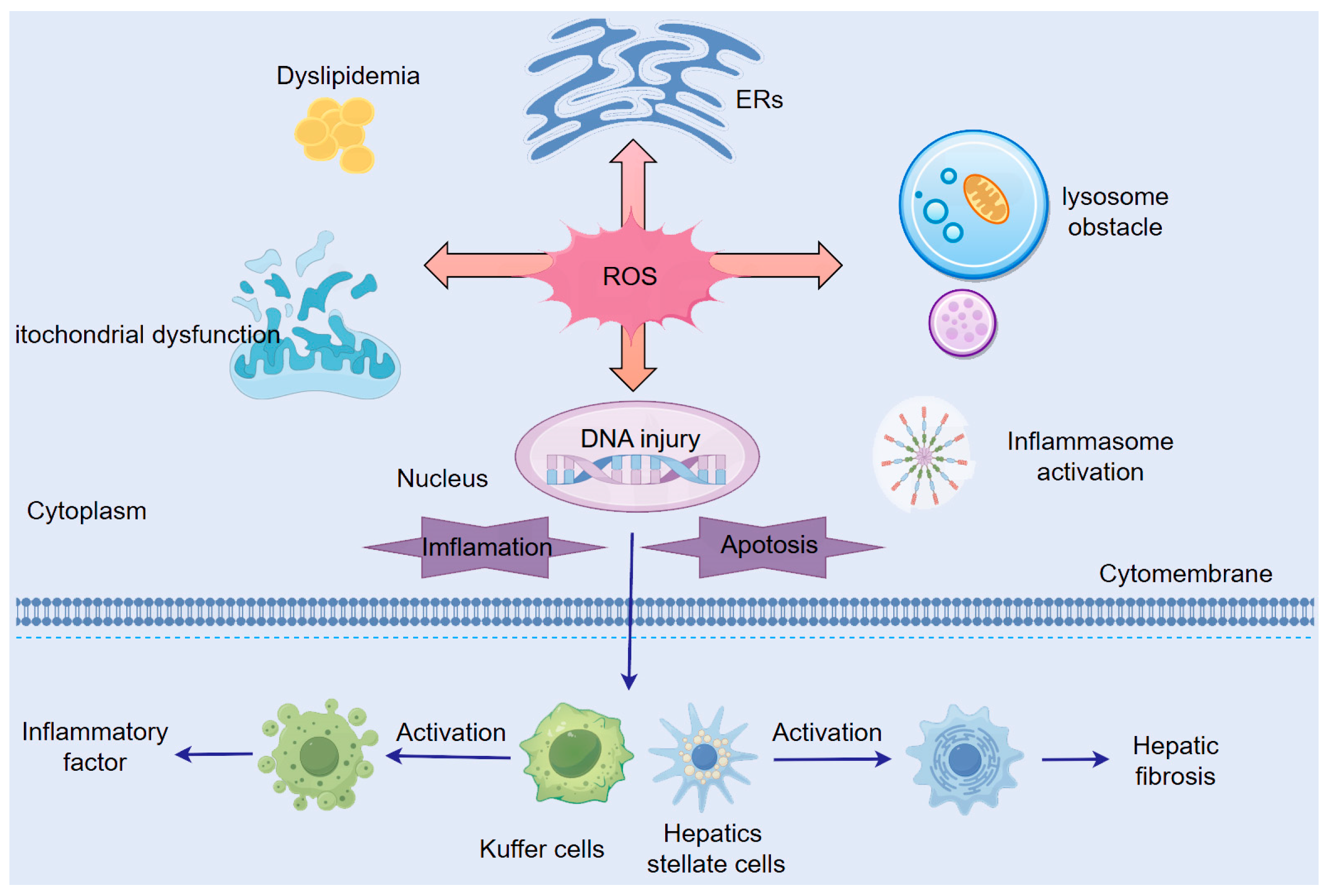
3.1.1. ROS-Induced Mitochondrial Dysfunction
3.1.2. ROS-Induced Endoplasmic Reticulum Stress (ER Stress)
- (1)
- IRE1α upregulates TNF-α and IL-6 via its downstream target, spliced X-box binding protein 1 (XBP1s) [35].
- (2)
- ER stress activates the IκB kinase (IKK) complex, inducing NF-κB nuclear translocation and subsequent TNF-α/IL-6 production [36].
- (3)
- IRE1α recruits TRAF2, activating the JNK pathway to amplify cytokine release [37].
- (1)
- Obesity and Insulin Resistance:
- (2)
- Lipid Metabolism Dysfunction:
3.1.3. ROS-Mediated Lysosomal Dysfunction
3.1.4. ROS-Mediated Activation of Inflammatory Signaling Pathways
3.1.5. ROS-Mediated Dysregulation of Lipid Metabolism
3.1.6. Additional Mechanisms of ROS Action in Hepatocytes
3.2. Differential Mechanisms of ROS in ALD Versus NAFLD
3.2.1. The Role of ROS in ALD
3.2.2. The Role of ROS in NAFLD
- (1)
- (2)
- Inhibition of fatty acid oxidation through various inflammatory signaling pathways, promoting lipid accumulation and worsening hepatic steatosis [86].
3.3. NAFLD with Concurrent ALD (MetALD)
4. Prevention and Therapeutic Approaches for ALD and NAFLD
4.1. Prevention of ALD and NAFLD
4.1.1. Abstinence from Alcohol
4.1.2. Comprehensive Lifestyle Management
- (1)
- Dietary Intervention
- (2)
- Exercise Regimen
- (3)
- Weight Control
4.1.3. Tobacco Control
4.2. Therapeutic Approaches for ALD and NAFLD
4.2.1. Antioxidant Therapy
4.2.2. Targeted Therapy
- (1)
- Mitochondria-Specific Protection: Novel mitochondria-targeted antioxidants (e.g., MitoQ and SkQ1) selectively accumulate in the mitochondrial matrix, neutralizing ROS leaked from the electron transport chain [130]. Anti-oxCIN4 improves NAFLD phenotypes in WD-fed mice through three primary mechanisms: A) enhancing mitochondrial function (fatty acid oxidation); B) stimulating the antioxidant defense system (enzymatic and non-enzymatic); and C) protecting against impaired autophagy. Collectively, these findings support the potential application of Anti-oxCIN4 in NAFLD prevention/therapy [131].
- (2)
- Redox Signaling Modulation: Selective regulation of NOX isoforms (e.g., inhibiting NOX4 while preserving NOX2-mediated immune function) maintains host defense while mitigating oxidative stress. Formononetin (FMN), a flavonoid with diverse bioactivities including antioxidant and anti-inflammatory effects, targets NOX4-based NADPH oxidase hyperactivity, enhances NADP/NADPH levels, and thereby promotes ferroptosis in activated HSCs, alleviating liver fibrosis [132].
- (3)
- Endoplasmic Reticulum-Targeted Therapy: 4-Acetylantroquinonol B (4-AAQB), a natural ubiquinone derivative extracted from Antrodia cinnamomea mycelia, significantly ameliorates ER stress and inflammation in NAFLD mouse models as well as in J774A.1 and RAW264.7 cells [133]. The lipid-lowering drug fenofibrate improves NAFLD in high-cholesterol diet-fed mice by suppressing ERN1 and XBP1 expression, reducing MAPK8 phosphorylation, and alleviating ER stress [134]. Additionally, the insulin sensitizer pioglitazone has been shown to inhibit hepatic ER stress and insulin resistance in diabetic mice [135].
- (4)
- Metal-Based Nanozyme Therapy: Nanomaterials with enzyme-like activities, termed nanozymes, feature metal-active centers as their key components. These centers effectively mimic catalytic redox processes, enabling them to emulate the activities of enzymes such as SOD and CAT [136]. The nanoparticles demonstrate robust ROS-scavenging capabilities, eliminating O₂⁻, H₂O₂, and ·OH while indirectly suppressing ONOO− formation. Animal studies have shown promising therapeutic outcomes [137].
4.2.3. Treatment of Comorbid Metabolic Disorders
4.2.4. Treatment of NAFLD with ALD Comorbidity
4.2.5. Therapeutic Distinctions Between ALD and NAFLD
5. Current Challenges and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 4-HNE | 4-hydroxynonenal |
| 4-PBA | 4-phenylbutyric acid |
| 8-OHdG | adduct 8-hydroxy-2′-deoxyguanosine |
| ADH | alcohol dehydrogenase |
| AKG | α-ketoglutarate |
| ALD | alcoholic fatty liver disease |
| ALDH2 | aldehyde dehydrogenase 2 |
| AMPK | AMP-activated protein kinase |
| ASH | alcohol-related hepatitis |
| ATF | activating transcription factor |
| DAG | diacylglycerols |
| ER | endoplasmic reticulum |
| ETC | electron transport chain |
| FFAs | free fatty acids |
| GSH | glutathione |
| GST | glutathione-S-transferase |
| HCC | hepatocellular carcinoma |
| HO-1 | heme oxygenase-1 |
| HSCs | hepatic stellate cell |
| IL-1β | interleukin |
| IRE1α | inositol-requiring enzyme 1α |
| MAPK | mitogen-activated protein kinase |
| MDA | malondialdehyde |
| MEOS | microsomal ethanol oxidizing system |
| mPTP | mitochondrial permeability transition pore |
| NAD+ | nicotinamide adenine dinucleotide |
| NAFLD | nonalcoholic fatty liver disease |
| NASH | nonalcoholic steatohepatitis |
| NLRP3 | Nod-like receptor pyrin-containing protein 3 |
| NOX | NADPH oxidase |
| Nrf2 | NF-E2 p45-related factor 2 |
| PERK | PKR-like ER kinase |
| PINK1 | PTEN-induced putative kinase 1 |
| PPARα | peroxisome proliferator-activated receptor α |
| PTX | pentoxifylline |
| ROS | reactive oxygen species |
| SAM | S-adenosylmethionine |
| SERCA | sarcoplasmic/endoplasmic reticulum calcium ATPase |
| SOD/GPx | superoxide dismutase/glutathione peroxidase |
| SREBP-1c | sterol regulatory element-binding protein 1c |
| TBE-31 | alkynyltricyclic dicyanenone |
| TCA | tricarboxylic acid cycle |
| TNF-α | tumor necrosis factor |
| TRAF2 | TNF receptor-associated factor 2 |
| TUDCA | taurine-conjugated ursodeoxycholic acid |
| UPR | unfolded protein response |
| VLDLs | very-low-density lipoproteins |
| XBP1s | X-box binding protein 1 |
References
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Prim. 2018, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Devarbhavi, H.; Asrani, S.K.; Arab, J.P.; Nartey, Y.A.; Pose, E.; Kamath, P.S. Global burden of liver disease: 2023 update. J. Hepatol. 2023, 79, 516–537. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yuan, G.; Zhong, F.; He, S. Roles of the complement system in alcohol-induced liver disease. Clin. Mol. Hepatol. 2020, 26, 677–685. [Google Scholar] [CrossRef]
- Rives, C.; Fougerat, A.; Ellero-Simatos, S.; Loiseau, N.; Guillou, H.; Gamet-Payrastre, L.; Wahli, W. Oxidative Stress in NAFLD: Role of Nutrients and Food Contaminants. Biomolecules 2020, 10, 1702. [Google Scholar] [CrossRef]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef]
- Sun, T.; Xiao, S.; Wang, M.; Xie, Q.; Zhang, L.; Gong, M.; Zhang, D.; Zhou, C. Reactive Oxygen Species Scavenging Nanozymes: Emerging Therapeutics for Acute Liver Injury Alleviation. Int. J. Nanomed. 2023, 18, 7901–7922. [Google Scholar] [CrossRef] [PubMed]
- Azzi, A. Oxidative Stress: What Is It? Can It Be Measured? Where Is It Located? Can It Be Good or Bad? Can It Be Prevented? Can It Be Cured? Antioxidants 2022, 11, 1431. [Google Scholar] [CrossRef]
- Jomova, K.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Several lines of antioxidant defense against oxidative stress: Antioxidant enzymes, nanomaterials with multiple enzyme-mimicking activities, and low-molecular-weight antioxidants. Arch. Toxicol. 2024, 98, 1323–1367. [Google Scholar] [CrossRef]
- Mittal, S.; El-Serag, H.B.; Sada, Y.H.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans is Associated With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2016, 14, 124–131.e1. [Google Scholar] [CrossRef]
- Sinha, R.A.; Singh, B.K.; Yen, P.M. Direct effects of thyroid hormones on hepatic lipid metabolism. Nat. Rev. Endocrinol. 2018, 14, 259–269. [Google Scholar] [CrossRef]
- Sakurai, Y.; Kubota, N.; Yamauchi, T.; Kadowaki, T. Role of Insulin Resistance in MAFLD. Int. J. Mol. Sci. 2021, 22, 4156. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Romero-Gomez, M.; SZelber-Sagi Trenell, M. Treatment of NAFLD with diet, physical activity and exercise. J. Hepatol. 2017, 67, 829–846. [Google Scholar] [CrossRef] [PubMed]
- DeBose-Boyd, R.A.; Ye, J. SREBPs in Lipid Metabolism, Insulin Signaling, and Beyond. Trends Biochem. Sci. 2018, 43, 358–368. [Google Scholar] [CrossRef]
- Mansouri, A.; Gattolliat, C.H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef]
- Lee, K.C.; Wu, P.S.; Lin, H.C. Pathogenesis and treatment of non-alcoholic steatohepatitis and its fibrosis. Clin. Mol. Hepatol. 2023, 29, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Qiu, T.; Yao, X.; Jiang, L.; Wang, N.; Jiang, J.; Jia, X.; Wei, S.; Zhang, J.; Zhu, Y.; et al. IRE1alpha/NOX4 signaling pathway mediates ROS-dependent activation of hepatic stellate cells in NaAsO(2)-induced liver fibrosis. J. Cell. Physiol. 2021, 236, 1469–1480. [Google Scholar] [CrossRef]
- Celik, C.; Lee, S.Y.T.; Yap, W.S.; Thibault, G. Endoplasmic reticulum stress and lipids in health and diseases. Prog. Lipid Res. 2023, 89, 101198. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S.; et al. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef] [PubMed]
- Kopp, M.C.; Larburu, N.; Durairaj, V.; Adams, C.J.; Ali, M.M.U. UPR proteins IRE1 and PERK switch BiP from chaperone to ER stress sensor. Nat. Struct. Mol. Biol. 2019, 26, 1053–1062. [Google Scholar] [CrossRef]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef] [PubMed]
- Rosso, C.; Kazankov, K.; Younes, R.; Esmaili, S.; Marietti, M.; Sacco, M.; Carli, F.; Gaggini, M.; Salomone, F.; Møller, H.J.; et al. Crosstalk between adipose tissue insulin resistance and liver macrophages in non-alcoholic fatty liver disease. J. Hepatol. 2019, 71, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Barreby, E.; Chen, P.; Aouadi, M. Macrophage functional diversity in NAFLD—more than inflammation. Nat. Rev. Endocrinol. 2022, 18, 461–472. [Google Scholar] [CrossRef]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Publisher Correction: Alcoholic liver disease. Nat. Rev. Dis. Prim. 2018, 4, 18. [Google Scholar] [CrossRef]
- Matsushita, H.; Takaki, A. Alcohol and hepatocellular carcinoma. BMJ Open Gastroenterol. 2019, 6, e000260. [Google Scholar] [CrossRef] [PubMed]
- Hyun, J.; Han, J.; Lee, C.; Yoon, M.; Jung, Y. Pathophysiological Aspects of Alcohol Metabolism in the Liver. Int. J. Mol. Sci. 2021, 22, 5717. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, C.; Chen, J.; Sang, T.; Peng, H.; Lin, X.; Zhao, Q.; Chen, S.; Eling, T.; Wang, X. Overexpression of NAG-1/GDF15 prevents hepatic steatosis through inhibiting oxidative stress-mediated dsDNA release and AIM2 inflammasome activation. Redox Biol. 2022, 52, 102322. [Google Scholar] [CrossRef]
- Sun, X.; Seidman, J.S.; Zhao, P.; Troutman, T.D.; Spann, N.J.; Que, X.; Zhou, F.; Liao, Z.; Pasillas, M.; Yang, X.; et al. Neutralization of Oxidized Phospholipids Ameliorates Non-alcoholic Steatohepatitis. Cell Metab. 2020, 31, 189–206.e8. [Google Scholar] [CrossRef]
- Liu, Y.-N.; Zhu, H.-X.; Li, T.-Y.; Yang, X.; Li, X.-J.; Zhang, W.K. Lipid nanoparticle encapsulated oleic acid induced lipotoxicity to hepatocytes via ROS overload and the DDIT3/BCL2/BAX/Caspases signaling in vitro and in vivo. Free. Radic. Biol. Med. 2024, 222, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Guo, N.; Xu, H.; Li, Y.; Sun, T.; Jiang, X.; Fu, D.; You, T.; Diao, S.; Huang, Y.; et al. Caveolin-1 ameliorates hepatic injury in non-alcoholic fatty liver disease by inhibiting ferroptosis via the NOX4/ROS/GPX4 pathway. Biochem. Pharmacol. 2024, 230, 116594. [Google Scholar] [CrossRef] [PubMed]
- Guan, B.-J.; Krokowski, D.; Majumder, M.; Schmotzer, C.L.; Kimball, S.R.; Merrick, W.C.; Koromilas, A.E.; Hatzoglou, M. Translational Control during Endoplasmic Reticulum Stress beyond Phosphorylation of the Translation Initiation Factor eIF2α. J. Biol. Chem. 2014, 289, 12593–12611. [Google Scholar] [CrossRef]
- Wang, L.; Wang, C.-C. Oxidative protein folding fidelity and redoxtasis in the endoplasmic reticulum. Trends Biochem. Sci. 2022, 48, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.S.; Harrison, D.J.; Kisielewski, D.; Cassidy, D.M.; McNeilly, A.D.; Gallagher, J.R.; Walsh, S.V.; Honda, T.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; et al. Experimental Nonalcoholic Steatohepatitis and Liver Fibrosis Are Ameliorated by Pharmacologic Activation of Nrf2 (NF-E2 p45-Related Factor 2). Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 367–398. [Google Scholar] [CrossRef]
- Lee, K.P.; Dey, M.; Neculai, D.; Cao, C.; Dever, T.E.; Sicheri, F. Structure of the Dual Enzyme Ire1 Reveals the Basis for Catalysis and Regulation in Nonconventional RNA Splicing. Cell 2008, 132, 89–100. [Google Scholar] [CrossRef]
- Li, Y.; Chen, M.; Xu, Y.; Yu, X.; Xiong, T.; Du, M.; Sun, J.; Liu, L.; Tang, Y.; Yao, P.; et al. Iron-Mediated Lysosomal Membrane Permeabilization in Ethanol-Induced Hepatic Oxidative Damage and Apoptosis: Protective Effects of Quercetin. Oxidative Med. Cell. Longev. 2015, 2016, 4147610. [Google Scholar] [CrossRef]
- Tam, A.B.; Mercado, E.L.; Hoffmann, A.; Niwa, M. ER stress activates NF-κB by integrating functions of basal IKK activity, IRE1 and PERK. PLoS ONE 2012, 7, e45078. [Google Scholar] [CrossRef]
- Sabio, G.; Das, M.; Mora, A.; Zhang, Z.; Jun, J.Y.; Ko, H.J.; Barrett, T.; Kim, J.K.; Davis, R.J. A Stress Signaling Pathway in Adipose Tissue Regulates Hepatic Insulin Resistance. Science 2008, 322, 1539–1543. [Google Scholar] [CrossRef]
- Das, M.; Sabio, G.; Jiang, F.; Rincón, M.; Flavell, R.A.; Davis, R.J. Induction of Hepatitis by JNK-Mediated Expression of TNF-α. Cell 2009, 136, 249–260. [Google Scholar] [CrossRef]
- Sabio, G.; Kennedy, N.J.; Cavanagh-Kyros, J.; Jung, D.Y.; Ko, H.J.; Ong, H.; Barrett, T.; Kim, J.K.; Davis, R.J. Role of Muscle c-Jun NH2-Terminal Kinase 1 in Obesity-Induced Insulin Resistance. Mol. Cell. Biol. 2010, 30, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Sabio, G.; Cavanagh-Kyros, J.; Ko, H.J.; Jung, D.Y.; Gray, S.; Jun, J.Y.; Barrett, T.; Mora, A.; Kim, J.K.; Davis, R.J. Prevention of Steatosis by Hepatic JNK1. Cell Metab. 2009, 10, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Nikain, C.; Fortounas, K.I.; Amengual, J.; Tufanli, O.; La Forest, M.; Yu, Y.; Wang, M.C.; Watts, R.; Lehner, R.; et al. FITM2 deficiency results in ER lipid accumulation, ER stress, and reduced apolipoprotein B lipidation and VLDL triglyceride secretion in vitro and in mouse liver. Mol. Metab. 2024, 90, 102048. [Google Scholar] [CrossRef] [PubMed]
- Carotti, S.; Aquilano, K.; Zalfa, F.; Ruggiero, S.; Valentini, F.; Zingariello, M.; Francesconi, M.; Perrone, G.; Alletto, F.; Antonelli-Incalzi, R.; et al. Lipophagy Impairment Is Associated With Disease Progression in NAFLD. Front. Physiol. 2020, 11, 850. [Google Scholar] [CrossRef]
- Koga, H.; Kaushik, S.; Cuervo, A.M. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 2010, 24, 3052–3065. [Google Scholar] [CrossRef]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER–mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef]
- Shen, Q.; Shi, Y.; Liu, J.; Su, H.; Huang, J.; Zhang, Y.; Peng, C.; Zhou, T.; Sun, Q.; Wan, W.; et al. Acetylation of STX17 (syntaxin 17) controls autophagosome maturation. Autophagy 2020, 17, 1157–1169. [Google Scholar] [CrossRef]
- Tilg, H.; ARMoschen Szabo, G. Interleukin-1 and inflammasomes in alcoholic liver disease/acute alcoholic hepatitis and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2016, 64, 955–965. [Google Scholar] [CrossRef]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef]
- Ahmad, A.; Ahmad, R. Resveratrol mitigate structural changes and hepatic stellate cell activation in N′-nitrosodimethylamine-induced liver fibrosis via restraining oxidative damage. Chem. Interact. 2014, 221, 1–12. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Liu, Y.; Su, Y.; Ji, P.; Kong, L.; Sun, R.; Zhang, D.; Xu, H.; Li, W.; Li, W. Ginsenoside Rg1 attenuates lipopolysaccharide-induced chronic liver damage by activating Nrf2 signaling and inhibiting inflammasomes in hepatic cells. J. Ethnopharmacol. 2024, 324, 117794. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Ge, Y.; Liu, X.; Deng, S.; Li, J.; Tan, P.; Yang, Y.; Wu, Z. Exposure to the environmental pollutant chlorpyrifos induces hepatic toxicity through activation of the JAK/STAT and MAPK pathways. Sci. Total. Environ. 2024, 928, 171711. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Li, F.; Lu, S.; Ren, L.; Bian, S.; Liu, M.; Zhao, D.; Wang, S.; Wang, J. Ginseng root extract attenuates inflammation by inhibiting the MAPK/NF-κB signaling pathway and activating autophagy and p62-Nrf2-Keap1 signaling in vitro and in vivo. J. Ethnopharmacol. 2022, 283, 114739. [Google Scholar] [CrossRef]
- Iracheta-Vellve, A.; Petrasek, J.; Gyogyosi, B.; Bala, S.; Csak, T.; Kodys, K.; Szabo, G. Interleukin-1 inhibition facilitates recovery from liver injury and promotes regeneration of hepatocytes in alcoholic hepatitis in mice. Liver Int. 2017, 37, 968–973. [Google Scholar] [CrossRef]
- Dong, J.; Li, M.; Peng, R.; Zhang, Y.; Qiao, Z.; Sun, N. ACACA reduces lipid accumulation through dual regulation of lipid metabolism and mitochondrial function via AMPK- PPARα- CPT1A axis. J. Transl. Med. 2024, 22, 1–14. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, W.; Zhai, T.; You, J.; Chen, Y. Silibinin ameliorates hepatic lipid accumulation and oxidative stress in mice with non-alcoholic steatohepatitis by regulating CFLAR-JNK pathway. Acta Pharm. Sin. B 2019, 9, 745–757. [Google Scholar] [CrossRef]
- Ding, X.; Zhu, X.-L.; Xu, D.-H.; Li, S.; Yang, Q.; Feng, X.; Wei, Y.-G.; Li, H.; Yang, L.; Zhang, Y.-J.; et al. NPM promotes hepatotoxin-induced fibrosis by inhibiting ROS-induced apoptosis of hepatic stellate cells and upregulating lncMIAT-induced TGF-β2. Cell Death Dis. 2023, 14, 1–16. [Google Scholar] [CrossRef]
- Seki, S.; Kitada, T.; Sakaguchi, H.; Nakatani, K.; Wakasa, K. Pathological significance of oxidative cellular damage in human alcoholic liver disease. Histopathology 2003, 42, 365–371. [Google Scholar] [CrossRef]
- Kitada, T.; Seki, S.; Iwai, S.; Yamada, T.; Sakaguchi, H.; Wakasa, K. In situ detection of oxidative DNA damage, 8-hydroxydeoxyguanosine, in chronic human liver disease. J. Hepatol. 2001, 35, 613–618. [Google Scholar] [CrossRef]
- Gao, H.; Jiang, Y.; Zeng, G.; Huda, N.; Thoudam, T.; Yang, Z.; Liangpunsakul, S.; Ma, J. Cell-to-cell and organ-to-organ crosstalk in the pathogenesis of alcohol-associated liver disease. eGastroenterology 2024, 2, e100104. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS biology 2018, 16, e2006203. [Google Scholar] [CrossRef]
- Catalá, A.; Díaz, M. Editorial: Impact of Lipid Peroxidation on the Physiology and Pathophysiology of Cell Membranes. Front. Physiol. 2016, 7, 423. [Google Scholar] [CrossRef]
- Bartesaghi, S.; Radi, R. Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration. Redox Biol. 2018, 14, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef]
- Penaloza, C.G.; Cruz, M.; Germain, G.; Jabeen, S.; Javdan, M.; Lockshin, R.A.; Zakeri, Z. Higher sensitivity of female cells to ethanol: Methylation of DNA lowers Cyp2e1, generating more ROS. Cell Commun. Signal. 2020, 18, 1–15. [Google Scholar] [CrossRef]
- Zeng, T.; Zhang, C.-L.; Song, F.-Y.; Zhao, X.-L.; Yu, L.-H.; Zhu, Z.-P.; Xie, K.-Q. PI3K/Akt pathway activation was involved in acute ethanol-induced fatty liver in mice. Toxicology 2012, 296, 56–66. [Google Scholar] [CrossRef]
- Krycer, J.R.; Sharpe, L.J.; Luu, W.; Brown, A.J. The Akt–SREBP nexus: Cell signaling meets lipid metabolism. Trends Endocrinol. Metab. 2010, 21, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Sarsour, E.H.; Kumar, M.G.; Chaudhuri, L.; Kalen, A.L.; Goswami, P.C. Redox Control of the Cell Cycle in Health and Disease. Antioxidants Redox Signal. 2009, 11, 2985–3011. [Google Scholar] [CrossRef]
- Goldar, S.; Khaniani, M.S.; Derakhshan, S.M.; Baradaran, B. Molecular Mechanisms of Apoptosis and Roles in Cancer Development and Treatment. Asian Pac. J. Cancer Prev. 2015, 16, 2129–2144. [Google Scholar] [CrossRef]
- Taniai, M. Alcohol and hepatocarcinogenesis. Clin. Mol. Hepatol. 2020, 26, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.J.; Emdin, C.; Bick, A.G.; Zekavat, S.M.; Niroula, A.; Pirruccello, J.P.; Dichtel, L.; Griffin, G.; Uddin, M.; Gibson, C.J.; et al. Clonal haematopoiesis and risk of chronic liver disease. Nature 2023, 616, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.Q.; Terrault, N.A.; Tacke, F.; Gluud, L.L.; Arrese, M.; Bugianesi, E.; Loomba, R. Global epidemiology of cirrhosis - aetiology, trends and predictions. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 388–398. [Google Scholar] [CrossRef]
- Lin, L.; Yang, S.; Xiao, Z.; Hong, P.; Sun, S.; Zhou, C.; Qian, Z.J. The Inhibition Effect of the Seaweed Polyphenol, 7-Phloro-Eckol from Ecklonia Cava on Alcohol-Induced Oxidative Stress in HepG2/CYP2E1 Cells. Mar. Drugs 2021, 19, 158. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, X.H.; Cederbaum, A.I. Ethanol Induction of CYP2A5: Role of CYP2E1-ROS-Nrf2 Pathway. Toxicol. Sci. 2012, 128, 427–438. [Google Scholar] [CrossRef]
- Gonzalez, A.; Huerta-Salgado, C.; Orozco-Aguilar, J.; Aguirre, F.; Tacchi, F.; Simon, F.; Cabello-Verrugio, C.; Gasparovic, A.C. Role of Oxidative Stress in Hepatic and Extrahepatic Dysfunctions during Nonalcoholic Fatty Liver Disease (NAFLD). Oxidative Med. Cell. Longev. 2020, 2020, 1–16. [Google Scholar] [CrossRef]
- Seki, S.; Kitada, T.; Yamada, T.; Sakaguchi, H.; Nakatani, K.; Wakasa, K. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J. Hepatol. 2002, 37, 56–62. [Google Scholar] [CrossRef]
- Pirola, C.J.; Garaycoechea, M.; Flichman, D.; Castaño, G.O.; Sookoian, S. Liver mitochondrial DNA damage and genetic variability of Cytochrome b – a key component of the respirasome – drive the severity of fatty liver disease. J. Intern. Med. 2020, 289, 84–96. [Google Scholar] [CrossRef]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef]
- Wang, N.; Lu, X.; Wang, J.; Wang, H.; Zhang, B.; Zhao, W.; Zhang, J. Quasi-LD-Targeted and ONOO–-Responsive Fluorescent Probe for Investigating the Interaction of Nonalcoholic Fatty Liver with Drug-Induced Liver Injury. Anal. Chem. 2023, 95, 5967–5975. [Google Scholar] [CrossRef]
- Dias, K.A.; Oliveira, L.A.; Pereira, S.M.S.; Abrantes, L.C.S.; Vicente, L.C.O.d.S.; Gonçalves, R.V.; Della Lucia, C.M. Anti-inflammatory and antioxidant effects of anthocyanins in Nonalcoholic fatty liver disease (NAFLD): A systematic review of in vivo studies. Crit. Rev. Food Sci. Nutr. 2025, 1–18. [Google Scholar] [CrossRef]
- Greatorex, S.; Kaur, S.; Xirouchaki, C.E.; Goh, P.K.; Wiede, F.; Genders, A.J.; Tran, M.; Jia, Y.; Raajendiran, A.; Brown, W.A.; et al. Mitochondria- and NOX4-dependent antioxidant defense mitigates progression to nonalcoholic steatohepatitis in obesity. J. Clin. Investig. 2023, 134. [Google Scholar] [CrossRef] [PubMed]
- Spahis, S.; Delvin, E.; Borys, J.-M.; Levy, E. Oxidative Stress as a Critical Factor in Nonalcoholic Fatty Liver Disease Pathogenesis. Antioxidants Redox Signal. 2017, 26, 519–541. [Google Scholar] [CrossRef]
- Zhang, T.; Liu, J.; Shen, S.; Tong, Q.; Ma, X.; Lin, L. SIRT3 promotes lipophagy and chaperon-mediated autophagy to protect hepatocytes against lipotoxicity. Cell Death Differ. 2019, 27, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Bartesaghi, S.; Wallenius, K.; Hovdal, D.; Liljeblad, M.; Wallin, S.; Dekker, N.; Barlind, L.; Davies, N.; Seeliger, F.; Winzell, M.S.; et al. Subcutaneous delivery of FGF21 mRNA therapy reverses obesity, insulin resistance, and hepatic steatosis in diet-induced obese mice. Mol. Ther.-Nucleic Acids 2022, 28, 500–513. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.I.; Shankaran, M.; Yoshino, M.; Schweitzer, G.G.; Chondronikola, M.; Beals, J.W.; Okunade, A.L.; Patterson, B.W.; Nyangau, E.; Field, T.; et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J. Clin. Investig. 2020, 130, 1453–1460. [Google Scholar] [CrossRef]
- Fromenty, B.; Roden, M. Mitochondrial alterations in fatty liver diseases. J. Hepatol. 2022, 78, 415–429. [Google Scholar] [CrossRef]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef]
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef]
- Kalligeros, M.; Vassilopoulos, A.; Vassilopoulos, S.; Victor, D.W.; Mylonakis, E.; Noureddin, M. Prevalence of Steatotic Liver Disease (MASLD, MetALD, and ALD) in the United States: NHANES 2017-2020. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2024, 22, 1330–1332.e1334. [Google Scholar] [CrossRef]
- Marek, G.W.; Malhi, H. MetALD: Does it require a different therapeutic option? Hepatology 2024, 80, 1424–1440. [Google Scholar] [CrossRef]
- Thoma, C.; Day, C.P.; Trenell, M.I. Lifestyle interventions for the treatment of non-alcoholic fatty liver disease in adults: A systematic review. J. Hepatol. 2012, 56, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Louvet, A.; Labreuche, J.; Artru, F.; Bouthors, A.; Rolland, B.; Saffers, P.; Lollivier, J.; Lemaître, E.; Dharancy, S.; Lassailly, G.; et al. Main drivers of outcome differ between short term and long term in severe alcoholic hepatitis: A prospective study. Hepatology 2017, 66, 1464–1473. [Google Scholar] [CrossRef]
- Shojaee-Moradie, F.; Cuthbertson, D.J.; Barrett, M.; Jackson, N.C.; Herring, R.; Thomas, E.L.; Bell, J.; Kemp, G.J.; Wright, J.; Umpleby, A.M. Exercise Training Reduces Liver Fat and Increases Rates of VLDL Clearance But Not VLDL Production in NAFLD. J. Clin. Endocrinol. Metab. 2016, 101, 4219–4228. [Google Scholar] [CrossRef]
- Hofer, B.S.; Simbrunner, B.; Hartl, L.; Jachs, M.; Bauer, D.J.M.; Balcar, L.; Paternostro, R.; Schwabl, P.; Semmler, G.; Scheiner, B.; et al. Alcohol Abstinence Improves Prognosis Across All Stages of Portal Hypertension in Alcohol-Related Cirrhosis. Clin. Gastroenterol. Hepatol. 2022, 21, 2308–2317.e7. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Parola, M.; Alisi, A.; Marra, F.; Piemonte, F.; Mombello, C.; Sutti, S.; Povero, D.; Maina, V.; Novo, E.; et al. Oxidative stress parameters in paediatric non-alcoholic fatty liver disease. Int. J. Mol. Med. 2010, 26, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Fernández-Galilea, M.; Martínez-Fernández, L.; González-Muniesa, P.; Pérez-Chávez, A.; Martínez, J.A.; Moreno-Aliaga, M.J. Oxidative Stress and Non-Alcoholic Fatty Liver Disease: Effects of Omega-3 Fatty Acid Supplementation. Nutrients 2019, 11, 872. [Google Scholar] [CrossRef]
- Yki-Järvinen, H.; Luukkonen, P.K.; Hodson, L.; Moore, J.B. Dietary carbohydrates and fats in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 770–786. [Google Scholar] [CrossRef]
- Li, S.; Tan, H.-Y.; Wang, N.; Zhang, Z.-J.; Lao, L.; Wong, C.-W.; Feng, Y. The Role of Oxidative Stress and Antioxidants in Liver Diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015, 149, 367–378.e5. [Google Scholar] [CrossRef]
- Promrat, K.; Kleiner, D.E.; Niemeier, H.M.; Jackvony, E.; Kearns, M.; Wands, J.R.; Fava, J.L.; Wing, R.R. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology 2010, 51, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Ayares, G.; Idalsoaga, F.; Díaz, L.A.; Arnold, J.; Arab, J.P. Current Medical Treatment for Alcohol-Associated Liver Disease. J. Clin. Exp. Hepatol. 2022, 12, 1333–1348. [Google Scholar] [CrossRef]
- Marti-Aguado, D.; Clemente-Sanchez, A.; Bataller, R. Cigarette smoking and liver diseases. J. Hepatol. 2022, 77, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-M.; Zhang, J.-R.; Li, M.-X.; Hou, H.; Wang, H.; Huang, Y. Cigarette smoking and alcohol-related liver disease. Liver Res. 2024, 8, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Violi, F.; Cangemi, R. ; Cangemi, R. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 363, 1185–1186; author reply 1186. [Google Scholar]
- Chin, K.-Y.; Ekeuku, S.O.; Chew, D.C.H.; Trias, A. Tocotrienol in the Management of Nonalcoholic Fatty Liver Disease: A Systematic Review. Nutrients 2023, 15, 834. [Google Scholar] [CrossRef]
- Tang, G.; Xu, Y.; Zhang, C.; Wang, N.; Li, H.; Feng, Y. Green Tea and Epigallocatechin Gallate (EGCG) for the Management of Nonalcoholic Fatty Liver Diseases (NAFLD): Insights into the Role of Oxidative Stress and Antioxidant Mechanism. Antioxidants 2021, 10, 1076. [Google Scholar] [CrossRef] [PubMed]
- Sakata, R.; Nakamura, T.; Torimura, T.; Ueno, T.; Sata, M. Green tea with high-density catechins improves liver function and fat infiltration in non-alcoholic fatty liver disease (NAFLD) patients: A double-blind placebo-controlled study. Int. J. Mol. Med. 2013, 32, 989–994. [Google Scholar] [CrossRef]
- Dey, P.; Kim, J.B.; Chitchumroonchokchai, C.; Li, J.; Sasaki, G.Y.; Olmstead, B.D.; Stock, K.L.; Thomas-Ahner, J.M.; Clinton, S.K.; Bruno, R.S. Green tea extract inhibits early oncogenic responses in mice with nonalcoholic steatohepatitis. Food Funct. 2019, 10, 6351–6361. [Google Scholar] [CrossRef]
- Zein, C.O.; Lopez, R.; Fu, X.; Kirwan, J.P.; Yerian, L.M.; McCullough, A.J.; Hazen, S.L.; Feldstein, A.E. Pentoxifylline decreases oxidized lipid products in nonalcoholic steatohepatitis: New evidence on the potential therapeutic mechanism. Hepatology 2012, 56, 1291–1299. [Google Scholar] [CrossRef]
- Zein, C.O.; Yerian, L.M.; Gogate, P.; Lopez, R.; Kirwan, J.P.; Feldstein, A.E.; McCullough, A.J. Pentoxifylline improves nonalcoholic steatohepatitis: A randomized placebo-controlled trial. Hepatology 2011, 54, 1610–1619. [Google Scholar] [CrossRef] [PubMed]
- Mora, S.I.; García-Román, J.; Gómez-Ñañez, I.; García-Román, R. Chronic liver diseases and the potential use of S-adenosyl-l-methionine as a hepatoprotector. Eur. J. Gastroenterol. Hepatol. 2018, 30, 893–900. [Google Scholar] [CrossRef]
- Lieber, C.S. S-Adenosyl-l-methionine and alcoholic liver disease in animal models: Implications for early intervention in human beings. Alcohol 2002, 27, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Karaa, A.; Thompson, K.J.; McKillop, I.H.; Clemens, M.G.; Schrum, L.W. S-ADENOSYL-L-METHIONINE ATTENUATES OXIDATIVE STRESS AND HEPATIC STELLATE CELL ACTIVATION IN AN ETHANOL-LPS-INDUCED FIBROTIC RAT MODEL. Shock 2008, 30, 197–205. [Google Scholar] [CrossRef]
- Kharbanda, K.K. Alcoholic Liver Disease and Methionine Metabolism. Semin. Liver Dis. 2009, 29, 155–165. [Google Scholar] [CrossRef]
- Purohit, V.; Abdelmalek, M.F.; Barve, S.; Benevenga, N.J.; Halsted, C.H.; Kaplowitz, N.; Kharbanda, K.K.; Liu, Q.-Y.; Lu, S.C.; McClain, C.J.; et al. Role of S-adenosylmethionine, folate, and betaine in the treatment of alcoholic liver disease: Summary of a symposium. Am. J. Clin. Nutr. 2007, 86, 14–24. [Google Scholar] [CrossRef]
- Kharbanda, K.K.; Mailliard, M.E.; Baldwin, C.R.; Beckenhauer, H.C.; Sorrell, M.F.; Tuma, D.J. Betaine attenuates alcoholic steatosis by restoring phosphatidylcholine generation via the phosphatidylethanolamine methyltransferase pathway. J. Hepatol. 2007, 46, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Jung, Y.S.; Kwon, D.Y.; Kim, Y.C. Alleviation of acute ethanol-induced liver injury and impaired metabolomics of S-containing substances by betaine supplementation. Biochem. Biophys. Res. Commun. 2008, 368, 893–898. [Google Scholar] [CrossRef]
- Lombardi, R.; Onali, S.; Thorburn, D.; Davidson, B.R.; Gurusamy, K.S.; Tsochatzis, E. Pharmacological interventions for non-alcohol related fatty liver disease (NAFLD): An attempted network meta-analysis. Cochrane Database Syst Rev. 2017, 3, CD011640. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, T.; Paik, Y.-H.; Watanabe, S.; Laleu, B.; Gaggini, F.; Fioraso-Cartier, L.; Molango, S.; Heitz, F.; Merlot, C.; Szyndralewiez, C.; et al. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology 2012, 56, 2316–2327. [Google Scholar] [CrossRef]
- Paik, Y.H.; Iwaisako, K.; Seki, E.; Inokuchi, S.; Schnabl, B.; Osterreicher, C.H.; Kisseleva, T.; Brenner, D.A. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice. Hepatology 2011, 53, 1730–1741. [Google Scholar] [CrossRef]
- Frenette, C.; Kayali, Z.; Mena, E.; Mantry, P.S.; Lucas, K.J.; Neff, G.; Rodriguez, M.; Thuluvath, P.J.; Weinberg, E.; Bhandari, B.R.; et al. Emricasan to prevent new decompensation in patients with NASH-related decompensated cirrhosis. J. Hepatol. 2021, 74, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Barreyro, F.J.; Holod, S.; Finocchietto, P.V.; Camino, A.M.; Aquino, J.B.; Avagnina, A.; Carreras, M.C.; Poderoso, J.J.; Gores, G.J. The pan-caspase inhibitor Emricasan (IDN-6556) decreases liver injury and fibrosis in a murine model of non-alcoholic steatohepatitis. Liver Int. 2014, 35, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.J. Resmetirom: First Approval. Drugs 2024, 84, 729–735. [Google Scholar] [CrossRef]
- Harrison, S.A.; Bedossa, P.; Guy, C.D.; Schattenberg, J.M.; Loomba, R.; Taub, R.; Labriola, D.; Moussa, S.E.; Neff, G.W.; Rinella, M.E.; et al. A Phase 3, Randomized, Controlled Trial of Resmetirom in NASH with Liver Fibrosis. N. Engl. J. Med. 2024, 390, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Unagolla, J.M.; Das, S.; Flanagan, R.; Oehler, M.; Menon, J.U. Targeting chronic liver diseases: Molecular markers, drug delivery strategies and future perspectives. Int. J. Pharm. 2024, 660, 124381. [Google Scholar] [CrossRef]
- Sookoian, S.; Pirola, C.J. Resmetirom for treatment of MASH. Cell 2024, 187, 2897.e1. [Google Scholar] [CrossRef]
- Ratziu, V.; Scanlan, T.S.; Bruinstroop, E. Thyroid hormone receptor-β analogues for the treatment of metabolic dysfunction-associated steatohepatitis (MASH). J. Hepatol. 2024, 82, 375–387. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Ratziu, V.; Loomba, R.; Anstee, Q.M.; Kowdley, K.V.; Rinella, M.E.; Sheikh, M.Y.; Trotter, J.F.; Knapple, W.; Lawitz, E.J.; et al. Results from a new efficacy and safety analysis of the REGENERATE trial of obeticholic acid for treatment of pre-cirrhotic fibrosis due to non-alcoholic steatohepatitis. J. Hepatol. 2023, 79, 1110–1120. [Google Scholar] [CrossRef]
- Williamson, J.; Hughes, C.M.; Cobley, J.N.; Davison, G.W. The mitochondria-targeted antioxidant MitoQ, attenuates exercise-induced mitochondrial DNA damage. Redox Biol. 2020, 36, 101673. [Google Scholar] [CrossRef]
- Amorim, R.; Simões, I.C.M.; Teixeira, J.; Cagide, F.; Potes, Y.; Soares, P.; Carvalho, A.; Tavares, L.C.; Benfeito, S.; Pereira, S.P.; et al. Mitochondria-targeted anti-oxidant AntiOxCIN(4) improved liver steatosis in Western diet-fed mice by preventing lipid accumulation due to upregulation of fatty acid oxidation, quality control mechanism and antioxidant defense systems. Redox Biol. 2022, 55, 102400. [Google Scholar] [CrossRef]
- Liu, M.; Gu, Y.; Nie, W.; Zhu, X.; Qi, M.; Zhao, R.; Zhu, W.; Zhang, X. Formononetin Induces Ferroptosis in Activated Hepatic Stellate Cells to Attenuate Liver Fibrosis by Targeting NADPH Oxidase 4. Phytotherapy Res. 2024, 38, 5988–6003. [Google Scholar] [CrossRef] [PubMed]
- Yen, I.-C.; Tu, Q.-W.; Chang, T.-C.; Lin, P.-H.; Li, Y.-F.; Lee, S.-Y. 4-Acetylantroquinonol B ameliorates nonalcoholic steatohepatitis by suppression of ER stress and NLRP3 inflammasome activation. Biomed. Pharmacother. 2021, 138, 111504. [Google Scholar] [CrossRef]
- Zhang, N.; Lu, Y.; Shen, X.; Bao, Y.; Cheng, J.; Chen, L.; Li, B.; Zhang, Q. Fenofibrate Treatment Attenuated Chronic Endoplasmic Reticulum Stress in the Liver of Nonalcoholic Fatty Liver Disease Mice. Pharmacology 2015, 95, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Yoshiuchi, K.; Kaneto, H.; Matsuoka, T.-A.; Kasami, R.; Kohno, K.; Iwawaki, T.; Nakatani, Y.; Yamasaki, Y.; Shimomura, I.; Matsuhisa, M. Pioglitazone Reduces ER Stress in the Liver: Direct Monitoring of in vivo ER Stress Using ER Stress-activated Indicator Transgenic Mice. Endocr. J. 2009, 56, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Chen, D.; Wang, Y.; Li, H.; Zhang, Y.; Chen, H.; Li, X.; Huo, M. Nanozymes-recent development and biomedical applications. J. Nanobiotechnol. 2022, 20, 1–18. [Google Scholar] [CrossRef]
- Hao, C.; Qu, A.; Xu, L.; Sun, M.; Zhang, H.; Xu, C.; Kuang, H. Chiral Molecule-mediated Porous CuxO Nanoparticle Clusters with Antioxidation Activity for Ameliorating Parkinson’s Disease. J. Am. Chem. Soc. 2019, 141, 1091–1099. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Abouda, G.; A Aldersley, M.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef]
- Pruett, J.E.; Everman, S.J.; Hoang, N.H.; Salau, F.; Taylor, L.C.; Edwards, K.S.; Hosler, J.P.; Huffman, A.M.; Romero, D.G.; Cardozo, L.L.Y. Mitochondrial function and oxidative stress in white adipose tissue in a rat model of PCOS: Effect of SGLT2 inhibition. Biol. Sex Differ. 2022, 13, 1–17. [Google Scholar] [CrossRef]
- Zannad, F.; Ferreira, J.P.; Butler, J.; Filippatos, G.; Januzzi, J.L.; Sumin, M.; Zwick, M.; Saadati, M.; Pocock, S.J.; Sattar, N.; et al. Effect of empagliflozin on circulating proteomics in heart failure: Mechanistic insights into the EMPEROR programme. Eur. Hear. J. 2022, 43, 4991–5002. [Google Scholar] [CrossRef]
- Gawrieh, S.; Noureddin, M.; Loo, N.; Mohseni, R.; Awasty, V.; Cusi, K.; Kowdley, K.V.; Lai, M.; Schiff, E.; Parmar, D.; et al. Saroglitazar, a PPAR-α/γ Agonist, for Treatment of NAFLD: A Randomized Controlled Double-Blind Phase 2 Trial. Hepatology (Baltimore, Md.) 2021, 74, 1809–1824. [Google Scholar] [CrossRef]
- Kokkorakis, M.; Muzurović, E.; Volčanšek, Š.; Chakhtoura, M.; Hill, M.A.; Mikhailidis, D.P.; Mantzoros, C.S. Steatotic Liver Disease: Pathophysiology and Emerging Pharmacotherapies. Pharmacol. Rev. 2024, 76, 454–499. [Google Scholar] [CrossRef] [PubMed]
- Gratacós-Ginès, J.; Ariño, S.; Sancho-Bru, P.; Bataller, R.; Pose, E. MetALD: Clinical aspects, pathophysiology and treatment. JHEP Rep. Innov. Hepatol. 2025, 7, 101250. [Google Scholar] [CrossRef]
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.-S.; Harrison, S.A. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Aranäs, C.; Edvardsson, C.E.; Shevchouk, O.T.; Zhang, Q.; Witley, S.; Sköldheden, S.B.; Zentveld, L.; Vallöf, D.; Tufvesson-Alm, M.; Jerlhag, E. Semaglutide reduces alcohol intake and relapse-like drinking in male and female rats. EBioMedicine 2023, 93, 104642. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Volkow, N.D.; Berger, N.A.; Davis, P.B.; Kaelber, D.C.; Xu, R. Associations of semaglutide with incidence and recurrence of alcohol use disorder in real-world population. Nat. Commun. 2024, 15, 1–13. [Google Scholar] [CrossRef]
- Volkow, N.D.; Wise, R.A.; Baler, R. The dopamine motive system: Implications for drug and food addiction. Nat. Rev. Neurosci. 2017, 18, 741–752. [Google Scholar] [CrossRef]
- A Harrison, S.; Frias, J.P.; Neff, G.; A Abrams, G.; Lucas, K.J.; Sanchez, W.; Gogia, S.; Sheikh, M.Y.; Behling, C.; Bedossa, P.; et al. Safety and efficacy of once-weekly efruxifermin versus placebo in non-alcoholic steatohepatitis (HARMONY): A multicentre, randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Gastroenterol. Hepatol. 2023, 8, 1080–1093. [Google Scholar] [CrossRef]
- Loomba, R.; Sanyal, A.J.; Kowdley, K.V.; Bhatt, D.L.; Alkhouri, N.; Frias, J.P.; Bedossa, P.; Harrison, S.A.; Lazas, D.; Barish, R.; et al. Randomized, Controlled Trial of the FGF21 Analogue Pegozafermin in NASH. N. Engl. J. Med. 2023, 389, 998–1008. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef]
- Francque, S.M.; Bedossa, P.; Ratziu, V.; Anstee, Q.M.; Bugianesi, E.; Sanyal, A.J.; Loomba, R.; Harrison, S.A.; Balabanska, R.; Mateva, L.; et al. A Randomized, Controlled Trial of the Pan-PPAR Agonist Lanifibranor in NASH. N. Engl. J. Med. 2021, 385, 1547–1558. [Google Scholar] [CrossRef]
- Hassanein, T.; McClain, C.J.; Vatsalya, V.; Stein, L.L.; Flamm, S.L.; Martin, P.; Cave, M.C.; Mitchell, M.; Barton, B.; Nagy, L.; et al. Safety, Pharmacokinetics, and Efficacy Signals of Larsucosterol (DUR-928) in Alcohol-Associated Hepatitis. Am. J. Gastroenterol. 2023, 119, 107–115. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ALD | NAFLD | ||
|---|---|---|---|
| Similarities | Main sources of ROS Endoplasmic reticulum stress Inflammation simulation | Alcohol metabolism and FFA metabolism can lead to increased mitochondrial ETC electron leakage and ROS overproduction. | |
| This results in an imbalance of calcium homeostasis and induces mitochondrial ROS production. | |||
| The activation of Kupffer cells and the release of inflammatory factors (such as TNF-α and IL-6) promote ROS production. | |||
| Difference | Triggers | Alcohol metabolism is the main source of ROS production. | Insulin resistance and lipid metabolism disorders are the main drivers of ROS production. |
| Generation pathways | CYP2E1 pathway: production of large amounts of O2− and H2O2. | Insulin resistance and lipid metabolism disorders are the main drivers of ROS production. | |
| ADH/ALDH2 pathway: Alcohol metabolism produces NADH, which inhibits the mitochondrial respiratory chain and increases electron leakage. | Mitochondrial fatty acid beta oxidation: FFA overload leads to overload of the electron transport chain and increased electron leakage. | ||
| ERS: FFA-induced Ca2+ leakage and mitochondrial ROS production. | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Yang, H.; Han, F.; Guo, P. Reactive Oxygen Species as Key Molecules in the Pathogenesis of Alcoholic Fatty Liver Disease and Nonalcoholic Fatty Liver Disease: Future Perspectives. Curr. Issues Mol. Biol. 2025, 47, 464. https://doi.org/10.3390/cimb47060464
Zhang Z, Yang H, Han F, Guo P. Reactive Oxygen Species as Key Molecules in the Pathogenesis of Alcoholic Fatty Liver Disease and Nonalcoholic Fatty Liver Disease: Future Perspectives. Current Issues in Molecular Biology. 2025; 47(6):464. https://doi.org/10.3390/cimb47060464
Chicago/Turabian StyleZhang, Zhiqing, Hong Yang, Fei Han, and Peng Guo. 2025. "Reactive Oxygen Species as Key Molecules in the Pathogenesis of Alcoholic Fatty Liver Disease and Nonalcoholic Fatty Liver Disease: Future Perspectives" Current Issues in Molecular Biology 47, no. 6: 464. https://doi.org/10.3390/cimb47060464
APA StyleZhang, Z., Yang, H., Han, F., & Guo, P. (2025). Reactive Oxygen Species as Key Molecules in the Pathogenesis of Alcoholic Fatty Liver Disease and Nonalcoholic Fatty Liver Disease: Future Perspectives. Current Issues in Molecular Biology, 47(6), 464. https://doi.org/10.3390/cimb47060464