
Mechanisms of Immune Evasion in HIV-1: The Role of Virus-Host Protein Interactions



Abstract
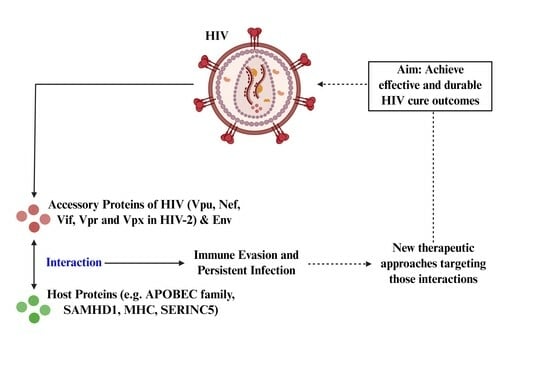
1. Introduction
1.1. HIV Virion and Genome
1.2. HIV-1 Life Cycle
1.3. HIV-1 Reservoir Cells
1.4. Immune Evasion
2. HIV Virus-Host Cell Proteins Interactions
2.1. Host Proteins That Interact with Viral Vpu
2.2. Host Proteins That Interact with Viral Nef
2.3. Host Proteins That Interact with Viral Vif
2.4. Host Proteins That Interact with Viral Vpr
2.5. Host Proteins That Interact with Viral Env
2.6. Host Proteins That Interact with Viral Vpx in HIV-2
3. Advances in Host-Targeted Therapeutic Strategies for HIV Infection and Persistence
4. Conclusions—Perspectives
Funding
Conflicts of Interest
Abbreviations
| ABCA1 | ATP Binding Cassette Transporter 1 |
| ADCC | Antibody-Dependent Cell-mediated Cytotoxicity |
| AIDS | Acquired Immunodeficiency Syndrome |
| ALIX | ALG-2 Interacting protein X |
| AMP | Adenosine Monophosphate |
| AP | Adaptor Protein Complex |
| APOBEC | Apolipoprotein B mRNA Editing Catalytic Polypeptide-like |
| Arf | ADP-Ribosylation Factor |
| ART | Antiretroviral Therapy |
| BCL-2 | B-cell leukemia/lymphoma 2 protein |
| bNAbs | Broadly Neutralizing Antibodies |
| BST-2 | Bone Marrow Stromal Antigen 2/Tetherin |
| BTRC | Beta-Transducin Repeat Containing |
| CBFβ | Core Binding Factor β |
| CCR | C-C Chemokine Receptor Type |
| CD | Cluster of Differentiation |
| cGAS | Cyclic GMP-AMP synthase |
| CHMP4 | Charged Multivesicular Body Protein 1 |
| CK-2 | Casein Kinase 2 |
| CRISPR-Cas9 | Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated protein 9 |
| CTL | Cytotoxic T-cell |
| CUL | Cullin |
| CXCR4 | C-X-C Chemokine Receptor Type 4 |
| DC | Dendritic Cell |
| DCAF1 | DDB1–CUL4-associated factor 1 |
| dCas9 | catalytically dead Cas9 |
| DDB1 | DNA Damage Binding Protein 1 |
| Elo | Elongin |
| Env | HIV Envelope Glycoprotein |
| ER | Endoplasmic Reticulum |
| ERAD | ER-associated degradation |
| ESCRT | Endosomal Sorting Complex Required for Transport |
| EV | Extracellular vesicle |
| EXOC | Exocyst Complex |
| Gag | Group-specific antigen |
| GBP5 | Guanylate Binding Protein 5 |
| GDP | Guanosine Diphosphate |
| GMP | Guanosine Monophosphate |
| gp | Glycoprotein |
| GPI | Glycosylphosphatidylinositol |
| GTP | Guanosine Triphosphate |
| GuavaH | Genomic Utility for Association and Viral Analysis in HIV |
| Hck | Hematopoietic Cell Kinase |
| HIV | Human Immunodeficiency Virus |
| HUSH | Human Silencing Hub |
| IFITM | Interferon Induced Transmembrane protein |
| IFN | Interferon |
| IL | Interleukin |
| ILCs | Innate Lymphoid Cells |
| IRF3 | Interferon Regulatory Factor 3 |
| Lck | Lymphocyte-specific protein tyrosine kinase |
| LEDGF | Lens Epithelium-Derived Growth Factor |
| LRAs | Latency Reversing Agents |
| LTR | Long Terminal Repeats |
| Lyn | Lck/Yes novel tyrosine kinase |
| MDMs | Monocyte Derived Macrophages |
| MHC | Major Histocompatibility Complex |
| MPP8 | M-phase Phosphoprotein 8 |
| MVB | Multivesicular Body |
| Nef | Negative Regulatory Factor |
| NF-κB | Nuclear Factor kappa B |
| NK | Natural-Killer |
| NKT | Natural-Killer T |
| NTB-A | NK-T-B antigen |
| NUP153 | Nucleoporin 153 |
| PACS | Phosphofurin Acidic Cluster Sorting Protein |
| PAK2 | p21-activated kinase 2 |
| PAMPs | Pathogen-Associated Molecular Patterns |
| PI3K | Phosphoinositide 3-kinase |
| pol | HIV Polymerase |
| PSGL-1 | P-Selectin Glycoprotein Ligand 1 |
| REAF | RNA-associated Early-stage Antiviral Factor |
| Rev | Regulator of Expression of Virion Proteins |
| ROC1 | Regulator of Cullins 1 |
| SAMHD1 | Sterile Alpha Motif and Histidine Aspartate domain-containing protein 1 |
| SCF | SKP1-cullin-F-box |
| SCID-hu | Severe Combined Immunodeficiency Humanized |
| SERINC | Serin Incorporator |
| SH3 Domain | SRC Homology 3 Domain |
| SIV | Simian Immunodeficiency Virus |
| Skp1 | S-phase kinase-associated protein 1 |
| SOCS | Suppressor of Cytokine Signaling proteins |
| STING | Stimulator of Interferon Genes |
| TALENs | Transcription Activator-Like Effector Nucleases |
| TASOR | Transcription Activation Suppressor |
| TAR | Transactivation Response Element |
| Tat | Transactivator of Transcription |
| TCR | T cell receptor |
| TET2 | Tet Methylcytosine Dioxygenase 2 |
| TGN | Trans-Golgi Network |
| THP-1 | Human Leukemia Monocytic Cell Line 1 |
| TSG101 | Tumor Susceptibility Gene 101 |
| UTR | Untranslated Region |
| Vif | Viral Infectivity Factor |
| Vpr | Viral Protein R |
| Vpu | Viral Protein U |
| Vpx | Viral Protein X |
| ZFNs | Zinc-Finger Nucleases |
References
- UNAIDS. Global HIV & AIDS statistics—2023 fact sheet. In UNAIDS Factsheet; UNAIDS: Geneva, Switzerland, 2023. [Google Scholar]
- Centers for Disease Control and Prevention (CDC). HIV Basics. In CDC HIV Basics; CDC: Atlanta, GA, USA, 2024. [Google Scholar]
- German Advisory Committee Blood (Arbeitskreis Blut), Subgroup ‘Assessment of Pathogens Transmissible by Blood’. Human Immunodeficiency Virus (HIV). Transfus. Med. Hemotherapy Off. Organ Der Dtsch. Ges. Fur Transfusionsmedizin Immunhamatol. 2016, 43, 203–222. [Google Scholar] [CrossRef]
- World Health Organization (WHO). HIV/AIDS. In WHO HIV/AIDS; WHO: Geneva, Switzerland, 2022. [Google Scholar]
- Woodham, A.W.; Skeate, J.G.; Sanna, A.M.; Taylor, J.R.; Da Silva, D.M.; Cannon, P.M.; Kast, W.M. Human Immunodeficiency Virus Immune Cell Receptors, Coreceptors, and Cofactors: Implications for Prevention and Treatment. AIDS Patient Care STDs 2016, 30, 291–306. [Google Scholar] [CrossRef]
- Wensing, A.M.; Calvez, V.; Ceccherini-Silberstein, F.; Charpentier, C.; Günthard, H.F.; Paredes, R.; Shafer, R.W.; Richman, D.D. 2022 update of the drug resistance mutations in HIV-1. Top. Antivir. Med. 2022, 30, 559–574. [Google Scholar] [PubMed]
- Foka, F.E.T.; Mufhandu, H.T. Current ARTs, Virologic Failure, and Implications for AIDS Management: A Systematic Review. Viruses 2023, 15, 1732. [Google Scholar] [CrossRef]
- Sorci, G.; Cornet, S.; Faivre, B. Immune evasion, immunopathology and the regulation of the immune system. Pathogens 2013, 2, 71–91. [Google Scholar] [CrossRef]
- Ploegh, H.L. Viral strategies of immune evasion. Science 1998, 280, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Finlay, B.B.; McFadden, G. Anti-immunology: Evasion of the host immune system by bacterial and viral pathogens. Cell 2006, 124, 767–782. [Google Scholar] [CrossRef] [PubMed]
- van Heuvel, Y.; Schatz, S.; Rosengarten, J.F.; Stitz, J. Infectious RNA: Human Immunodeficiency Virus (HIV) Biology, Therapeutic Intervention, and the Quest for a Vaccine. Toxins 2022, 14, 138. [Google Scholar] [CrossRef]
- Fanales-Belasio, E.; Raimondo, M.; Suligoi, B.; Buttò, S. HIV virology and pathogenetic mechanisms of infection: A brief overview. Ann. Dell’istituto Super. Sanita 2010, 46, 5–14. [Google Scholar] [CrossRef]
- Chen, J.; Zhou, T.; Zhang, Y.; Luo, S.; Chen, H.; Chen, D.; Li, C.; Li, W. The reservoir of latent HIV. Front. Cell. Infect. Microbiol. 2022, 12, 945956. [Google Scholar] [CrossRef]
- Kulpa, D.A.; Paiardini, M.; Silvestri, G. Immune-mediated strategies to solving the HIV reservoir problem. Nat. Rev. Immunol. 2025, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Guha, D.; Ayyavoo, V. Innate immune evasion strategies by human immunodeficiency virus type 1. Int. Sch. Res. Not. 2013, 2013, 954806. [Google Scholar] [CrossRef] [PubMed]
- Serra-Moreno, R.; Jia, B.; Breed, M.; Alvarez, X.; Evans, D.T. Compensatory changes in the cytoplasmic tail of gp41 confer resistance to tetherin/BST-2 in a pathogenic nef-deleted SIV. Cell Host Microbe 2011, 9, 46–57. [Google Scholar] [CrossRef]
- Salazar-Gonzalez, J.F.; Salazar, M.G.; Keele, B.F.; Learn, G.H.; Giorgi, E.E.; Li, H.; Decker, J.M.; Wang, S.; Baalwa, J.; Kraus, M.H.; et al. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med. 2009, 206, 1273–1289. [Google Scholar] [CrossRef]
- Castro-Gonzalez, S.; Colomer-Lluch, M.; Serra-Moreno, R. Barriers for HIV Cure: The Latent Reservoir. AIDS Res. Hum. Retroviruses 2018, 34, 739–759. [Google Scholar] [CrossRef] [PubMed]
- Justiz Vaillant, A.A.; Naik, R. HIV-1–Associated Opportunistic Infections. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Khan, N.; Geiger, J.D. Role of Viral Protein U (Vpu) in HIV-1 Infection and Pathogenesis. Viruses 2021, 13, 1466. [Google Scholar] [CrossRef]
- Schubert, U.; Schneider, T.; Henklein, P.; Hoffmann, K.; Berthold, E.; Hauser, H.; Pauli, G.; Porstmann, T. Human-immunodeficiency-virus-type-1-encoded Vpu protein is phosphorylated by casein kinase II. Eur. J. Biochem. 1992, 204, 875–883. [Google Scholar] [CrossRef]
- Vincent, M.J.; Abdul Jabbar, M. The human immunodeficiency virus type 1 Vpu protein: A potential regulator of proteolysis and protein transport in the mammalian secretory pathway. Virology 1995, 213, 639–649. [Google Scholar] [CrossRef]
- Hussain, A.; Wesley, C.; Khalid, M.; Chaudhry, A.; Jameel, S. Human immunodeficiency virus type 1 Vpu protein interacts with CD74 and modulates major histocompatibility complex class II presentation. J. Virol. 2008, 82, 893–902. [Google Scholar] [CrossRef]
- Verma, S.; Ali, A.; Arora, S.; Banerjea, A.C. Inhibition of β-TrcP-dependent ubiquitination of p53 by HIV-1 Vpu promotes p53-mediated apoptosis in human T cells. Blood 2011, 117, 6600–6607. [Google Scholar] [CrossRef]
- Ramirez, P.W.; Famiglietti, M.; Sowrirajan, B.; DePaula-Silva, A.B.; Rodesch, C.; Barker, E.; Bosque, A.; Planelles, V. Downmodulation of CCR7 by HIV-1 Vpu results in impaired migration and chemotactic signaling within CD4⁺ T cells. Cell Rep. 2014, 7, 2019–2030. [Google Scholar] [CrossRef]
- Robinson, M.S.; Antrobus, R.; Sanger, A.; Davies, A.K.; Gershlick, D.C. The role of the AP-1 adaptor complex in outgoing and incoming membrane traffic. J. Cell Biol. 2024, 223, e202310071. [Google Scholar] [CrossRef]
- Jia, X.; Weber, E.; Tokarev, A.; Lewinski, M.; Rizk, M.; Suarez, M.; Guatelli, J.; Xiong, Y. Structural basis of HIV-1 Vpu-mediated BST2 antagonism via hijacking of the clathrin adaptor protein complex 1. eLife 2014, 3, e02362. [Google Scholar] [CrossRef] [PubMed]
- Rashid, F.; Zaongo, S.D.; Iqbal, H.; Harypursat, V.; Song, F.; Chen, Y. Interactions between HIV proteins and host restriction factors: Implications for potential therapeutic intervention in HIV infection. Front. Immunol. 2024, 15, 1390650. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fu, Y.; Wang, Q.; Li, M.; Zhou, Z.; Dabbagh, D.; Fu, C.; Zhang, H.; Li, S.; Zhang, T.; et al. Proteomic profiling of HIV-1 infection of human CD4+ T cells identifies PSGL-1 as an HIV restriction factor. Nat. Microbiol. 2019, 4, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.P.; Patel, K.; Johnson, K.R.; Maric, D.; Calabresi, P.A.; Hasbun, R.; Nath, A. Induction of IL-17 and nonclassical T-cell activation by HIV-Tat protein. Proc. Natl. Acad. Sci. USA 2013, 110, 13588–13593. [Google Scholar] [CrossRef]
- Hotter, D.; Sauter, D.; Kirchhoff, F. Guanylate binding protein 5: Impairing virion infectivity by targeting retroviral envelope glycoproteins. Small GTPases 2017, 8, 31–37. [Google Scholar] [CrossRef]
- Staudt, R.P.; Alvarado, J.J.; Emert-Sedlak, L.A.; Shi, H.; Shu, S.T.; Wales, T.E.; Engen, J.R.; Smithgall, T.E. Structure, function, and inhibitor targeting of HIV-1 Nef-effector kinase complexes. J. Biol. Chem. 2020, 295, 15158–15171. [Google Scholar] [CrossRef]
- Jäger, S.; Cimermancic, P.; Gulbahce, N.; Johnson, J.R.; McGovern, K.E.; Clarke, S.C.; Shales, M.; Mercenne, G.; Pache, L.; Li, K.; et al. Global landscape of HIV-human protein complexes. Nature 2011, 481, 365–370. [Google Scholar] [CrossRef]
- Chaudhuri, R.; Lindwasser, O.W.; Smith, W.J.; Hurley, J.H.; Bonifacino, J.S. Downregulation of CD4 by human immunodeficiency virus type 1 Nef is dependent on clathrin and involves direct interaction of Nef with the AP2 clathrin adaptor. J. Virol. 2007, 81, 3877–3890. [Google Scholar] [CrossRef]
- Jin, Y.J.; Cai, C.Y.; Mezei, M.; Ohlmeyer, M.; Sanchez, R.; Burakoff, S.J. Identification of a novel binding site between HIV type 1 Nef C-terminal flexible loop and AP2 required for Nef-mediated CD4 downregulation. AIDS Res. Hum. Retroviruses 2013, 29, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Preusser, A.; Briese, L.; Baur, A.S.; Willbold, D. Direct in vitro binding of full-length human immunodeficiency virus type 1 Nef protein to CD4 cytoplasmic domain. J. Virol. 2001, 75, 3960–3964. [Google Scholar] [CrossRef]
- Amorim, N.A.; da Silva, E.M.; de Castro, R.O.; da Silva-Januário, M.E.; Mendonça, L.M.; Bonifacino, J.S.; da Costa, L.J.; daSilva, L.L. Interaction of HIV-1 Nef protein with the host protein Alix promotes lysosomal targeting of CD4 receptor. J. Biol. Chem. 2014, 289, 27744–27756. [Google Scholar] [CrossRef] [PubMed]
- DaSilva, L.L.; Sougrat, R.; Burgos, P.V.; Janvier, K.; Mattera, R.; Bonifacino, J.S. Human immunodeficiency virus type 1 Nef protein targets CD4 to the multivesicular body pathway. J. Virol. 2009, 83, 6578–6590. [Google Scholar] [CrossRef]
- Katoh, K.; Shibata, H.; Suzuki, H.; Nara, A.; Ishidoh, K.; Kominami, E.; Yoshimori, T.; Maki, M. The ALG-2-interacting protein Alix associates with CHMP4b, a human homologue of yeast Snf7 that is involved in multivesicular body sorting. J. Biol. Chem. 2003, 278, 39104–39113. [Google Scholar] [CrossRef] [PubMed]
- Piguet, V.; Gu, F.; Foti, M.; Demaurex, N.; Gruenberg, J.; Carpentier, J.L.; Trono, D. Nef-induced CD4 degradation: A diacidic-based motif in Nef functions as a lysosomal targeting signal through the binding of beta-COP in endosomes. Cell 1999, 97, 63–73. [Google Scholar] [CrossRef]
- Schaefer, M.R.; Wonderlich, E.R.; Roeth, J.F.; Leonard, J.A.; Collins, K.L. HIV-1 Nef targets MHC-I and CD4 for degradation via a final common beta-COP-dependent pathway in T cells. PLoS Pathog. 2008, 4, e1000131. [Google Scholar] [CrossRef]
- Leonard, J.A.; Filzen, T.; Carter, C.C.; Schaefer, M.; Collins, K.L. HIV-1 Nef disrupts intracellular trafficking of major histocompatibility complex class I, CD4, CD8, and CD28 by distinct pathways that share common elements. J. Virol. 2011, 85, 6867–6881. [Google Scholar] [CrossRef]
- Williams, M.; Roeth, J.F.; Kasper, M.R.; Fleis, R.I.; Przybycin, C.G.; Collins, K.L. Direct binding of human immunodeficiency virus type 1 Nef to the major histocompatibility complex class I (MHC-I) cytoplasmic tail disrupts MHC-I trafficking. J. Virol. 2002, 76, 12173–12184. [Google Scholar] [CrossRef]
- Noviello, C.M.; Benichou, S.; Guatelli, J.C. Cooperative binding of the class I major histocompatibility complex cytoplasmic domain and human immunodeficiency virus type 1 Nef to the endosomal AP-1 complex via its mu subunit. J. Virol. 2008, 82, 1249–1258. [Google Scholar] [CrossRef]
- Atkins, K.M.; Thomas, L.; Youker, R.T.; Harriff, M.J.; Pissani, F.; You, H.; Thomas, G. HIV-1 Nef binds PACS-2 to assemble a multikinase cascade that triggers major histocompatibility complex class I (MHC-I) down-regulation: Analysis using short interfering RNA and knock-out mice. J. Biol. Chem. 2008, 283, 11772–11784. [Google Scholar] [CrossRef]
- Dikeakos, J.D.; Atkins, K.M.; Thomas, L.; Emert-Sedlak, L.; Byeon, I.J.; Jung, J.; Ahn, J.; Wortman, M.D.; Kukull, B.; Saito, M.; et al. Small molecule inhibition of HIV-1-induced MHC-I down-regulation identifies a temporally regulated switch in Nef action. Mol. Biol. Cell 2010, 21, 3279–3292. [Google Scholar] [CrossRef] [PubMed]
- Dikeakos, J.D.; Thomas, L.; Kwon, G.; Elferich, J.; Shinde, U.; Thomas, G. An interdomain binding site on HIV-1 Nef interacts with PACS-1 and PACS-2 on endosomes to down-regulate MHC-I. Mol. Biol. Cell 2012, 23, 2184–2197. [Google Scholar] [CrossRef]
- Collette, Y.; Dutartre, H.; Benziane, A.; Ramos-Morales, F.; Benarous, R.; Harris, M.; Olive, D. Physical and functional interaction of Nef with Lck. HIV-1 Nef-induced T-cell signaling defects. J. Biol. Chem. 1996, 271, 6333–6341. [Google Scholar] [CrossRef] [PubMed]
- Trible, R.P.; Emert-Sedlak, L.; Smithgall, T.E. HIV-1 Nef selectively activates Src family kinases Hck, Lyn, and c-Src through direct SH3 domain interaction. J. Biol. Chem. 2006, 281, 27029–27038. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, A.A.; Kulkarni, A.; Tolstova, A.P.; Anashkina, A.A.; Sviridov, D.; Makarov, A.A.; Bukrinsky, M.I. Direct interaction between ABCA1 and HIV-1 Nef: Molecular modeling and virtual screening for inhibitors. Comput. Struct. Biotechnol. J. 2021, 19, 3876–3884. [Google Scholar] [CrossRef]
- Jennelle, L.; Hunegnaw, R.; Dubrovsky, L.; Pushkarsky, T.; Fitzgerald, M.L.; Sviridov, D.; Popratiloff, A.; Brichacek, B.; Bukrinsky, M. HIV-1 protein Nef inhibits activity of ATP-binding cassette transporter A1 by targeting endoplasmic reticulum chaperone calnexin. J. Biol. Chem. 2014, 289, 28870–28884. [Google Scholar] [CrossRef]
- Mujawar, Z.; Rose, H.; Morrow, M.P.; Pushkarsky, T.; Dubrovsky, L.; Mukhamedova, N.; Fu, Y.; Dart, A.; Orenstein, J.M.; Bobryshev, Y.V.; et al. Human immunodeficiency virus impairs reverse cholesterol transport from macrophages. PLoS Biol. 2006, 4, e365. [Google Scholar] [CrossRef]
- Imle, A.; Abraham, L.; Tsopoulidis, N.; Hoflack, B.; Saksela, K.; Fackler, O.T. Association with PAK2 Enables Functional Interactions of Lentiviral Nef Proteins with the Exocyst Complex. MBio 2015, 6, e01309-15. [Google Scholar] [CrossRef]
- Stolp, B.; Reichman-Fried, M.; Abraham, L.; Pan, X.; Giese, S.I.; Hannemann, S.; Goulimari, P.; Raz, E.; Grosse, R.; Fackler, O.T. HIV-1 Nef interferes with host cell motility by deregulation of Cofilin. Cell Host Microbe 2009, 6, 174–186. [Google Scholar] [CrossRef]
- Stolp, B.; Fackler, O.T. How HIV takes advantage of the cytoskeleton in entry and replication. Viruses 2011, 3, 293–311. [Google Scholar] [CrossRef] [PubMed]
- Renkema, G.H.; Pulkkinen, K.; Saksela, K. Cdc42/Rac1-mediated activation primes PAK2 for superactivation by tyrosine phosphorylation. Mol. Cell. Biol. 2002, 22, 6719–6725. [Google Scholar] [CrossRef]
- Fu, Y.; He, S.; Waheed, A.A.; Dabbagh, D.; Zhou, Z.; Trinité, B.; Wang, Z.; Yu, J.; Wang, D.; Li, F.; et al. PSGL-1 restricts HIV-1 infectivity by blocking virus particle attachment to target cells. Proc. Natl. Acad. Sci. USA 2020, 117, 9537–9545. [Google Scholar] [CrossRef]
- Lee, W.J.; Fu, R.M.; Liang, C.; Sloan, R.D. IFITM proteins inhibit HIV-1 protein synthesis. Sci. Rep. 2018, 8, 14551. [Google Scholar] [CrossRef] [PubMed]
- Brass, A.L.; Huang, I.C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; van der Weyden, L.; Fikrig, E.; et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Pan, Q.; Rong, L.; Liu, S.-L.; Liang, C. The IFITM proteins inhibit HIV-1 infection. J. Virol. 2011, 85, 2126–2137. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.L.; Wilson, H.; Iyer, S.S.; Coss, K.; Doores, K.; Smith, S.; Kellam, P.; Finzi, A.; Borrow, P.; Hahn, B.H.; et al. Resistance of Transmitted Founder HIV-1 to IFITM-Mediated Restriction. Cell Host Microbe 2016, 20, 429–442. [Google Scholar] [CrossRef]
- da Silva-Januário, M.E.; da Costa, C.S.; Tavares, L.A.; Oliveira, A.K.; Januário, Y.C.; de Carvalho, A.N.; Cassiano, M.H.A.; Rodrigues, R.L.; Miller, M.E.; Palameta, S.; et al. HIV-1 Nef Changes the Proteome of T Cells Extracellular Vesicles Depleting IFITMs and Other Antiviral Factors. Mol. Cell. Proteom. MCP 2023, 22, 100676. [Google Scholar] [CrossRef]
- Zhang, H.; Pomerantz, R.J.; Dornadula, G.; Sun, Y. Human immunodeficiency virus type 1 Vif protein is an integral component of an mRNP complex of viral RNA and could be involved in the viral RNA folding and packaging process. J. Virol. 2000, 74, 8252–8261. [Google Scholar] [CrossRef]
- Takaori-Kondo, A.; Shindo, K. HIV-1 Vif: A guardian of the virus that opens up a new era in the research field of restriction factors. Front. Microbiol. 2013, 4, 34. [Google Scholar] [CrossRef]
- Marin, M.; Rose, K.M.; Kozak, S.L.; Kabat, D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat. Med. 2003, 9, 1398–1403. [Google Scholar] [CrossRef] [PubMed]
- Mehle, A.; Strack, B.; Ancuta, P.; Zhang, C.; McPike, M.; Gabuzda, D. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J. Biol. Chem. 2004, 279, 7792–7798. [Google Scholar] [CrossRef]
- Stopak, K.; de Noronha, C.; Yonemoto, W.; Greene, W.C. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol. Cell 2003, 12, 591–601. [Google Scholar] [CrossRef]
- Mehle, A.; Goncalves, J.; Santa-Marta, M.; McPike, M.; Gabuzda, D. Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev. 2004, 18, 2861–2866. [Google Scholar] [CrossRef]
- Evans, S.L.; Schön, A.; Gao, Q.; Han, X.; Zhou, X.; Freire, E.; Yu, X.F. HIV-1 Vif N-terminal motif is required for recruitment of Cul5 to suppress APOBEC3. Retrovirology 2014, 11, 4. [Google Scholar] [CrossRef]
- Yu, Y.; Xiao, Z.; Ehrlich, E.S.; Yu, X.; Yu, X.F. Selective assembly of HIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complex through a novel SOCS box and upstream cysteines. Genes Dev. 2004, 18, 2867–2872. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, J.R.; Huthoff, H.; Veselkov, D.A.; Beavil, R.L.; Simpson, P.J.; Matthews, S.J.; Malim, M.H.; Sanderson, M.R. The SOCS-box of HIV-1 Vif interacts with ElonginBC by induced-folding to recruit its Cul5-containing ubiquitin ligase complex. PLoS Pathog. 2010, 6, e1000925. [Google Scholar] [CrossRef] [PubMed]
- da Costa, K.S.; Leal, E.; dos Santos, A.M.; Lima e Lima, A.H.; Alves, C.N.; Lameira, J. Structural analysis of viral infectivity factor of HIV type 1 and its interaction with A3G, EloC and EloB. PLoS ONE 2014, 9, e89116. [Google Scholar] [CrossRef]
- Shirakawa, K.; Takaori-Kondo, A.; Kobayashi, M.; Tomonaga, M.; Izumi, T.; Fukunaga, K.; Sasada, A.; Abudu, A.; Miyauchi, Y.; Akari, H.; et al. Ubiquitination of APOBEC3 proteins by the Vif-Cullin5-ElonginB-ElonginC complex. Virology 2006, 344, 263–266. [Google Scholar] [CrossRef]
- Zhang, W.; Du, J.; Evans, S.L.; Yu, Y.; Yu, X.F. T-cell differentiation factor CBF-β regulates HIV-1 Vif-mediated evasion of host restriction. Nature 2011, 481, 376–379. [Google Scholar] [CrossRef]
- Matsui, Y.; Shindo, K.; Nagata, K.; Io, K.; Tada, K.; Iwai, F.; Kobayashi, M.; Kadowaki, N.; Harris, R.S.; Takaori-Kondo, A. Defining HIV-1 Vif residues that interact with CBFβ by site-directed mutagenesis. Virology 2014, 449, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Marno, K.M.; Ogunkolade, B.W.; Pade, C.; Oliveira, N.M.; O’Sullivan, E.; McKnight, Á. Novel restriction factor RNA-associated early-stage anti-viral factor (REAF) inhibits human and simian immunodeficiency viruses. Retrovirology 2014, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, J.M.; Marno, K.M.; Pike, R.; Lee, W.J.; Jones, C.E.; Ogunkolade, B.W.; Pardieu, C.; Bryan, A.; Fu, R.M.; Warnes, G.; et al. HIV-1 Accessory Protein Vpr Interacts with REAF/RPRD2 To Mitigate Its Antiviral Activity. J. Virol. 2020, 94, e01591-19. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Wang, Q.; Xu, Y.; Tsao, L.C.; Nakagawa, T.; Guo, H.; Su, L.; Xiong, Y. Vpr Targets TET2 for Degradation by CRL4VprBP E3 Ligase to Sustain IL-6 Expression and Enhance HIV-1 Replication. Mol. Cell 2018, 70, 961–970.e5. [Google Scholar] [CrossRef]
- Khan, H.; Sumner, R.P.; Rasaiyaah, J.; Tan, C.P.; Rodriguez-Plata, M.T.; Van Tulleken, C.; Fink, D.; Zuliani-Alvarez, L.; Thorne, L.; Stirling, D.; et al. HIV-1 Vpr antagonizes innate immune activation by targeting karyopherin-mediated NF-κB/IRF3 nuclear transport. eLife 2020, 9, e60821. [Google Scholar] [CrossRef]
- Miyatake, H.; Sanjoh, A.; Murakami, T.; Murakami, H.; Matsuda, G.; Hagiwara, K.; Yokoyama, M.; Sato, H.; Miyamoto, Y.; Dohmae, N.; et al. Molecular Mechanism of HIV-1 Vpr for Binding to Importin-α. J. Mol. Biol. 2016, 428, 2744–2757. [Google Scholar] [CrossRef]
- Vodicka, M.A.; Koepp, D.M.; Silver, P.A.; Emerman, M. HIV-1 Vpr interacts with the nuclear transport pathway to promote macrophage infection. Genes Dev. 1998, 12, 175–185. [Google Scholar] [CrossRef]
- Nitahara-Kasahara, Y.; Kamata, M.; Yamamoto, T.; Zhang, X.; Miyamoto, Y.; Muneta, K.; Iijima, S.; Yoneda, Y.; Tsunetsugu-Yokota, Y.; Aida, Y. Novel nuclear import of Vpr promoted by importin alpha is crucial for human immunodeficiency virus type 1 replication in macrophages. J. Virol. 2007, 81, 5284–5293. [Google Scholar] [CrossRef]
- Arrildt, K.T.; Joseph, S.B.; Swanstrom, R. The HIV-1 env protein: A coat of many colors. Curr. HIV/AIDS Rep. 2012, 9, 52–63. [Google Scholar] [CrossRef]
- Le Tortorec, A.; Neil, S.J. Antagonism to and intracellular sequestration of human tetherin by the human immunodeficiency virus type 2 envelope glycoprotein. J. Virol. 2009, 83, 11966–11978. [Google Scholar] [CrossRef]
- Usami, Y.; Wu, Y.; Göttlinger, H.G. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef]
- Beitari, S.; Ding, S.; Pan, Q.; Finzi, A.; Liang, C. Effect of HIV-1 Env on SERINC5 Antagonism. J. Virol. 2017, 91, e02214-16. [Google Scholar] [CrossRef] [PubMed]
- Planelles, V.; Barker, E. Roles of Vpr and Vpx in modulating the virus-host cell relationship. Mol. Asp. Med. 2010, 31, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Lahouassa, H.; Daddacha, W.; Hofmann, H.; Ayinde, D.; Logue, E.C.; Dragin, L.; Bloch, N.; Maudet, C.; Bertrand, M.; Gramberg, T.; et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 2012, 13, 223–228. [Google Scholar] [CrossRef]
- Antonucci, J.M.; St Gelais, C.; de Silva, S.; Yount, J.S.; Tang, C.; Ji, X.; Shepard, C.; Xiong, Y.; Kim, B.; Wu, L. SAMHD1-mediated HIV-1 restriction in cells does not involve ribonuclease activity. Nat. Med. 2016, 22, 1072–1074. [Google Scholar] [CrossRef]
- Goujon, C.; Arfi, V.; Pertel, T.; Luban, J.; Lienard, J.; Rigal, D.; Darlix, J.L.; Cimarelli, A. Characterization of simian immunodeficiency virus SIVSM/human immunodeficiency virus type 2 Vpx function in human myeloid cells. J. Virol. 2008, 82, 12335–12345. [Google Scholar] [CrossRef] [PubMed]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Ségéral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Ayinde, D.; Bruel, T.; Cardinaud, S.; Porrot, F.; Prado, J.G.; Moris, A.; Schwartz, O. SAMHD1 Limits HIV-1 Antigen Presentation by Monocyte-Derived Dendritic Cells. J. Virol. 2015, 89, 6994–7006. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Rui, Y.; Lou, M.; Yin, L.; Xiong, H.; Zhou, Z.; Shen, S.; Chen, T.; Zhang, Z.; Zhao, N.; et al. HIV-2/SIV Vpx targets a novel functional domain of STING to selectively inhibit cGAS-STING-mediated NF-κB signalling. Nat. Microbiol. 2019, 4, 2552–2564. [Google Scholar] [CrossRef]
- Chougui, G.; Munir-Matloob, S.; Matkovic, R.; Martin, M.M.; Morel, M.; Lahouassa, H.; Leduc, M.; Ramirez, B.C.; Etienne, L.; Margottin-Goguet, F. HIV-2/SIV viral protein X counteracts HUSH repressor complex. Nat. Microbiol. 2018, 3, 891–897. [Google Scholar] [CrossRef]
- Wilkin, T.J.; Gulick, R.M. CCR5 antagonism in HIV infection: Current concepts opportunities. Annu. Rev. Med. 2012, 63, 81–93. [Google Scholar] [CrossRef]
- Allers, K.; Schneider, T. CCR5Δ32 mutation and HIV infection: Basis for curative HIV therapy. Curr. Opin. Virol. 2015, 14, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Grande, F.; Occhiuzzi, M.A.; Rizzuti, B.; Ioele, G.; De Luca, M.; Tucci, P.; Svicher, V.; Aquaro, S.; Garofalo, A. CCR5/CXCR4 Dual Antagonism for the Improvement of HIV Infection Therapy. Molecules 2019, 24, 550. [Google Scholar] [CrossRef] [PubMed]
- Christ, F.; Debyser, Z. The LEDGF/p75 integrase interaction, a novel target for anti-HIV. Virology 2013, 435, 102–109. [Google Scholar] [CrossRef]
- Caskey, M. Broadly neutralizing antibodies for the treatment and prevention of HIV infection. Curr Opin HIV AIDS 2020, 85, 49–55. [Google Scholar] [CrossRef]
- Caskey, M.; Klein, F.; Lorenzi, J.C.; Seaman, M.S.; West, A.P., Jr.; Buckley, N.; Kremer, G.; Nogueira, L.; Braunschweig, M.; Scheid, J.F.; et al. Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature 2015, 522, 487–491. [Google Scholar] [CrossRef]
- Ledgerwood, J.E.; Coates, E.E.; Yamshchikov, G.; Saunders, J.G.; Holman, L.; Enama, M.E.; DeZure, A.; Lynch, R.M.; Gordon, I.; Plummer, S.; et al. Safety, pharmacokinetics and neutralization of the broadly neutralizing HIV-1 human monoclonal antibody VRC01 in healthy adults. Clin. Exp. Immunol. 2015, 182, 289–301. [Google Scholar]
- Lynch, R.M.; Boritz, E.; Coates, E.E.; DeZure, A.; Madden, P.; Costner, P.; Enama, M.E.; Plummer, S.; Holman, L.; Hendel, C.S.; et al. Virologic effects of broadly neutralizing antibody VRC01 administration during chronic HIV-1 infection. Sci. Transl. Med. 2015, 7, 319ra206. [Google Scholar] [CrossRef] [PubMed]
- Caskey, M.; Schoofs, T.; Gruell, H.; Settler, A.; Karagounis, T.; Kreider, E.F.; Murrell, B.; Pfeifer, N.; Nogueira, L.; Oliveira, T.Y.; et al. Antibody 10–1074 suppresses viremia in HIV-1-infected individuals. Nat. Med. 2017, 23, 185–191. [Google Scholar] [CrossRef]
- Caskey, M.; Klein, F.; Nussenzweig, M.C. Broadly neutralizing anti-HIV-1 monoclonal antibodies in the clinic. Nat. Med. 2019, 25, 547–553. [Google Scholar] [CrossRef]
- Huang, Y.; Yu, J.; Lanzi, A.; Yao, X.; Andrews, C.D.; Tsai, L.; Gajjar, M.R.; Sun, M.; Seaman, M.S.; Padte, N.N.; et al. Engineered Bispecific Antibodies with Exquisite HIV-1-Neutralizing Activity. Cell 2016, 165, 1621–1631. [Google Scholar] [CrossRef]
- Xu, L.; Pegu, A.; Rao, E.; Doria-Rose, N.; Beninga, J.; McKee, K.; Lord, D.M.; Wei, R.R.; Deng, G.; Louder, M.; et al. Trispecific broadly neutralizing HIV antibodies mediate potent SHIV protection in macaques. Science 2017, 358, 85–90. [Google Scholar] [CrossRef]
- Mayer, K.H.; Seaton, K.E.; Huang, Y.; Grunenberg, N.; Isaacs, A.; Allen, M.; Ledgerwood, J.E.; Frank, I.; Sobieszczyk, M.E.; Baden, L.R.; et al. Safety, pharmacokinetics, and immunological activities of multiple intravenous or subcutaneous doses of an anti-HIV monoclonal antibody, VRC01, administered to HIV-uninfected adults: Results of a phase 1 randomized trial. PLoS Med. 2017, 14, e1002435. [Google Scholar] [CrossRef]
- Margot, N.A.; Naik, V.; VanderVeen, L.; Anoshchenko, O.; Singh, R.; Dvory-Sobol, H.; Rhee, M.S.; Callebaut, C. Resistance analyses in highly treatment-experienced people with human immunodeficiency virus (HIV) treated with the novel capsid HIV inhibitor lenacapavir. J. Infect. Dis. 2022, 226, 1985–1991. [Google Scholar] [CrossRef]
- Reeves, J.; Zheng, Y.; Olefsky, M.; Lie, Y.; Burke, L.; Taiwo, B. Susceptibility to bNAbs is concordant in pre-ART plasma and on-ART PBMCs: ACTG NWC413. In Proceedings of the Conference on Retroviruses and Opportunistic Infections, Seattle, WA, USA, 4–9 March 2019. [Google Scholar]
- Cohen, Y.Z.; Butler, A.L.; Millard, K.; Witmer-Pack, M.; Levin, R.; Unson-O’brien, C.; Patel, R.; Shimeliovich, I.; Lorenzi, J.C.C.; Horowitz, J.; et al. Safety, pharmacokinetics, and immunogenicity of the combination of the broadly neutralizing anti-HIV-1 antibodies 3BNC117 and 10-1074 in healthy adults: A randomized, phase 1 study. PLoS ONE 2019, 14, e0219142. [Google Scholar] [CrossRef]
- Mendoza, P.; Gruell, H.; Nogueira, L.; Pai, J.A.; Butler, A.L.; Millard, K.; Lehmann, C.; Suarez, I.; Oliveira, T.Y.; Lorenzi, J.C.C.; et al. Combination therapy with anti-HIV-1 antibodies maintains viral suppression. Nature 2018, 561, 479–484. [Google Scholar] [CrossRef]
- Huang, Y.; Karuna, S.; Carpp, L.N.; Reeves, D.; Pegu, A.; Seaton, K.; Mayer, K.; Schiffer, J.; Mascola, J.; Gilbert, P.B. Modeling cumulative overall prevention efficacy for the VRC01 phase 2b efficacy trials. Hum. Vaccin. Immunother. 2018, 14, 2116–2127. [Google Scholar] [CrossRef]
- Horwitz, J.A.; Bar-On, Y.; Lu, C.L.; Fera, D.; Lockhart, A.A.K.; Lorenzi, J.C.C.; Nogueira, L.; Golijanin, J.; Scheid, J.F.; Seaman, M.S.; et al. Non-neutralizing Antibodies Alter the Course of HIV-1 Infection In Vivo. Cell 2017, 170, 637–648.e610. [Google Scholar] [CrossRef]
- Halper-Stromberg, A.; Lu, C.L.; Klein, F.; Horwitz, J.A.; Bournazos, S.; Nogueira, L.; Eisenreich, T.R.; Liu, C.; Gazumyan, A.; Schaefer, U.; et al. Broadly neutralizing antibodies and viral inducers decrease rebound from HIV-1 latent reservoirs in humanized mice. Cell 2014, 158, 989–999. [Google Scholar] [CrossRef]
- Bournazos, S.; Klein, F.; Pietzsch, J.; Seaman, M.S.; Nussenzweig, M.C.; Ravetch, J.V. Broadly neutralizing anti-HIV-1 antibodies require Fc effector functions for in vivo activity. Cell 2014, 158, 1243–1253. [Google Scholar] [CrossRef]
- Rosás-Umbert, M.; Gunst, J.D.; Pahus, M.H.; Olesen, R.; Schleimann, M.; Denton, P.W.; Ramos, V.; Ward, A.; Kinloch, N.N.; Copertino, D.C.; et al. Administration of broadly neutralizing anti-HIV-1 antibodies at ART initiation maintains long-term CD8+ T cell immunity. Nat. Commun. 2022, 13, 6473. [Google Scholar] [CrossRef]
- Eron, J.J.; Little, S.J.; Crofoot, G.; Cook, P.; Ruane, P.J.; Jayaweera, D.; VanderVeen, L.A.; DeJesus, E.; Zheng, Y.; Mills, A.; et al. Safety of teropavimab and zinlirvimab with lenacapavir once every 6 months for HIV treatment: A phase 1b, randomised, proof-of-concept study. Lancet HIV 2024, 11, e146–e155. [Google Scholar] [CrossRef]
- Xiao, Q.; Guo, D.; Chen, S. Application of CRISPR/Cas9-Based Gene Editing in HIV-1/AIDS Therapy. Front. Cell. Infect. Microbiol. 2019, 9, 69. [Google Scholar] [CrossRef]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef]
- Yin, L.; Hu, S.; Mei, S.; Sun, H.; Xu, F.; Li, J.; Zhu, W.; Liu, X.; Zhao, F.; Zhang, D.; et al. CRISPR/Cas9 inhibits multiple steps of HIV-1 infection. Hum. Gene Ther. 2018, 29, 1264–1276. [Google Scholar] [CrossRef]
- Yu, A.Q.; Ding, Y.; Lu, Z.Y.; Hao, Y.Z.; Teng, Z.P.; Yan, S.R.; Li, D.S.; Zeng, Y. TALENs-mediated homozygous CCR5Delta32 mutations endow CD4+ U87 cells with resistance against HIV1 infection. Mol. Med. Rep. 2018, 17, 243–249. [Google Scholar] [CrossRef]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef]
- Ebina, H.; Misawa, N.; Kanemura, Y.; Koyanagi, Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 2013, 3, 2510. [Google Scholar] [CrossRef]
- Hu, W.; Kaminski, R.; Yang, F.; Zhang, Y.; Cosentino, L.; Li, F.; Luo, B.; Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Karn, J.; et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc. Natl. Acad. Sci. USA 2014, 111, 11461–11466. [Google Scholar] [CrossRef]
- Liao, H.K.; Gu, Y.; Diaz, A.; Marlett, J.; Takahashi, Y.; Li, M.; Suzuki, K.; Xu, R.; Hishida, T.; Chang, C.J.; et al. Use of the CRISPR/Cas9 system as an intracellular defense against HIV-1 infection in human cells. Nat. Commun. 2015, 6, 6413. [Google Scholar] [CrossRef]
- Yin, C.; Zhang, T.; Qu, X.; Zhang, Y.; Putatunda, R.; Xiao, X.; Li, F.; Xiao, W.; Zhao, H.; Dai, S.; et al. In vivo excision of HIV-1 provirus by saCas9 and multiplex single-guide RNAs in animal models. Mol. Ther. 2017, 25, 1168–1186. [Google Scholar] [CrossRef]
- Wilen, C.B.; Wang, J.; Tilton, J.C.; Miller, J.C.; Kim, K.A.; Rebar, E.J.; Sherrill-Mix, S.A.; Patro, S.C.; Secreto, A.J.; Jordan, A.P.O.; et al. Engineering HIV-resistant human CD4+ T cells with CXCR4-specific zinc-finger nucleases. PLoS Pathog. 2011, 7, e1002020. [Google Scholar] [CrossRef]
- Li, C.; Guan, X.; Du, T.; Jin, W.; Wu, B.; Liu, Y.; Wang, P.; Hu, B.; Griffin, G.E.; Shattock, R.J.; et al. Inhibition of HIV-1 infection of primary CD4+ T-cells by gene editing of CCR5 using adenovirus-delivered CRISPR/Cas9. J. Gen. Virol. 2015, 96, 2381–2393. [Google Scholar] [CrossRef]
- Hou, P.; Chen, S.; Wang, S.; Yu, X.; Chen, Y.; Jiang, M.; Zhuang, K.; Ho, W.; Hou, W.; Huang, J.; et al. Genome editing of CXCR4 by CRISPR/cas9 confers cells resistant to HIV-1 infection. Sci. Rep. 2015, 5, 15577. [Google Scholar] [CrossRef]
- Huang, J.; Wang, F.; Argyris, E.; Chen, K.; Liang, Z.; Tian, H.; Huang, W.; Squires, K.; Verlinghieri, G.; Zhang, H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat. Med. 2007, 13, 1241–1247. [Google Scholar] [CrossRef]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.; Strain, M.C.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef]
- Bullen, C.K.; Laird, G.M.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nat. Med. 2014, 20, 425–429. [Google Scholar] [CrossRef]
- Kim, V.; Mears, B.M.; Powell, B.H.; Witwer, K.W. Mutant Cas9-transcriptional activator activates HIV-1 in U1 cells in the presence and absence of LTR-specific guide RNAs. Matters 2017, 2017, 1–4. [Google Scholar] [CrossRef][Green Version]
- Bialek, J.K.; Dunay, G.A.; Voges, M.; Schafer, C.; Spohn, M.; Stucka, R.; Hauber, J.; Lange, U.C. Targeted HIV-1 latency reversal using CRISPR/Cas9-derived transcriptional activator systems. PLoS ONE 2016, 11, e0158294. [Google Scholar] [CrossRef]
- Limsirichai, P.; Gaj, T.; Schaffer, D.V. CRISPR-mediated activation of latent HIV-1 expression. Mol. Ther. 2016, 24, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Jiang, Z.; Lu, P.; Ma, L.; Li, C.; Pan, H.; Fu, Z.; Qu, X.; Wang, P.; Deng, J.; et al. Specific reactivation of latent HIV-1 by dCas9-suntag-VP64-mediated guide RNA targeting the HIV-1 promoter. Mol. Ther. 2016, 24, 508–521. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Viral Protein | Host Protein | Function | Ref. |
|---|---|---|---|
| Vpu | BST-2 | Inhibits the antiviral effect of BST-2 | [20] |
| BTRC | Acts like a SCF E3 ubiquitin-protein ligase complex for the BST-2 degradation | [20] | |
| AP-1 | Hijacks AP-dependent trafficking pathways | [27] | |
| CD4 | Promotes CD4 downregulation and degradation | [20] | |
| PSGL-1 | Promotes the ubiquitination and degradation of PSGL-1 | [29] | |
| GBP5 | Counteracts GBP5 indirectly | [31] |
| Viral Protein | Host Protein | Function | Ref. |
|---|---|---|---|
| Nef | CD4 | Promotes CD4 downregulation and degradation | [36] |
| ALIX | Promotes lysosomal targeting of CD4 receptor | [37] | |
| AP-1 | Hijacks AP-dependent trafficking pathways | [44] | |
| AP-2 | [34,35] | ||
| SERINC5 | Promotes the downregulation of SERINC5 and its degradation through the endolysosomal pathway | [32] | |
| MHC-I | Promotes the disruption of MHC-I trafficking to the plasma membrane | [43] | |
| ABCA1 | Promotes the dysregulation of cholesterol metabolism | [50] | |
| Calnexin | Promotes ABCA1 activity inhibition | [51] | |
| PACS-1 | Association triggers MHC-I downregulation | [47] | |
| PACS-2 | [45,47] | ||
| ARF1 | [32] | ||
| EXOC | Promotes the inhibition of actin remodeling and interference with proximal signaling triggered by TCR engagement | [53] | |
| PAK2 | [53] | ||
| Lck | Influences the outcome of T-cell activation | [48] | |
| Hck | Promotes MCH-I downregulation | [46] | |
| Lyn | [46] | ||
| PSGL-1 | Promotes the downregulation of PSGL-1 and its possible redirection to intracellular compartments | [57] | |
| IFITM (1-3) | Alters the intracellular distribution of IFITM | [58,62] |
| Viral Protein | Host Protein | Function | Ref. |
|---|---|---|---|
| Vif | APOBEC3 family | Prevents hypermutations in viral DNA via degradation | [73] |
| APOBEC3G | [66,67] | ||
| EloB | Part of the E3 ligase complex for the degradation of APOBEC proteins | [71] | |
| CUL5 | [70] | ||
| EloC | [72] | ||
| CBF-β | [75] |
| Viral Protein | Host Protein | Function | Ref. |
|---|---|---|---|
| Vpr | REAF | Promotes the degradation of REAF, enhancing viral replication | [77] |
| TET2 | Promotes the degradation of TET2, enhancing viral replication | [78] | |
| Karyopherins | Blocks the IRF3 and NF-κB nuclear translocation | [79] |
| Viral Protein | Host Protein | Function | Ref. |
|---|---|---|---|
| Env | BST-2 | Confines BST-2 to the trans-Golgi network (HIV-2) | [84] |
| SERINC5 | Unknown counteract mechanism | [85,86] |
| Viral Protein | Host Protein | Function | Ref. |
|---|---|---|---|
| Vpx (HIV-2) | SAMHD1 | Leads SAMHD1 to proteasomal degradation | [87] |
| STING | Interacts with STING and inhibits the activation of NF-κB | [90] | |
| HUSH | Promotes the degradation of HUSH, enabling viral gene expression | [91] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mouzakis, A.; Petrakis, V.; Tryfonopoulou, E.; Panopoulou, M.; Panagopoulos, P.; Chlichlia, K. Mechanisms of Immune Evasion in HIV-1: The Role of Virus-Host Protein Interactions. Curr. Issues Mol. Biol. 2025, 47, 367. https://doi.org/10.3390/cimb47050367
Mouzakis A, Petrakis V, Tryfonopoulou E, Panopoulou M, Panagopoulos P, Chlichlia K. Mechanisms of Immune Evasion in HIV-1: The Role of Virus-Host Protein Interactions. Current Issues in Molecular Biology. 2025; 47(5):367. https://doi.org/10.3390/cimb47050367
Chicago/Turabian StyleMouzakis, Antonios, Vasileios Petrakis, Eleni Tryfonopoulou, Maria Panopoulou, Periklis Panagopoulos, and Katerina Chlichlia. 2025. "Mechanisms of Immune Evasion in HIV-1: The Role of Virus-Host Protein Interactions" Current Issues in Molecular Biology 47, no. 5: 367. https://doi.org/10.3390/cimb47050367
APA StyleMouzakis, A., Petrakis, V., Tryfonopoulou, E., Panopoulou, M., Panagopoulos, P., & Chlichlia, K. (2025). Mechanisms of Immune Evasion in HIV-1: The Role of Virus-Host Protein Interactions. Current Issues in Molecular Biology, 47(5), 367. https://doi.org/10.3390/cimb47050367