
Mechanistic Insights into the Anti-Hepatocellular Carcinoma Effects of ACY-1215: p53 Acetylation and Ubiquitination Regulation

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Lines and Cell Culture
2.3. CCK-8 Assay
2.4. Colony Formation Assay
2.5. Transwell Migration and Invasion Assay
2.6. Immunoblotting Analysis
2.7. Quantitative Reverse Transcription PCR
2.8. Protein Stability Assay
2.9. Coimmunoprecipitation
2.10. Mass Spectrometry
2.11. Statistical Analysis
3. Results
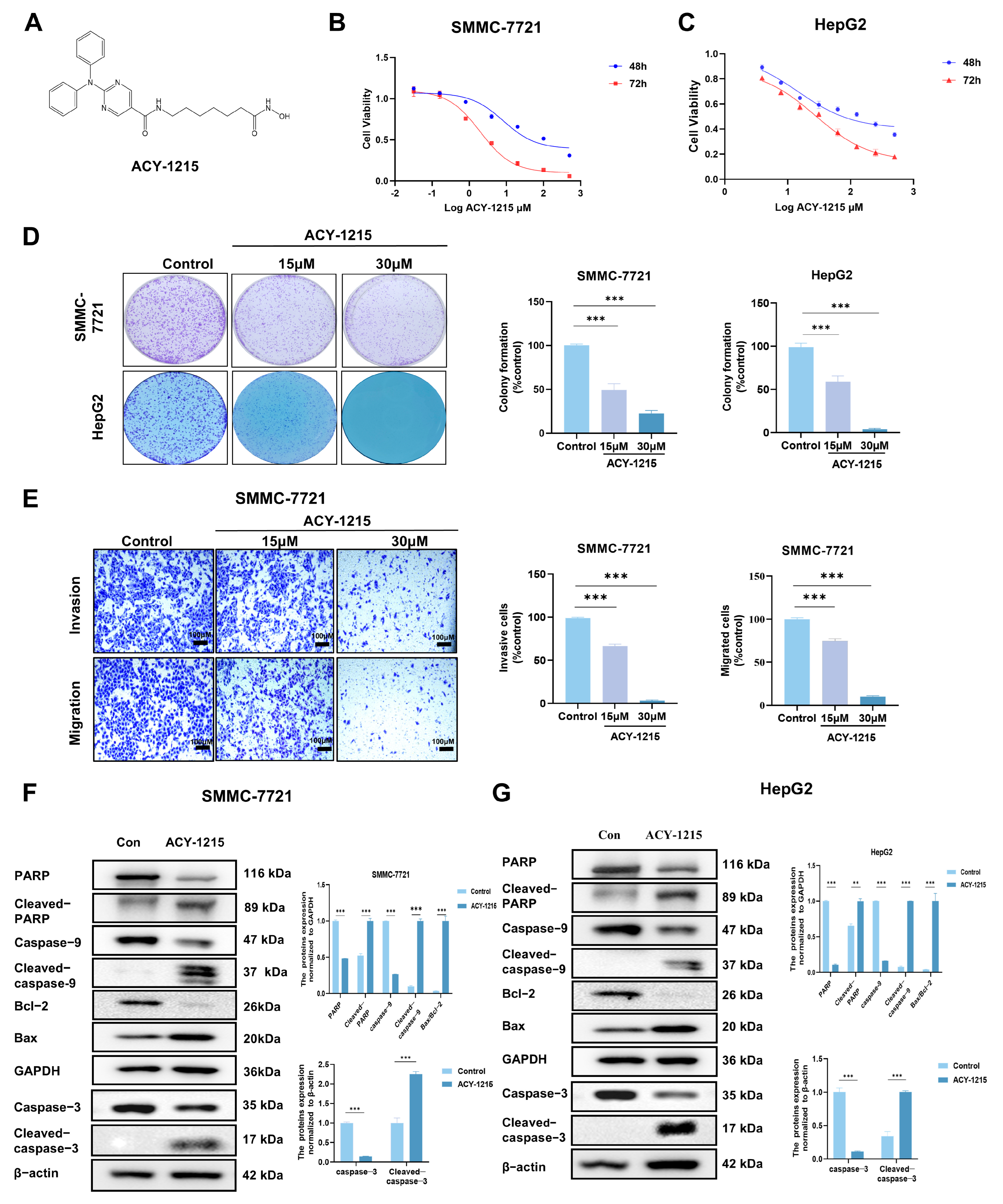
3.1. ACY-1215 Inhibits Cell Proliferation, Invasion, and Migration, and Induces Apoptosis in HCC Cells
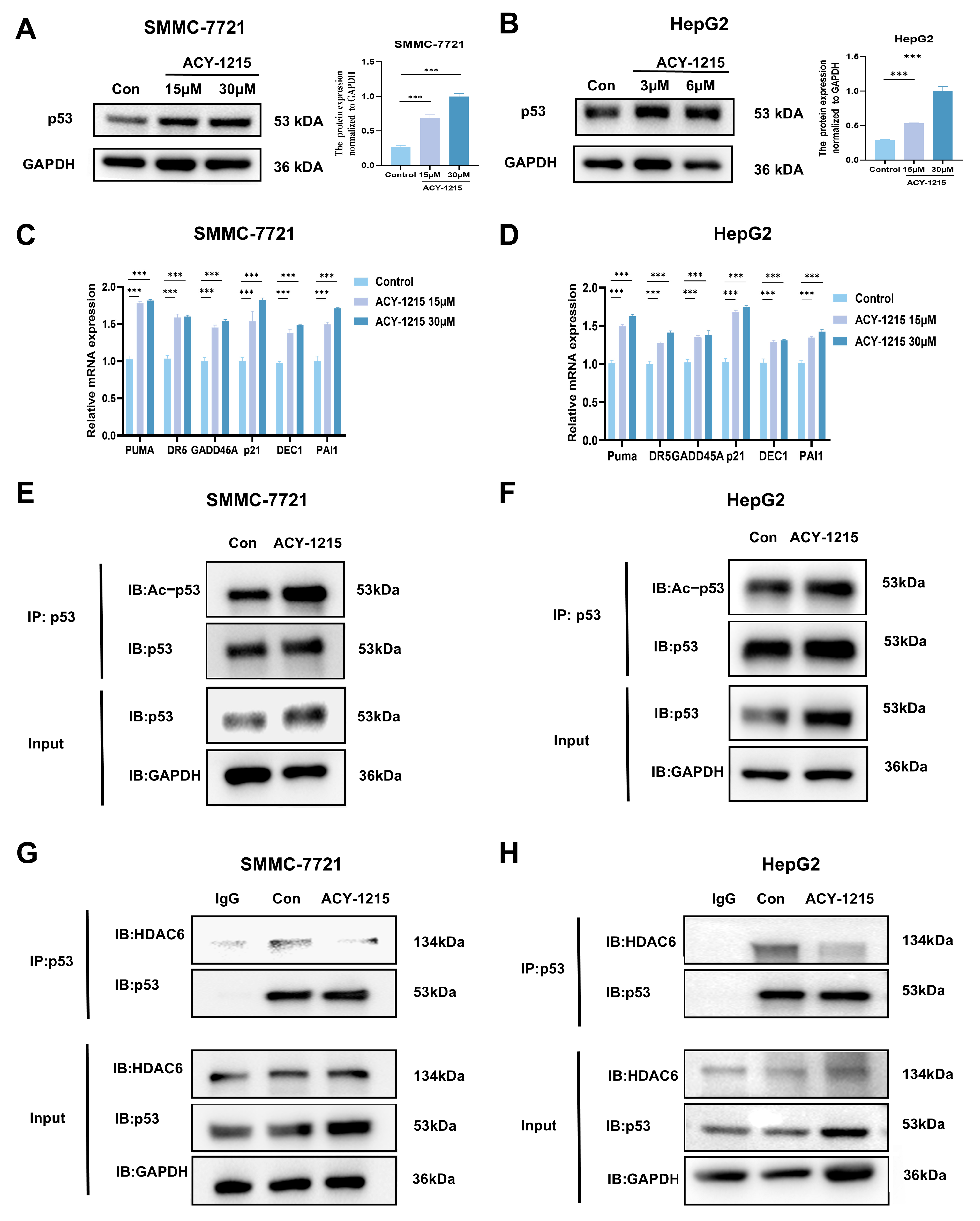
3.2. ACY-1215 Enhances the Expression Level of p53 and Inhibits HDAC6-Mediated Deacetylation to Activate the Transcription of Its Downstream Target Genes
3.3. ACY-1215 Inhibits the Ubiquitination Level of p53 and Disrupts the Interaction Between MDM2 and p53
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Bianco, C.; Jamialahmadi, O.; Pelusi, S.; Baselli, G.; Dongiovanni, P.; Zanoni, I.; Santoro, L.; Maier, S.; Liguori, A.; Meroni, M.; et al. Non-invasive stratification of hepatocellular carcinoma risk in non-alcoholic fatty liver using polygenic risk scores. J. Hepatol. 2021, 74, 775–782. [Google Scholar] [CrossRef]
- Fujiwara, N.; Kubota, N.; Crouchet, E.; Koneru, B.; Marquez, C.A.; Jajoriya, A.K.; Panda, G.; Qian, T.; Zhu, S.; Goossens, N.; et al. Molecular signatures of long-term hepatocellular carcinoma risk in nonalcoholic fatty liver disease. Sci. Transl. Med. 2022, 14, eabo4474. [Google Scholar] [CrossRef] [PubMed]
- Feng, F.; Zhao, Y. Hepatocellular Carcinoma: Prevention, Diagnosis, and Treatment. Med. Princ. Pract. 2024, 33, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, H. Heterogeneity of liver cancer and personalized therapy. Cancer Lett. 2016, 379, 191–197. [Google Scholar] [CrossRef]
- Vécsei, L. Nature Reviews Drug Discovery: Editorial article of neuroscientists from Szeged about kynurenine (IF: 33.078). Ideggyogy. Szle. 2014, 67, 70. [Google Scholar]
- Cheng, C.; Hsu, S.K.; Chen, Y.C.; Liu, W.; Shu, E.D.; Chien, C.M.; Chiu, C.C.; Chang, W.T. Burning down the house: Pyroptosis in the tumor microenvironment of hepatocellular carcinoma. Life Sci. 2024, 347, 122627. [Google Scholar] [CrossRef]
- Rahimi-Farsi, N.; Bostanian, F.; Shahbazi, T.; Shamsinejad, F.S.; Bolideei, M.; Mohseni, P.; Zangooie, A.; Boustani, F.; Shoorei, H. Novel oncogenes and tumor suppressor genes in Hepatocellular Carcinoma: Carcinogenesis, progression, and therapeutic targets. Gene 2025, 941, 149229. [Google Scholar] [CrossRef]
- Patil, M.R.; Bihari, A. A comprehensive study of p53 protein. J. Cell Biochem. 2022, 123, 1891–1937. [Google Scholar] [CrossRef]
- Soussi, T.; Wiman, K.G. Shaping Genetic Alterations in Human Cancer: The p53 Mutation Paradigm. Cancer Cell 2007, 12, 303–312. [Google Scholar] [CrossRef]
- Miyashita, T.; Krajewski, S.; Krajewska, M.; Wang, H.G.; Lin, H.K.; Liebermann, D.A.; Hoffman, B.; Reed, J.C. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene 1994, 9, 1799–1805. [Google Scholar]
- Zhao, J.; Chen, H.Q.; Yang, H.F.; Li, Y.; Chen, D.J.; Huang, Y.J.; He, L.X.; Zheng, C.F.; Wang, L.Q.; Wang, J.; et al. Epigenetic silencing of ALX4 regulates microcystin-LR induced hepatocellular carcinoma through the P53 pathway. Sci. Total Env. 2019, 683, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Bode, A.M.; Dong, Z. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 2004, 4, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Kon, N.; Gu, A.P.; Tavana, O.; Gu, W. Deciphering the acetylation code of p53 in transcription regulation and tumor suppression. Oncogene 2022, 41, 3039–3050. [Google Scholar] [CrossRef]
- Oliveira-Silva, J.M.; de Oliveira, L.S.; Tagliéri, J.V.M.; Lopes, L.B.; de Souza, C.V.E.; Batistão, H.K.A.; Castro-Gamero, A.M. HDAC6: Tumor Progression and Beyond. Curr. Cancer Drug Targets 2025. [Google Scholar] [CrossRef]
- Lee, S.I.; Seo, Y.; Oanh, H.T.; Vo, T.T.H.; Go, H.; Kim, M.H.; Lee, J.Y. HDAC6 preserves BNIP3 expression and mitochondrial integrity by deacetylating p53 at lysine 320. Biochem. Biophys. Res. Commun. 2024, 691, 149320. [Google Scholar] [CrossRef]
- Meng, X.; Franklin, D.A.; Dong, J.; Zhang, Y. MDM2-p53 pathway in hepatocellular carcinoma. Cancer Res. 2014, 74, 7161–7167. [Google Scholar] [CrossRef] [PubMed]
- Loredana, S.; Teru, H.; Andrew, L.K.; Jen-Chieh, T.; David, T.; Min, Y.; Matthew, J.; John, H.v.D.; Ralph, M.; Walter, C.O.; et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012, 119, 2579–2589. [Google Scholar] [CrossRef]
- Shifeng, F.; Mengmeng, X.; Jianglei, L.; Meihong, Y.; Siyi, W.; Liu, H.; Rong, L.; Feihong, D.; Hailing, P.; Deliang, L.; et al. HDAC6 inhibitor ACY-1215 protects from nonalcoholic fatty liver disease via inhibiting CD14/TLR4/MyD88/MAPK/NFκB signal pathway. Heliyon 2024, 10, e33740. [Google Scholar] [CrossRef]
- Wen-Bin, Z.; Hai-Yue, Z.; Fang-Zhou, J.; Lu-Wen, W.; Hong, Z.; Zuo-Jiong, G. Histone deacetylase 6 inhibitor ACY-1215 protects against experimental acute liver failure by regulating the TLR4-MAPK/NF-κB pathway. Biomed. Pharmacother. 2017, 97. [Google Scholar] [CrossRef]
- Li, W.; Yang, C.; Shi, Z.; Long, Q.; Cheng, Z.; He, S.; Dong, J.; Liu, T.; Wang, C. Caffeic Acid Phenethyl Ester Inhibits Ubiquitination and Degradation of p53 and Blocks Cervical Cancer Cell Growth. Curr. Mol. Med. 2023, 23, 960–970. [Google Scholar] [CrossRef] [PubMed]
- Ding, G.; Liu, H.; Huang, Q.; Liang, H.; Ding, Z.; Liao, Z.; Huang, G. HDAC6 promotes hepatocellular carcinoma progression by inhibiting P53 transcriptional activity. FEBS Lett. 2013, 587, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Guan, Y.S.; La, Z.; Yang, L.; He, Q.; Li, P. p53 gene in treatment of hepatic carcinoma: Status quo. World J. Gastroenterol. 2007, 13, 985–992. [Google Scholar] [CrossRef]
- Liu, Y.; Tavana, O.; Gu, W. p53 modifications: Exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 2019, 11, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Hyun-Wook, R.; Dong-Hee, S.; Dong Hoon, L.; Junjeong, C.; Gyoonhee, H.; Kang Young, L.; So Hee, K. HDAC6 deacetylates p53 at lysines 381/382 and differentially coordinates p53-induced apoptosis. Cancer Lett. 2017, 391, 162–171. [Google Scholar] [CrossRef]
- Wu, S.Y.; Chiang, C.M. Crosstalk between sumoylation and acetylation regulates p53-dependent chromatin transcription and DNA binding. Embo J. 2009, 28, 1246–1259. [Google Scholar] [CrossRef]
- Li, J.; Yu, M.; Fu, S.; Liu, D.; Tan, Y. Role of Selective Histone Deacetylase 6 Inhibitor ACY-1215 in Cancer and Other Human Diseases. Front. Pharmacol. 2022, 13, 907981. [Google Scholar] [CrossRef]
- Henningsen, K.M.; Manzini, V.; Magerhans, A.; Gerber, S.; Dobbelstein, M. MDM2-Driven Ubiquitination Rapidly Removes p53 from Its Cognate Promoters. Biomolecules 2021, 12. [Google Scholar] [CrossRef]
- Grigoreva, T.; Sagaidak, A.; Novikova, D.; Tribulovich, V. New Insights into Chemoresistance Mediated by Mdm2 Inhibitors: The Benefits of Targeted Therapy over Common Cytostatics. Biomedicines 2024, 12, 547. [Google Scholar] [CrossRef]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Ghate, N.B.; Kim, S.; Mehmood, R.; Shin, Y.; Kim, K.; An, W. VprBP/DCAF1 regulates p53 function and stability through site-specific phosphorylation. Oncogene 2023, 42, 1405–1416. [Google Scholar] [CrossRef] [PubMed]
- Santo, L.; Hideshima, T.; Kung, A.L.; Jarpe, M.; Cirstea, D.; Patel, K.; Pozzi, S.; Tseng, J.C.; Rodig, S.J.; Bradner, J.; et al. Selective Inhibition of HDAC6 with a New Prototype Inhibitor (ACY-1215) Overcomes Bortezomib Resistance In Multiple Myeloma (MM). Blood 2010, 116, 2997. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, Y.; Du, Y.; Xu, Y.; Zhu, Z.; Hu, Y.; Xu, L.; Yang, K.; Chen, T.; Shi, Y.; Wang, C.; et al. Mechanistic Insights into the Anti-Hepatocellular Carcinoma Effects of ACY-1215: p53 Acetylation and Ubiquitination Regulation. Curr. Issues Mol. Biol. 2025, 47, 338. https://doi.org/10.3390/cimb47050338
Yin Y, Du Y, Xu Y, Zhu Z, Hu Y, Xu L, Yang K, Chen T, Shi Y, Wang C, et al. Mechanistic Insights into the Anti-Hepatocellular Carcinoma Effects of ACY-1215: p53 Acetylation and Ubiquitination Regulation. Current Issues in Molecular Biology. 2025; 47(5):338. https://doi.org/10.3390/cimb47050338
Chicago/Turabian StyleYin, Yi, Yutong Du, Yiting Xu, Zhuan Zhu, Yu Hu, Lingling Xu, Kunming Yang, Tian Chen, Yuyang Shi, Chengcheng Wang, and et al. 2025. "Mechanistic Insights into the Anti-Hepatocellular Carcinoma Effects of ACY-1215: p53 Acetylation and Ubiquitination Regulation" Current Issues in Molecular Biology 47, no. 5: 338. https://doi.org/10.3390/cimb47050338
APA StyleYin, Y., Du, Y., Xu, Y., Zhu, Z., Hu, Y., Xu, L., Yang, K., Chen, T., Shi, Y., Wang, C., & Zhang, Y. (2025). Mechanistic Insights into the Anti-Hepatocellular Carcinoma Effects of ACY-1215: p53 Acetylation and Ubiquitination Regulation. Current Issues in Molecular Biology, 47(5), 338. https://doi.org/10.3390/cimb47050338