

Protein Misfolding and Aggregation as a Mechanistic Link Between Chronic Pain and Neurodegenerative Diseases

 , ,
, ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
3. Protein Misfolding and Aggregation: Molecular Mechanisms
3.1. Protein Folding and Molecular Chaperones
3.2. Proteostasis and Protein Quality Control System
3.3. Unfolded Protein Response (UPR) and ER Stress
3.4. Key Disease-Associated Proteins and Prion-like Propagation
3.5. Neuroinflammation and Oxidative Stress in Protein Misfolding
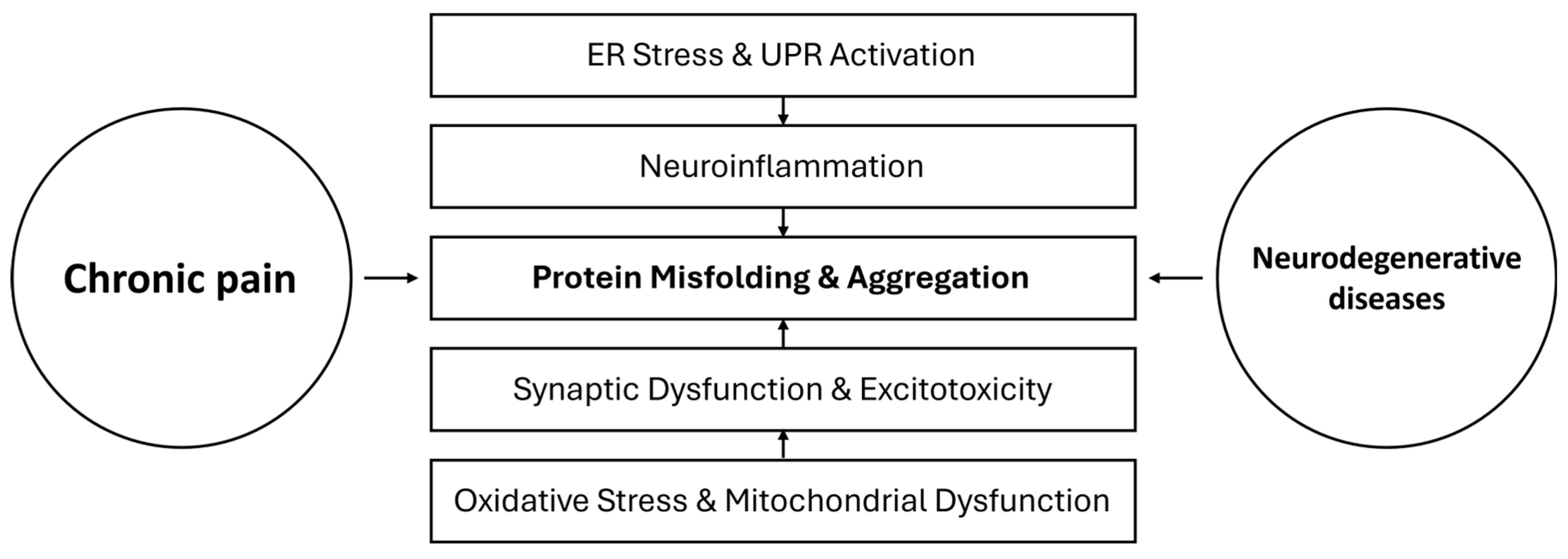
4. Chronic Pain and Protein Misfolding: Mechanistic Insights
4.1. Evidence of Protein Aggregation in Chronic Pain Conditions
4.2. Proteostasis Dysfunction and Neuronal Stress in Chronic Pain
4.3. Chronic Pain as a Risk Factor for Protein Misfolding
5. Neurodegeneration and Chronic Pain: Common Pathways
5.1. Shared Neuroinflammatory and Neuroimmune Mechanisms
5.2. Dysregulation of Protein Homeostasis in Pain and NDs
5.3. Prion-like Propagation of Protein Aggregates in Pain and NDs
5.4. Impact of Chronic Pain on Neurodegenerative Progression
6. Potential Therapeutic Implications
6.1. Chaperone Inhibitors
6.2. Modulation of Autophagy and Proteasomal Degradation
6.3. Glial Inhibitors
6.4. Antioxidant Therapy
6.5. Future Directions
7. Conclusions
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Treede, R.D.; Rief, W.; Barke, A.; Aziz, Q.; Bennett, M.I.; Benoliel, R.; Cohen, M.; Evers, S.; Finnerup, N.B.; First, M.B.; et al. A classification of chronic pain for ICD-11. Pain 2015, 156, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, D.S.; McGee, S.J. Pain as a global public health priority. BMC Public Health 2011, 11, 770. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.; Ewing, J.; Garrett, A.; Harrison, E.K.; Lwin, K.K.; Wheeler, D.W. The nature and prevalence of chronic pain in homeless persons: An observational study. F1000Research 2013, 2, 164. [Google Scholar] [CrossRef] [PubMed]
- Dydyk, A.M.; Conermann, T. Chronic Pain. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- GBD 2021 Low Back Pain Collaborators. Global, regional, and national burden of low back pain, 1990–2020, its attributable risk factors, and projections to 2050: A systematic analysis of the Global Burden of Disease Study 2021. Lancet Rheumatol. 2023, 5, e316–e329. [Google Scholar] [CrossRef]
- De La Rosa, J.S.; Brady, B.R.; Ibrahim, M.M.; Herder, K.E.; Wallace, J.S.; Padilla, A.R.; Vanderah, T.W. Co-occurrence of chronic pain and anxiety/depression symptoms in U.S. adults: Prevalence, functional impacts, and opportunities. Pain 2024, 165, 666–673. [Google Scholar] [CrossRef]
- Gaskin, D.J.; Richard, P. The economic costs of pain in the United States. J. Pain 2012, 13, 715–724. [Google Scholar] [CrossRef]
- De Marchi, F.; Munitic, I.; Vidatic, L.; Papić, E.; Rački, V.; Nimac, J.; Jurak, I.; Novotni, G.; Rogelj, B.; Vuletic, V.; et al. Overlapping Neuroimmune Mechanisms and Therapeutic Targets in Neurodegenerative Disorders. Biomedicines 2023, 11, 2793. [Google Scholar] [CrossRef]
- Niedowicz, D.M.; Nelson, P.T.; Murphy, M.P. Alzheimer’s disease: Pathological mechanisms and recent insights. Curr. Neuropharmacol. 2011, 9, 674–684. [Google Scholar] [CrossRef]
- Dong-Chen, X.; Yong, C.; Yang, X.; Chen-Yu, S.; Li-Hua, P. Signaling pathways in Parkinson’s disease: Molecular mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 73. [Google Scholar] [CrossRef]
- Le Gall, L.; Anakor, E.; Connolly, O.; Vijayakumar, U.G.; Duddy, W.J.; Duguez, S. Molecular and Cellular Mechanisms Affected in ALS. J. Pers. Med. 2020, 10, 101. [Google Scholar] [CrossRef]
- Polymenidou, M.; Cleveland, D.W. Prion-like spread of protein aggregates in neurodegeneration. J. Exp. Med. 2012, 209, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Fisher, D.W.; Yu, T.; Dong, H. The link between chronic pain and Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 204. [Google Scholar] [CrossRef] [PubMed]
- Handy, C.R.; Krudy, C.; Boulis, N.; Federici, T. Pain in amyotrophic lateral sclerosis: A neglected aspect of disease. Neurol. Res. Int. 2011, 2011, 403808. [Google Scholar] [CrossRef]
- Innes, K.E.; Sambamoorthi, U. The potential contribution of chronic pain and common chronic pain conditions to subsequent cognitive decline, new onset cognitive impairment, and incident dementia: A systematic review and conceptual model for future research. J. Alzheimer’s Dis. 2020, 78, 1177–1195. [Google Scholar] [CrossRef]
- Whitlock, E.L.; Diaz-Ramirez, L.G.; Glymour, M.M.; Boscardin, W.J.; Covinsky, K.E.; Smith, A.K. Association between persistent pain and memory decline and dementia in a longitudinal cohort of elders. J. Am. Med. Assoc. Intern. Med. 2017, 177, 1146–1153. [Google Scholar] [CrossRef]
- Guerreiro, S.R.; Guimarães, M.R.; Silva, J.M.; Dioli, C.; Vamvaka-Iakovou, A.; Sousa, R.; Gomes, P.; Megalokonomou, A.; Campos-Marques, C.; Cunha, A.M.; et al. Chronic pain causes Tau-mediated hippocampal pathology and memory deficits. Mol. Psychiatry 2022, 27, 4385–4393. [Google Scholar] [CrossRef] [PubMed]
- Eratne, D.; Kang, M.J.Y.; Lewis, C.; Dang, C.; Malpas, C.B.; Keem, M.; Grewal, J.; Marinov, V.; Coe, A.; Kaylor-Hughes, C.; et al. Plasma and CSF neurofilament light chain distinguish neurodegenerative from primary psychiatric conditions in a clinical setting. Alzheimers Dement. 2024, 20, 7989–8001. [Google Scholar] [CrossRef]
- Loggia, M.L.; Chonde, D.B.; Akeju, O.; Arabasz, G.; Catana, C.; Edwards, R.R.; Hill, E.; Hsu, S.; Izquierdo-Garcia, D.; Ji, R.R.; et al. Evidence for brain glial activation in chronic pain patients. Brain 2015, 138 Pt 3, 604–615. [Google Scholar] [CrossRef]
- Marques, A.; Brefel-Courbon, C. Chronic pain in Parkinson’s disease: Clinical and pathophysiological aspects. Rev. Neurol. 2021, 177, 394–399. [Google Scholar] [CrossRef]
- Soto, C.; Estrada, L.D. Protein misfolding and neurodegeneration. Arch. Neurol. 2008, 65, 184–189. [Google Scholar] [CrossRef]
- Díaz-Villanueva, J.F.; Díaz-Molina, R.; García-González, V. Protein folding and mechanisms of proteostasis. Int. J. Mol. Sci. 2015, 16, 17193–17230. [Google Scholar] [CrossRef]
- Saibil, H. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Radons, J. The human HSP70 family of chaperones: Where do we stand? Cell Stress Chaperones 2016, 21, 379–404. [Google Scholar] [CrossRef] [PubMed]
- Hoter, A.; El-Sabban, M.E.; Naim, H.Y. The HSP90 family: Structure, regulation, function, and implications in health and disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef] [PubMed]
- Shorter, J. The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS ONE 2011, 6, e26319. [Google Scholar] [CrossRef]
- Hu, C.; Yang, J.; Qi, Z.; Wu, H.; Wang, B.; Zou, F.; Mei, H.; Liu, J.; Wang, W.; Liu, Q. Heat shock proteins: Biological functions, pathological roles, and therapeutic opportunities. MedComm 2022, 3, e161. [Google Scholar] [CrossRef]
- Lindquist, S.L.; Kelly, J.W. Chemical and biological approaches for adapting proteostasis to ameliorate protein misfolding and aggregation diseases: Progress and prognosis. Cold Spring Harb. Perspect. Biol. 2011, 3, a004507. [Google Scholar] [CrossRef]
- Li, Y.; Li, S.; Wu, H. Ubiquitination-proteasome system (UPS) and autophagy two main protein degradation machineries in response to cell stress. Cells 2022, 11, 851. [Google Scholar] [CrossRef]
- Kandel, R.; Jung, J.; Neal, S. Proteotoxic stress and the ubiquitin proteasome system. Semin. Cell Dev. Biol. 2024, 156, 107–120. [Google Scholar] [CrossRef]
- Rosser, M.F.; Washburn, E.; Muchowski, P.J.; Patterson, C.; Cyr, D.M. Chaperone functions of the E3 ubiquitin ligase CHIP. J. Biol. Chem. 2007, 282, 22267–22277. [Google Scholar] [CrossRef]
- Rao, G.; Croft, B.; Teng, C.; Awasthi, V. Ubiquitin-Proteasome System in Neurodegenerative Disorders. J. Drug Metab. Toxicol. 2015, 6, 187. [Google Scholar] [CrossRef] [PubMed]
- Carozzi, V.A.; Renn, C.L.; Bardini, M.; Fazio, G.; Chiorazzi, A.; Meregalli, C.; Oggioni, N.; Shanks, K.; Quartu, M.; Serra, M.P.; et al. Bortezomib-induced painful peripheral neuropathy: An electrophysiological, behavioral, morphological, and mechanistic study in the mouse. PLoS ONE 2013, 8, e72995. [Google Scholar] [CrossRef]
- Khandia, R.; Dadar, M.; Munjal, A.; Dhama, K.; Karthik, K.; Tiwari, R.; Yatoo, M.I.; Iqbal, H.M.N.; Singh, K.P.; Joshi, S.K.; et al. A Comprehensive Review of Autophagy and Its Various Roles in Infectious, Non-Infectious, and Lifestyle Diseases: Current Knowledge and Prospects for Disease Prevention, Novel Drug Design, and Therapy. Cells 2019, 8, 674. [Google Scholar] [CrossRef]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [CrossRef]
- Metcalf, D.J.; García-Arencibia, M.; Hochfeld, W.E.; Rubinsztein, D.C. Autophagy and misfolded proteins in neurodegeneration. Exp. Neurol. 2012, 238, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Kan, Y.Y.; Chang, Y.S.; Liao, W.C.; Chao, T.N.; Hsieh, Y.L. Roles of Neuronal Protein Kinase Cε on Endoplasmic Reticulum Stress and Autophagic Formation in Diabetic Neuropathy. Mol. Neurobiol. 2024, 61, 2481–2495. [Google Scholar] [CrossRef] [PubMed]
- Zapun, A.; Jakob, C.A.; Thomas, D.Y.; Bergeron, J.J. Protein folding in a specialized compartment: The endoplasmic reticulum. Structure 1999, 7, R173–R182. [Google Scholar] [CrossRef]
- Nakatsukasa, K.; Brodsky, J.L. The recognition and retrotranslocation of misfolded proteins from the endoplasmic reticulum. Traffic 2008, 9, 861–870. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 2007, 18, 716–731. [Google Scholar] [CrossRef]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef]
- Beilankouhi, E.A.V.; Sajadi, M.A.; Alipourfard, I.; Hassani, P.; Valilo, M.; Safaralizadeh, R. Role of the ER-induced UPR pathway, apoptosis, and autophagy in colorectal cancer. Pathol. Res. Pract. 2023, 248, 154706. [Google Scholar] [CrossRef] [PubMed]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Louessard, M.; Bardou, I.; Lemarchand, E.; Thiebaut, A.M.; Parcq, J.; Leprince, J.; Terrisse, A.; Carraro, V.; Fafournoux, P.; Bruhat, A.; et al. Activation of cell surface GRP78 decreases endoplasmic reticulum stress and neuronal death. Cell Death Differ. 2017, 24, 1518–1529. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Li, W.; Jin, K.; Luo, J.; Xu, W.; Wu, Y.; Zhou, J.; Wang, Y.; Xu, R.; Jiao, L.; Wang, T.; et al. NF-κB and its crosstalk with endoplasmic reticulum stress in atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 988266. [Google Scholar] [CrossRef]
- Owen, C.R.; Kumar, R.; Zhang, P.; McGrath, B.C.; Cavener, D.R.; Krause, G.S. PERK is responsible for the increased phosphorylation of eIF2alpha and the severe inhibition of protein synthesis after transient global brain ischemia. J. Neurochem. 2005, 94, 1235–1242. [Google Scholar] [CrossRef]
- Blais, J.D.; Filipenko, V.; Bi, M.; Harding, H.P.; Ron, D.; Koumenis, C.; Wouters, B.G.; Bell, J.C. Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol. Cell Biol. 2004, 24, 7469–7482. [Google Scholar] [CrossRef]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2019, 9, 3083. [Google Scholar] [CrossRef]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- McKimpson, W.M.; Kitsis, R.N. A New Role for the ER Unfolded Protein Response Mediator ATF6: Induction of a Generalized Antioxidant Program. Circ. Res. 2017, 120, 759–761. [Google Scholar] [CrossRef]
- Hasan, S.A.M.; James, A.W.; Fazili, F.M.; Tarabishi, S.; Sheikh, N.M.; Shah, Z.A. Endoplasmic Reticulum Stress in Neurodegenerative Diseases. J. Dementia Alzheimer’s Dis. 2024, 1, 87–97. [Google Scholar] [CrossRef]
- Hoozemans, J.J.; van Haastert, E.S.; Nijholt, D.A.; Rozemuller, A.J.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am. J. Pathol. 2009, 174, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Stojkovska, I.; Wani, W.Y.; Zunke, F.; Belur, N.R.; Pavlenko, E.A.; Mwenda, N.; Sharma, K.; Francelle, L.; Mazzulli, J.R. Rescue of α-synuclein aggregation in Parkinson’s patient neurons by synergistic enhancement of ER proteostasis and protein trafficking. Neuron 2022, 110, 436–451.e11. [Google Scholar] [CrossRef] [PubMed]
- Kawanaka, R.; Jin, H.; Aoe, T. Unraveling the Connection: Pain and Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2024, 25, 4995. [Google Scholar] [CrossRef]
- Chow, V.W.; Mattson, M.P.; Wong, P.C.; Gleichmann, M. An overview of APP processing enzymes and products. Neuromol. Med. 2010, 12, 1–12. [Google Scholar] [CrossRef]
- Tolar, M.; Hey, J.; Power, A.; Abushakra, S. Neurotoxic Soluble Amyloid Oligomers Drive Alzheimer’s Pathogenesis and Represent a Clinically Validated Target for Slowing Disease Progression. Int. J. Mol. Sci. 2021, 22, 6355. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.X.; Grundke-Iqbal, I. Tau in Alzheimer Disease and Related Tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef]
- Zhang, H.; Wei, W.; Zhao, M.; Ma, L.; Jiang, X.; Pei, H.; Cao, Y.; Li, H. Interaction Between Aβ and Tau in the Pathogenesis of Alzheimer’s Disease. Int. J. Biol. Sci. 2021, 17, 2181–2192. [Google Scholar] [CrossRef]
- Scialò, C.; De Cecco, E.; Manganotti, P.; Legname, G. Prion and Prion-Like Protein Strains: Deciphering the Molecular Basis of Heterogeneity in Neurodegeneration. Viruses 2019, 11, 261. [Google Scholar] [CrossRef]
- Bolmont, T.; Clavaguera, F.; Meyer-Luehmann, M.; Herzig, M.C.; Radde, R.; Staufenbiel, M.; Lewis, J.; Hutton, M.; Tolnay, M.; Jucker, M. Induction of Tau Pathology by Intracerebral Infusion of Amyloid-Beta-Containing Brain Extract and by Amyloid-Beta Deposition in APP x Tau Transgenic Mice. Am. J. Pathol. 2007, 171, 2012–2020. [Google Scholar] [CrossRef]
- Gao, V.; Briano, J.A.; Komer, L.E.; Burré, J. Functional and Pathological Effects of α-Synuclein on Synaptic SNARE Complexes. J. Mol. Biol. 2023, 435, 167714. [Google Scholar] [CrossRef]
- Stefanis, L. α-Synuclein in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [PubMed]
- Steiner, J.A.; Angot, E.; Brundin, P. A Deadly Spread: Cellular Mechanisms of α-Synuclein Transfer. Cell Death Differ. 2011, 18, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Seidel, K.; Mahlke, J.; Siswanto, S.; Krüger, R.; Heinsen, H.; Auburger, G.; Bouzrou, M.; Grinberg, L.T.; Wicht, H.; Korf, H.W.; et al. The Brainstem Pathologies of Parkinson’s Disease and Dementia with Lewy Bodies. Brain Pathol. 2015, 25, 121–135. [Google Scholar] [CrossRef]
- Buhidma, Y.; Hobbs, C.; Malcangio, M.; Duty, S. Periaqueductal Grey and Spinal Cord Pathology Contribute to Pain in Parkinson’s Disease. NPJ Park. Dis. 2023, 9, 69. [Google Scholar] [CrossRef]
- Suk, T.R.; Rousseaux, M.W.C. The Role of TDP-43 Mislocalization in Amyotrophic Lateral Sclerosis. Mol. Neurodegener. 2020, 15, 45. [Google Scholar] [CrossRef]
- Prudencio, M.; Borchelt, D.R. Superoxide Dismutase 1 Encoding Mutations Linked to ALS Adopts a Spectrum of Misfolded States. Mol. Neurodegener. 2011, 6, 77. [Google Scholar] [CrossRef]
- Nolan, M.; Talbot, K.; Ansorge, O. Pathogenesis of FUS-Associated ALS and FTD: Insights from Rodent Models. Acta Neuropathol. Commun. 2016, 4, 99. [Google Scholar] [CrossRef]
- Nordström, U.; Lang, L.; Ekhtiari Bidhendi, E.; Zetterström, P.; Oliveberg, M.; Danielsson, J.; Andersen, P.M.; Marklund, S.L. Mutant SOD1 Aggregates Formed in Vitro and in Cultured Cells Are Polymorphic and Differ from Those Arising in the CNS. J. Neurochem. 2023, 164, 77–93. [Google Scholar] [CrossRef]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of Neuroinflammation in Neurodegeneration Development. Signal Transduct. Target. Ther. 2023, 8, 267. [Google Scholar] [CrossRef]
- Wendimu, M.Y.; Hooks, S.B. Microglia Phenotypes in Aging and Neurodegenerative Diseases. Cells 2022, 11, 2091. [Google Scholar] [CrossRef] [PubMed]
- Merlo, S.; Spampinato, S.F.; Caruso, G.I.; Sortino, M.A. The Ambiguous Role of Microglia in Aβ Toxicity: Chances for Therapeutic Intervention. Curr. Neuropharmacol. 2020, 18, 446–455. [Google Scholar] [CrossRef]
- Jurcau, A. Insights into the Pathogenesis of Neurodegenerative Diseases: Focus on Mitochondrial Dysfunction and Oxidative Stress. Int. J. Mol. Sci. 2021, 22, 11847. [Google Scholar] [CrossRef]
- Dias, V.; Junn, E.; Mouradian, M.M. The Role of Oxidative Stress in Parkinson’s Disease. J. Parkinsons Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef]
- Lévy, E.; El Banna, N.; Baïlle, D.; Heneman-Masurel, A.; Truchet, S.; Rezaei, H.; Huang, M.E.; Béringue, V.; Martin, D.; Vernis, L. Causative Links between Protein Aggregation and Oxidative Stress: A Review. Int. J. Mol. Sci. 2019, 20, 3896. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.X.; Yan, S.D. Amyloid-Beta-Induced Mitochondrial Dysfunction. J. Alzheimer’s Dis. 2007, 12, 177–184. [Google Scholar] [CrossRef]
- Reeve, A.K.; Ludtmann, M.H.; Angelova, P.R.; Simcox, E.M.; Horrocks, M.H.; Klenerman, D.; Gandhi, S.; Turnbull, D.M.; Abramov, A.Y. Aggregated α-Synuclein and Complex I Deficiency: Exploration of Their Relationship in Differentiated Neurons. Cell Death Dis. 2015, 6, e1820. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.R.; Berta, T.; Nedergaard, M. Glia and Pain: Is Chronic Pain a Gliopathy? Pain 2013, 154, S10–S28. [Google Scholar] [CrossRef]
- Lee, H.G.; Wheeler, M.A.; Quintana, F.J. Function and Therapeutic Value of Astrocytes in Neurological Diseases. Nat. Rev. Drug Discov. 2022, 21, 339–358. [Google Scholar] [CrossRef]
- Arnold, F.J.; Nguyen, A.D.; Bedlack, R.S.; Bennett, C.L.; La Spada, A.R. Intercellular Transmission of Pathogenic Proteins in ALS: Exploring the Pathogenic Wave. Neurobiol. Dis. 2023, 184, 106218. [Google Scholar] [CrossRef]
- Simonetti, M.; Mauceri, D. Cellular and Molecular Mechanisms Underlying Pain Chronicity. Cells 2023, 12, 1126. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Zhang, Q.; Wang, L.; Li, M.; Li, T.; Wang, Y.; Cao, Z.; Jiang, X.; Luo, P. Progress in the Mechanisms of Pain Associated with Neurodegenerative Diseases. Ageing Res. Rev. 2024, 102, 102579. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, S.I.; Itokazu, T.; Nishibe, M.; Yamashita, T. Neuroplasticity Related to Chronic Pain and Its Modulation by Microglia. Inflamm. Regen. 2022, 42, 15. [Google Scholar] [CrossRef] [PubMed]
- Niederberger, E. Novel Insights into Molecular Mechanisms of Chronic Pain. Cells 2020, 9, 2220. [Google Scholar] [CrossRef]
- Cuomo, A.; Crispo, A.; Truini, A.; Natoli, S.; Zanetti, O.; Barone, P.; Cascella, M. Toward More Focused Multimodal and Multidisciplinary Approaches for Pain Management in Parkinson’s Disease. J. Pain Res. 2019, 12, 2201–2209. [Google Scholar] [CrossRef]
- Nardone, R.; Holler, Y.; Brigo, F.; Versace, V.; Sebastianelli, L.; Florea, C.; Schwenker, K.; Golaszewski, S.; Saltuari, L.; Trinka, E. Spinal Cord Involvement in Lewy Body-Related Alpha-Synucleinopathies. J. Spinal Cord Med. 2020, 43, 832–845. [Google Scholar] [CrossRef]
- Nardelli, D.; Gambioli, F.; De Bartolo, M.I.; Mancinelli, R.; Biagioni, F.; Carotti, S.; Falato, E.; Leodori, G.; Puglisi-Allegra, S.; Vivacqua, G.; et al. Pain in Parkinson’s Disease: A Neuroanatomy-Based Approach. Brain Commun. 2024, 6, fcae210. [Google Scholar] [CrossRef]
- Chesselet, M.F. In Vivo Alpha-Synuclein Overexpression in Rodents: A Useful Model of Parkinson’s Disease? Exp. Neurol. 2008, 209, 22–27. [Google Scholar] [CrossRef]
- McBenedict, B.; Goh, K.S.; Yau, R.C.C.; Elamin, S.; Yusuf, W.H.; Verly, G.; Thomas, A.; Alphonse, B.; Ouabicha, K.; Valentim, G.; et al. Neuropathic Pain Secondary to Multiple Sclerosis: A Narrative Review. Cureus 2024, 16, e61587. [Google Scholar] [CrossRef]
- Yousuf, M.S.; Samtleben, S.; Lamothe, S.M.; Friedman, T.N.; Catuneanu, A.; Thorburn, K.; Desai, M.; Tenorio, G.; Schenk, G.J.; Ballanyi, K.; et al. Endoplasmic Reticulum Stress in the Dorsal Root Ganglia Regulates Large-Conductance Potassium Channels and Contributes to Pain in a Model of Multiple Sclerosis. FASEB J. 2020, 34, 12577–12598. [Google Scholar] [CrossRef]
- Mhaille, A.N.; McQuaid, S.; Windebank, A.; Cunnea, P.; McMahon, J.; Samali, A.; FitzGerald, U. Increased Expression of Endoplasmic Reticulum Stress-Related Signaling Pathway Molecules in Multiple Sclerosis Lesions. J. Neuropathol. Exp. Neurol. 2008, 67, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Yi, M.H.; Shin, N.; Baek, H.; Kim, S.; Kim, E.; Kwon, K.; Lee, S.; Kim, H.W.; Chul Bae, Y.; et al. Endoplasmic Reticulum Stress Impairment in the Spinal Dorsal Horn of a Neuropathic Pain Model. Sci. Rep. 2015, 5, 11555. [Google Scholar] [CrossRef]
- Dong, Y.; Yang, S.; Fu, B.; Liu, F.; Zhou, S.; Ding, H.; Ma, W. Mechanism of Tauroursodeoxycholic Acid-Mediated Neuronal Protection After Acute Spinal Cord Injury Through AKT Signaling Pathway in Rats. Int. J. Clin. Exp. Pathol. 2020, 13, 2218–2227. [Google Scholar]
- Wu, S.; García-Rama, C.; Romero-Ramírez, L.; de Munter, J.P.J.M.; Wolters, E.C.; Kramer, B.W.; Mey, J. Tauroursodeoxycholic Acid Reduces Neuroinflammation but Does Not Support Long-Term Functional Recovery of Rats with Spinal Cord Injury. Biomedicines 2022, 10, 1501. [Google Scholar] [CrossRef] [PubMed]
- Askari, S.; Javadpour, P.; Rashidi, F.S.; Dargahi, L.; Kashfi, K.; Ghasemi, R. Behavioral and Molecular Effects of Thapsigargin-Induced Brain ER-Stress: Encompassing Inflammation, MAPK, and Insulin Signaling Pathway. Life 2022, 12, 1374. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.X.; Yazdi, C.; Islam, R.K.; Anwar, A.I.; Alvares-Amado, A.; Townsend, H.; Allen, K.E.; Plakotaris, E.; Hirsch, J.D.; Rieger, R.G.; et al. Diabetic Neuropathy: A Guide to Pain Management. Curr. Pain Headache Rep. 2024, 28, 1067–1072. [Google Scholar] [CrossRef]
- Cameron, N.E. Role of Endoplasmic Reticulum Stress in Diabetic Neuropathy. Diabetes 2013, 62, 696–697. [Google Scholar] [CrossRef]
- Barati, M.T.; Powell, D.W.; Kechavarzi, B.D.; Isaacs, S.M.; Zheng, S.; Epstein, P.N.; Cai, L.; Coventry, S.; Rane, M.J.; Klein, J.B. Differential Expression of Endoplasmic Reticulum Stress-Response Proteins in Different Renal Tubule Subtypes of OVE26 Diabetic Mice. Cell Stress Chaperones 2016, 21, 155–166. [Google Scholar] [CrossRef]
- Inceoglu, B.; Bettaieb, A.; Trindade da Silva, C.A.; Lee, K.S.; Haj, F.G.; Hammock, B.D. Endoplasmic Reticulum Stress in the Peripheral Nervous System is a Significant Driver of Neuropathic Pain. Proc. Natl. Acad. Sci. USA 2015, 112, 9082–9087. [Google Scholar] [CrossRef]
- Salaffi, F.; Giacobazzi, G.; Di Carlo, M. Chronic Pain in Inflammatory Arthritis: Mechanisms, Metrology, and Emerging Targets—A Focus on the JAK-STAT Pathway. Pain Res. Manag. 2018, 2018, 8564215. [Google Scholar] [CrossRef]
- Park, Y.J.; Yoo, S.A.; Kim, W.U. Role of Endoplasmic Reticulum Stress in Rheumatoid Arthritis Pathogenesis. J. Korean Med. Sci. 2014, 29, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Sadlon, A.; Takousis, P.; Ankli, B.; Alexopoulos, P.; Perneczky, R.; Alzheimer’s Disease Neuroimaging Initiative. Association of Chronic Pain with Biomarkers of Neurodegeneration, Microglial Activation, and Inflammation in Cerebrospinal Fluid and Impaired Cognitive Function. Ann. Neurol. 2024, 95, 195–206. [Google Scholar] [CrossRef]
- Bell, T.R.; Franz, C.E.; Thomas, K.R.; Williams, M.E.; Eyler, L.T.; Lerman, I.; Fennema-Notestine, C.; Puckett, O.K.; Dorros, S.M.; Panizzon, M.S.; et al. Elevated C-Reactive Protein in Older Men with Chronic Pain: Association with Plasma Amyloid Levels and Hippocampal Volume. J. Gerontol. A Biol. Sci. Med. Sci. 2024, 79, glae206. [Google Scholar] [CrossRef]
- Mezhov, V.; Guymer, E.; Littlejohn, G. Central Sensitivity and Fibromyalgia. Intern. Med. J. 2021, 51, 1990–1998. [Google Scholar] [CrossRef]
- Cordero, M.D.; de Miguel, M.; Carmona-López, I.; Bonal, P.; Campa, F.; Moreno-Fernández, A.M. Oxidative Stress and Mitochondrial Dysfunction in Fibromyalgia. Neuro Endocrinol. Lett. 2010, 31, 169–173. [Google Scholar]
- Thi Nguy, B.H.; Liu, W.T.; Chang, Y.T.; Lin, C.P.; Kang, J.H. Elevated Tau and β-Amyloid in the Serum of Fibromyalgia Patients. CNS Spectr. 2022, 27, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, D.S.; Forsberg, A.; Sandström, A.; Bergan, C.; Kadetoff, D.; Protsenko, E.; Lampa, J.; Lee, Y.C.; Höglund, C.O.; Catana, C.; et al. Brain Glial Activation in Fibromyalgia—A Multi-Site Positron Emission Tomography Investigation. Brain Behav. Immun. 2019, 75, 72–83. [Google Scholar] [CrossRef]
- Milligan, E.D.; Watkins, L.R. Pathological and Protective Roles of Glia in Chronic Pain. Nat. Rev. Neurosci. 2009, 10, 23–36. [Google Scholar] [CrossRef]
- Xiong, H.Y.; Hendrix, J.; Schabrun, S.; Wyns, A.; Campenhout, J.V.; Nijs, J.; Polli, A. The Role of the Brain-Derived Neurotrophic Factor in Chronic Pain: Links to Central Sensitization and Neuroinflammation. Biomolecules 2024, 14, 71. [Google Scholar] [CrossRef]
- Helwig, M.; Ulusoy, A.; Rollar, A.; O’Sullivan, S.A.; Lee, S.S.L.; Aboutalebi, H.; Pinto-Costa, R.; Jevans, B.; Klinkenberg, M.; Di Monte, D.A. Neuronal Hyperactivity-Induced Oxidant Stress Promotes In Vivo α-Synuclein Brain Spreading. Sci. Adv. 2022, 8, eabn0356. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative Stress, Mitochondrial Damage and Neurodegenerative Diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Silva Santos Ribeiro, P.; Willemen, H.L.D.M.; Eijkelkamp, N. Mitochondria and Sensory Processing in Inflammatory and Neuropathic Pain. Front. Pain Res. 2022, 3, 1013577. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wu, M.; Wang, M.; Zhang, X.; Zhu, J. The Regulatory Role of Endoplasmic Reticulum Chaperone Proteins in Neurodevelopment. Front. Neurosci. 2022, 16, 1032607. [Google Scholar] [CrossRef]
- Pontisso, I.; Ornelas-Guevara, R.; Chevet, E.; Combettes, L.; Dupont, G. Gradual ER Calcium Depletion Induces a Progressive and Reversible UPR Signaling. PNAS Nexus 2024, 3, pgae229. [Google Scholar] [CrossRef] [PubMed]
- Yousuf, M.S.; Maguire, A.D.; Simmen, T.; Kerr, B.J. Endoplasmic Reticulum-Mitochondria Interplay in Chronic Pain: The Calcium Connection. Mol. Pain 2020, 16, 1744806920946889. [Google Scholar] [CrossRef]
- Steinberger, A.E.; Tecos, M.E.; Phelps, H.M.; Rubin, D.C.; Davidson, N.O.; Guo, J.; Warner, B.W. A Novel Maladaptive Unfolded Protein Response as a Mechanism for Small Bowel Resection-Induced Liver Injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2022, 323, G165–G176. [Google Scholar] [CrossRef]
- Teske, B.F.; Wek, S.A.; Bunpo, P.; Cundiff, J.K.; McClintick, J.N.; Anthony, T.G.; Wek, R.C. The eIF2 Kinase PERK and the Integrated Stress Response Facilitate Activation of ATF6 During Endoplasmic Reticulum Stress. Mol. Biol. Cell 2011, 22, 4390–4405. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, Regulation and Functions of the Unfolded Protein Response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Gonçalves Dos Santos, G.; Delay, L.; Yaksh, T.L.; Corr, M. Neuraxial Cytokines in Pain States. Front. Immunol. 2020, 10, 3061. [Google Scholar] [CrossRef]
- Yap, J.; Chen, X.; Delmotte, P.; Sieck, G.C. TNFα Selectively Activates the IRE1α/XBP1 Endoplasmic Reticulum Stress Pathway in Human Airway Smooth Muscle Cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L483–L493. [Google Scholar] [CrossRef]
- Zen, K.; Guo, Y.; Bian, Z.; Lv, Z.; Zhu, D.; Ohnishi, H.; Matozaki, T.; Liu, Y. Inflammation-Induced Proteolytic Processing of the SIRPα Cytoplasmic ITIM in Neutrophils Propagates a Proinflammatory State. Nat. Commun. 2013, 4, 2436. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.G.; Zhou, L.J. Long-Term Potentiation at Spinal C-Fiber Synapses: A Target for Pathological Pain. Curr. Pharm. Des. 2015, 21, 895–905. [Google Scholar] [CrossRef]
- Wang, H.G.; Lu, F.M.; Jin, I.; Udo, H.; Kandel, E.R.; de Vente, J.; Walter, U.; Lohmann, S.M.; Hawkins, R.D.; Antonova, I. Presynaptic and Postsynaptic Roles of NO, cGK, and RhoA in Long-Lasting Potentiation and Aggregation of Synaptic Proteins. Neuron 2005, 45, 389–403. [Google Scholar] [CrossRef] [PubMed]
- An, K.; Zhen, C.; Liu, Z.H.; Zhao, Q.; Liu, H.P.; Zhong, X.L.; Huang, W.Q. Spinal Protein Kinase Mζ Contributes to the Maintenance of Peripheral Inflammation-Primed Persistent Nociceptive Sensitization After Plantar Incision. Eur. J. Pain 2015, 19, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Nascimento de Lima, A.P.; Zhang, H.; Chen, L.; Effraim, P.R.; Gomis-Perez, C.; Cheng, X.; Huang, J.; Waxman, S.G.; Dib-Hajj, S.D. Nav1.8 in Small Dorsal Root Ganglion Neurons Contributes to Vincristine-Induced Mechanical Allodynia. Brain 2024, 147, 3157–3170. [Google Scholar] [CrossRef]
- Wu, L.J.; Zhuo, M. Targeting the NMDA Receptor Subunit NR2B for the Treatment of Neuropathic Pain. Neurotherapeutics 2009, 6, 693–702. [Google Scholar] [CrossRef]
- Liu, X.D.; Yang, J.J.; Fang, D.; Cai, J.; Wan, Y.; Xing, G.G. Functional Upregulation of Nav1.8 Sodium Channels on the Membrane of Dorsal Root Ganglia Neurons Contributes to the Development of Cancer-Induced Bone Pain. PLoS ONE 2014, 9, e114623. [Google Scholar] [CrossRef]
- Bornier, N.; Mulliez, A.; Chenaf, C.; Elyn, A.; Teixeira, S.; Authier, N.; Bertin, C.; Kerckhove, N. Chronic Pain is a Risk Factor for Incident Alzheimer’s Disease: A Nationwide Propensity-Matched Cohort Using Administrative Data. Front. Aging Neurosci. 2023, 15, 1193108. [Google Scholar] [CrossRef]
- Tian, J.; Jones, G.; Lin, X.; Zhou, Y.; King, A.; Vickers, J.; Pan, F. Association Between Chronic Pain and Risk of Incident Dementia: Findings from a Prospective Cohort. BMC Med. 2023, 21, 169. [Google Scholar] [CrossRef]
- Guo, X.; Hou, C.; Tang, P.; Li, R. Chronic Pain, Analgesics, and Cognitive Status: A Comprehensive Mendelian Randomization Study. Anesth. Analg. 2023, 137, 896–905. [Google Scholar] [CrossRef]
- Vergne-Salle, P.; Bertin, P. Chronic Pain and Neuroinflammation. Jt. Bone Spine 2021, 88, 105222. [Google Scholar] [CrossRef] [PubMed]
- Koszła, O.; Sołek, P. Misfolding and Aggregation in Neurodegenerative Diseases: Protein Quality Control Machinery as Potential Therapeutic Clearance Pathways. Cell Commun. Signal. 2024, 22, 421. [Google Scholar] [CrossRef]
- Wang, L.M.; Wu, Q.; Kirk, R.A.; Horn, K.P.; Ebada Salem, A.H.; Hoffman, J.M.; Yap, J.T.; Sonnen, J.A.; Towner, R.A.; Bozza, F.A.; et al. Lipopolysaccharide Endotoxemia Induces Amyloid-β and p-Tau Formation in the Rat Brain. Am. J. Nucl. Med. Mol. Imaging 2018, 8, 86–99. [Google Scholar]
- Hilal, S.; Ikram, M.A.; Verbeek, M.M.; Franco, O.H.; Stoops, E.; Vanderstichele, H.; Niessen, W.J.; Vernooij, M.W. C-Reactive Protein, Plasma Amyloid-β Levels, and Their Interaction with Magnetic Resonance Imaging Markers. Stroke 2018, 49, 2692–2698. [Google Scholar] [CrossRef] [PubMed]
- Litwiniuk, A.; Bik, W.; Kalisz, M.; Baranowska-Bik, A. Inflammasome NLRP3 Potentially Links Obesity-Associated Low-Grade Systemic Inflammation and Insulin Resistance with Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 5603. [Google Scholar] [CrossRef]
- Buongiorno, M.; Granell, E.; Caruana, G.; Sansa, G.; Vives-Gilabert, Y.; Cullell, N.; Molina-Seguin, J.; Almeria, M.; Artero, C.; Sánchez-Benavides, G.; et al. Impairments in Sleep and Brain Molecular Clearance in People with Cognitive Deterioration and Biological Evidence of AD: A Report of Four Cases. BMC Neurol. 2023, 23, 417. [Google Scholar] [CrossRef]
- Lee, M.T.; Peng, W.H.; Kan, H.W.; Wu, C.C.; Wang, D.W.; Ho, Y.C. Neurobiology of Depression: Chronic Stress Alters the Glutamatergic System in the Brain—Focusing on AMPA Receptor. Biomedicines 2022, 10, 1005. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.T.; Qu, J.; Wang, C.Y.; Yang, X.; Hu, F.; Hu, L.; Wu, X.F.; Jiang, C.Y.; Liu, W.T.; Han, Y. Rescue of HSP70 in Spinal Neurons Alleviates Opioid-Induced Hyperalgesia via the Suppression of Endoplasmic Reticulum Stress in Rodents. Front. Cell Dev. Biol. 2020, 8, 269. [Google Scholar] [CrossRef]
- Maravis, M.Y.; Siafaka, I.; Vadalouca, A.; Georgoudis, G. Role of Microglia in Neuropathic Pain. Cureus 2023, 15, e43555. [Google Scholar] [CrossRef]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in Neurodegenerative Diseases: Mechanism and Potential Therapeutic Targets. Signal Transduct. Target Ther. 2023, 8, 359. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, Y.Q.; Qadri, Y.J.; Serhan, C.N.; Ji, R.R. Microglia in Pain: Detrimental and Protective Roles in Pathogenesis and Resolution of Pain. Neuron 2018, 100, 1292–1311. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.J.; Gao, Y.J. Astrocytes in Chronic Pain: Cellular and Molecular Mechanisms. Neurosci. Bull. 2023, 39, 425–439. [Google Scholar] [CrossRef]
- Fan, Z.; Brooks, D.J.; Okello, A.; Edison, P. An Early and Late Peak in Microglial Activation in Alzheimer’s Disease Trajectory. Brain 2017, 140, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Clarke, B.E.; Patani, R. The Microglial Component of Amyotrophic Lateral Sclerosis. Brain 2020, 143, 3526–3539. [Google Scholar] [CrossRef]
- Rosenström, A.H.C.; Konsman, J.P.; Kosek, E. Cytokines in Cerebrospinal Fluid and Chronic Pain in Humans: Past, Present, and Future. Neuroimmunomodulation 2024, 31, 157–172. [Google Scholar] [CrossRef]
- Pinho-Ribeiro, F.A.; Verri, W.A., Jr.; Chiu, I.M. Nociceptor Sensory Neuron-Immune Interactions in Pain and Inflammation. Trends Immunol. 2017, 38, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Plantone, D.; Pardini, M.; Righi, D.; Manco, C.; Colombo, B.M.; De Stefano, N. The Role of TNF-α in Alzheimer’s Disease: A Narrative Review. Cells 2024, 13, 54. [Google Scholar] [CrossRef]
- Majbour, N.K.; Aasly, J.O.; Hustad, E.; Thomas, M.A.; Vaikath, N.N.; Elkum, N.; van de Berg, W.D.J.; Tokuda, T.; Mollenhauer, B.; Berendse, H.W.; et al. CSF Total and Oligomeric α-Synuclein along with TNF-α as Risk Biomarkers for Parkinson’s Disease: A Study in LRRK2 Mutation Carriers. Transl. Neurodegener. 2020, 9, 15. [Google Scholar] [CrossRef]
- Brázda, V.; Klusáková, I.; Svíženská, I.H.; Dubový, P. Dynamic Response to Peripheral Nerve Injury Detected by In Situ Hybridization of IL-6 and Its Receptor mRNAs in the Dorsal Root Ganglia is Not Strictly Correlated with Signs of Neuropathic Pain. Mol. Pain 2013, 9, 42. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, J.; Wu, Y.; Xie, M.; Tao, S.; Lv, Q.; Wang, Q. Plasma IL-6 Levels and Their Association with Brain Health and Dementia Risk: A Population-Based Cohort Study. Brain Behav. Immun. 2024, 120, 430–438. [Google Scholar] [CrossRef]
- Ryu, S.; Liu, X.; Guo, T.; Guo, Z.; Zhang, J.; Cao, Y.Q. Peripheral CCL2-CCR2 Signalling Contributes to Chronic Headache-Related Sensitization. Brain 2023, 146, 4274–4291. [Google Scholar] [CrossRef]
- Arfaei, R.; Mikaeili, N.; Daj, F.; Boroumand, A.; Kheyri, A.; Yaraghi, P.; Shirzad, Z.; Keshavarz, M.; Hassanshahi, G.; Jafarzadeh, A.; et al. Decoding the Role of the CCL2/CCR2 Axis in Alzheimer’s Disease and Innovating Therapeutic Approaches: Keeping All Options Open. Int. Immunopharmacol. 2024, 135, 112328. [Google Scholar] [CrossRef]
- Szabo-Pardi, T.A.; Barron, L.R.; Lenert, M.E.; Burton, M.D. Sensory Neuron TLR4 Mediates the Development of Nerve-Injury Induced Mechanical Hypersensitivity in Female Mice. Brain Behav. Immun. 2021, 97, 42–60. [Google Scholar] [CrossRef]
- Yamasoba, D.; Tsubota, M.; Domoto, R.; Sekiguchi, F.; Nishikawa, H.; Liu, K.; Nishibori, M.; Ishikura, H.; Yamamoto, T.; Taga, A.; et al. Peripheral HMGB1-Induced Hyperalgesia in Mice: Redox State-Dependent Distinct Roles of RAGE and TLR4. J. Pharmacol. Sci. 2016, 130, 139–142. [Google Scholar] [PubMed]
- Zhou, Y.; Chen, Y.; Xu, C.; Zhang, H.; Lin, C. TLR4 Targeting as a Promising Therapeutic Strategy for Alzheimer Disease Treatment. Front. Neurosci. 2020, 14, 602508. [Google Scholar] [CrossRef]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-Like Receptor 4 is Required for α-Synuclein Dependent Activation of Microglia and Astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef]
- Zhao, W.; Beers, D.R.; Henkel, J.S.; Zhang, W.; Urushitani, M.; Julien, J.P.; Appel, S.H. Extracellular Mutant SOD1 Induces Microglial-Mediated Motoneuron Injury. Glia 2010, 58, 231–243. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Silva Santos Ribeiro, P.; Willemen, H.L.D.M.; Versteeg, S.; Martin Gil, C.; Eijkelkamp, N. NLRP3 Inflammasome Activation in Sensory Neurons Promotes Chronic Inflammatory and Osteoarthritis Pain. Immunother. Adv. 2023, 3, ltad022. [Google Scholar] [CrossRef]
- McManus, R.M.; Latz, E. NLRP3 Inflammasome Signalling in Alzheimer’s Disease. Neuropharmacology 2024, 252, 109941. [Google Scholar] [CrossRef]
- Heid, M.E.; Keyel, P.A.; Kamga, C.; Shiva, S.; Watkins, S.C.; Salter, R.D. Mitochondrial Reactive Oxygen Species Induces NLRP3-Dependent Lysosomal Damage and Inflammasome Activation. J. Immunol. 2013, 191, 5230–5238. [Google Scholar] [CrossRef]
- Guo, J.S.; Jing, P.B.; Wang, J.A.; Zhang, R.; Jiang, B.C.; Gao, Y.J.; Zhang, Z.J. Increased Autophagic Activity in Dorsal Root Ganglion Attenuates Neuropathic Pain Following Peripheral Nerve Injury. Neurosci. Lett. 2015, 599, 158–163. [Google Scholar] [CrossRef]
- Fernandes, S.M.; Mayer, J.; Nilsson, P.; Shimozawa, M. How Close is Autophagy-Targeting Therapy for Alzheimer’s Disease to Clinical Use? A Summary of Autophagy Modulators in Clinical Studies. Front. Cell Dev. Biol. 2025, 12, 1520949. [Google Scholar] [CrossRef] [PubMed]
- Rui, Q.; Ni, H.; Li, D.; Gao, R.; Chen, G. The Role of LRRK2 in Neurodegeneration of Parkinson Disease. Curr. Neuropharmacol. 2018, 16, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Beckers, J.; Tharkeshwar, A.K.; Van Damme, P. C9orf72 ALS-FTD: Recent Evidence for Dysregulation of the Autophagy-Lysosome Pathway at Multiple Levels. Autophagy 2021, 17, 3306–3322. [Google Scholar] [CrossRef] [PubMed]
- Jahrling, J.B.; Laberge, R.M. Age-Related Neurodegeneration Prevention Through mTOR Inhibition: Potential Mechanisms and Remaining Questions. Curr. Top. Med. Chem. 2015, 15, 2139–2151. [Google Scholar] [CrossRef]
- Yeo, J.H.; Roh, D.H. The mTOR Inhibitor Rapamycin Suppresses Trigeminal Neuropathic Pain and p-MKK4/p-p38 Mitogen-Activated Protein Kinase-Mediated Microglial Activation in the Trigeminal Nucleus Caudalis of Mice with Infraorbital Nerve Injury. Front. Mol. Neurosci. 2023, 16, 1013577. [Google Scholar] [CrossRef]
- Sharma, A.; Tajerian, M.; Berner, J. Rapamycin Augmentation of Chronic Ketamine as a Novel Treatment for Complex Regional Pain Syndrome. Cureus 2023, 15, e43715. [Google Scholar] [CrossRef]
- Um, S.W.; Kim, M.J.; Leem, J.W.; Bai, S.J.; Lee, B.H. Pain-Relieving Effects of mTOR Inhibitor in the Anterior Cingulate Cortex of Neuropathic Rats. Mol. Neurobiol. 2019, 56, 2482–2494. [Google Scholar] [CrossRef]
- Cheng, J.; Deng, Y.; Zhou, J. Role of the Ubiquitin System in Chronic Pain. Front. Mol. Neurosci. 2021, 14, 674914. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, B.; Lan, H.; Sun, J.; Wei, G. Bortezomib-Induced Peripheral Neuropathy: Clinical Features, Molecular Basis, and Therapeutic Approach. Crit. Rev. Oncol. Hematol. 2024, 197, 104353. [Google Scholar] [CrossRef]
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A Common Mechanism of Proteasome Impairment by Neurodegenerative Disease-Associated Oligomers. Nat. Commun. 2018, 9, 1097. [Google Scholar] [CrossRef]
- Deas, E.; Wood, N.W.; Plun-Favreau, H. Mitophagy and Parkinson’s Disease: The PINK1-Parkin Link. Biochim. Biophys. Acta 2011, 1813, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Huang, J.; Zhang, Z.; Zhang, R.; Zhang, Z.; Liu, Y.; Ma, B. Translocator Protein 18 kDa (TSPO) as a Novel Therapeutic Target for Chronic Pain. Neural Plast. 2022, 2022, 8057854. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S.H.; Kam, T.I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.e7. [Google Scholar] [CrossRef] [PubMed]
- Edwards, G., III; Zhao, J.; Dash, P.K.; Soto, C.; Moreno-Gonzalez, I. Traumatic Brain Injury Induces Tau Aggregation and Spreading. J. Neurotrauma 2020, 37, 80–92. [Google Scholar] [CrossRef] [PubMed]
- D’Agnelli, S.; Gerra, M.C.; Bignami, E.; Arendt-Nielsen, L. Exosomes as a New Pain Biomarker Opportunity. Mol. Pain 2020, 16, 1744806920957800. [Google Scholar] [CrossRef]
- Huber, C.C.; Wang, H. Pathogenic and Therapeutic Role of Exosomes in Neurodegenerative Disorders. Neural Regen. Res. 2024, 19, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Pascual, M.; Ibáñez, F.; Guerri, C. Exosomes as Mediators of Neuron-Glia Communication in Neuroinflammation. Neural Regen. Res. 2020, 15, 796–801. [Google Scholar] [CrossRef]
- Liang, T.; Wu, Z.; Li, J.; Wu, S.; Shi, W.; Wang, L. The Emerging Double-Edged Sword Role of Exosomes in Alzheimer’s Disease. Front. Aging Neurosci. 2023, 15, 1209115. [Google Scholar] [CrossRef]
- Pinnell, J.R.; Cui, M.; Tieu, K. Exosomes in Parkinson Disease. J. Neurochem. 2021, 157, 413–428. [Google Scholar] [CrossRef]
- Kalani, A.; Tyagi, A.; Tyagi, N. Exosomes: Mediators of Neurodegeneration, Neuroprotection and Therapeutics. Mol. Neurobiol. 2014, 49, 590–600. [Google Scholar] [CrossRef]
- Heydari, R.; Koohi, F.; Rasouli, M.; Rezaei, K.; Abbasgholinejad, E.; Bekeschus, S.; Doroudian, M. Exosomes as Rheumatoid Arthritis Diagnostic Biomarkers and Therapeutic Agents. Vaccines 2023, 11, 687. [Google Scholar] [CrossRef]
- Ramanathan, S.; Douglas, S.R.; Alexander, G.M.; Shenoda, B.B.; Barrett, J.E.; Aradillas, E.; Sacan, A.; Ajit, S.K. Exosome MicroRNA Signatures in Patients with Complex Regional Pain Syndrome Undergoing Plasma Exchange. J. Transl. Med. 2019, 17, 81. [Google Scholar] [CrossRef]
- Bai, S.; Wang, Z.; Wang, M.; Li, J.; Wei, Y.; Xu, R.; Du, J. Tumor-Derived Exosomes Modulate Primary Site Tumor Metastasis. Front. Cell Dev. Biol. 2022, 10, 752818. [Google Scholar] [CrossRef]
- Khan, M.I.; Jeong, E.S.; Khan, M.Z.; Shin, J.H.; Kim, J.D. Stem Cells-Derived Exosomes Alleviate Neurodegeneration and Alzheimer’s Pathogenesis by Ameliorating Neuroinflammation and Regulating the Associated Molecular Pathways. Sci. Rep. 2023, 13, 15731. [Google Scholar] [CrossRef]
- Ren, J.; Liu, N.; Sun, N.; Zhang, K.; Yu, L. Mesenchymal Stem Cells and Their Exosomes: Promising Therapeutics for Chronic Pain. Curr. Stem Cell Res. Ther. 2019, 14, 644–653. [Google Scholar] [CrossRef]
- Choi, S.M.; Kim, B.C.; Jung, H.J.; Yoon, G.J.; Kang, K.W.; Choi, K.H.; Kim, J.T.; Lee, S.H.; Park, M.S.; Kim, M.K.; et al. Impact of Pain and Pain Subtypes on the Quality of Life of Patients with Parkinson’s Disease. J. Clin. Neurosci. 2017, 45, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Apkarian, A.V.; Sosa, Y.; Sonty, S.; Levy, R.M.; Harden, R.N.; Parrish, T.B.; Gitelman, D.R. Chronic Back Pain is Associated with Decreased Prefrontal and Thalamic Gray Matter Density. J. Neurosci. 2004, 24, 10410–10415. [Google Scholar] [CrossRef]
- Zhao, W.; Zhao, L.; Chang, X.; Lu, X.; Tu, Y. Elevated Dementia Risk, Cognitive Decline, and Hippocampal Atrophy in Multisite Chronic Pain. Proc. Natl. Acad. Sci. USA 2023, 120, e2215192120. [Google Scholar] [CrossRef]
- Meisner, J.G.; Marsh, A.D.; Marsh, D.R. Loss of GABAergic Interneurons in Laminae I-III of the Spinal Cord Dorsal Horn Contributes to Reduced GABAergic Tone and Neuropathic Pain After Spinal Cord Injury. J. Neurotrauma 2010, 27, 729–737. [Google Scholar] [CrossRef]
- Jiang, Y.; Rumble, J.L.; Gleixner, A.M.; Unnithan, A.S.; Pulugulla, S.H.; Posimo, J.M.; Choi, H.J.; Crum, T.S.; Pant, D.B.; Leak, R.K. N-Acetyl Cysteine Blunts Proteotoxicity in a Heat Shock Protein Dependent Manner. Neuroscience 2013, 255, 19–32. [Google Scholar] [CrossRef]
- Gao, J.; Liu, Y.; Si, C.; Guo, R.; Hou, S.; Liu, X.; Long, H.; Liu, D.; Xu, D.; Zhang, Z.R.; et al. Aspirin inhibits proteasomal degradation and promotes α-synuclein aggregate clearance through K63 ubiquitination. Nat. Commun. 2025, 16, 1438. [Google Scholar] [CrossRef] [PubMed]
- Krukowski, K.; Nijboer, C.H.; Huo, X.; Kavelaars, A.; Heijnen, C.J. Prevention of Chemotherapy-Induced Peripheral Neuropathy by the Small-Molecule Inhibitor Pifithrin-μ. Pain 2015, 156, 2184–2192. [Google Scholar] [CrossRef] [PubMed]
- Stine, C.; Coleman, D.L.; Flohrschutz, A.T.; Thompson, A.L.; Mishra, S.; Blagg, B.S.; Largent-Milnes, T.M.; Lei, W.; Streicher, J.M. Heat Shock Protein 90 Inhibitors Block the Antinociceptive Effects of Opioids in Mouse Chemotherapy-Induced Neuropathy and Cancer Bone Pain Models. Pain 2020, 161, 1798–1807. [Google Scholar] [CrossRef]
- Tikka, T.; Fiebich, B.L.; Goldsteins, G.; Keinanen, R.; Koistinaho, J. Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J. Neurosci 2001, 21, 2580–2588. [Google Scholar] [CrossRef]
- Lisi, L.; Navarra, P.; Cirocchi, R.; Sharp, A.; Stigliano, E.; Feinstein, D.L.; Dello Russo, C. Rapamycin reduces clinical signs and neuropathic pain in a chronic model of experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2012, 243, 43–51. [Google Scholar] [CrossRef]
- Kraft, V.; Schmitz, K.; Wilken-Schmitz, A.; Geisslinger, G.; Sisignano, M.; Tegeder, I. Trehalose reduces nerve injury-induced nociception in mice but negatively affects alertness. Nutrients 2021, 13, 2953. [Google Scholar] [CrossRef] [PubMed]
- Murphy, P.J.; Galigniana, M.D.; Morishima, Y.; Harrell, J.M.; Kwok, R.P.; Ljungman, M.; Pratt, W.B. Pifithrin-Alpha Inhibits p53 Signaling after Interaction of the Tumor Suppressor Protein with Hsp90 and Its Nuclear Translocation. J. Biol. Chem. 2004, 279, 30195–30201. [Google Scholar] [CrossRef] [PubMed]
- Maj, M.A.; Ma, J.; Krukowski, K.N.; Kavelaars, A.; Heijnen, C.J. Inhibition of Mitochondrial p53 Accumulation by PFT-μ Prevents Cisplatin-Induced Peripheral Neuropathy. Front. Mol. Neurosci. 2017, 10, 108. [Google Scholar] [CrossRef]
- Caponegro, M.D.; Torres, L.F.; Rastegar, C.; Rath, N.; Anderson, M.E.; Robinson, J.K.; Tsirka, S.E. Pifithrin-μ Modulates Microglial Activation and Promotes Histological Recovery Following Spinal Cord Injury. CNS Neurosci. Ther. 2019, 25, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, M.R.; Ramos, K.M.; Loram, L.C.; Wieseler, J.; Sholar, P.W.; Kearney, J.J.; Lewis, M.T.; Crysdale, N.Y.; Zhang, Y.; Harrison, J.A.; et al. Evidence for a Role of Heat Shock Protein-90 in Toll-Like Receptor 4 Mediated Pain Enhancement in Rats. Neuroscience 2009, 164, 1821–1832. [Google Scholar] [CrossRef]
- Miles, V.N.; Patel, R.K.; Smith, A.G.; McCall, R.P.; Wu, J.; Lei, W. The Effect of Heat Shock Protein 90 Inhibitor on Pain in Cancer Patients: A Systematic Review and Meta-Analysis. Medicina 2020, 57, 5. [Google Scholar] [CrossRef]
- Tanaka, K.; Matsuda, N. Proteostasis and Neurodegeneration: The Roles of Proteasomal Degradation and Autophagy. Biochim. Biophys. Acta 2014, 1843, 197–204. [Google Scholar] [CrossRef]
- Li, Y.Y.; Qin, Z.H.; Sheng, R. The Multiple Roles of Autophagy in Neural Function and Diseases. Neurosci. Bull. 2024, 40, 363–382. [Google Scholar] [CrossRef]
- Liu, X.; Zhu, M.; Ju, Y.; Li, A.; Sun, X. Autophagy Dysfunction in Neuropathic Pain. Neuropeptides 2019, 75, 41–48. [Google Scholar] [CrossRef]
- Huang, H.C.; Chen, L.; Zhang, H.X.; Li, S.F.; Liu, P.; Zhao, T.Y.; Li, C.X. Autophagy Promotes Peripheral Nerve Regeneration and Motor Recovery Following Sciatic Nerve Crush Injury in Rats. J. Mol. Neurosci. 2016, 58, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Yang, Y.; Zhou, F.; Xiao, Y.; Shi, L. Inhibition of PI3K/AKT/mTOR Signaling Pathway Promotes Autophagy and Relieves Hyperalgesia in Diabetic Rats. Neuroreport 2020, 31, 644–649. [Google Scholar] [CrossRef]
- Yuan, J.; Fei, Y. Lidocaine Activates Autophagy of Astrocytes and Ameliorates Chronic Constriction Injury-Induced Neuropathic Pain. J. Biochem. 2021, 170, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Chen, X.; Cheng, J.; Kong, F.; Xia, H.; Wu, J. Mammalian Target of Rapamycin Signaling Pathway Is Involved in Synaptic Plasticity of the Spinal Dorsal Horn and Neuropathic Pain in Rats by Regulating Autophagy. Neuroreport 2021, 32, 925–935. [Google Scholar] [CrossRef]
- Ge, Y.; Huang, M.; Yao, Y.M. Autophagy and proinflammatory cytokines: Interactions and clinical implications. Cytokine Growth Factor Rev. 2018, 43, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, L.; Wang, R.; Gao, Y.; Che, H.; Pan, Y.; Fu, P. Evaluating the effectiveness of GTM-1, rapamycin, and carbamazepine on autophagy and Alzheimer disease. Med. Sci. Monit. 2017, 23, 801–808. [Google Scholar] [CrossRef]
- Malagelada, C.; Jin, Z.H.; Jackson-Lewis, V.; Przedborski, S.; Greene, L.A. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci. 2010, 30, 1166–1175. [Google Scholar] [CrossRef]
- Baeza-Flores, G.D.C.; Guzmán-Priego, C.G.; Parra-Flores, L.I.; Murbartián, J.; Torres-López, J.E.; Granados-Soto, V. Metformin: A prospective alternative for the treatment of chronic pain. Front. Pharmacol. 2020, 11, 558474. [Google Scholar] [CrossRef]
- Ossipov, M.H.; Bazov, I.; Gardell, L.R.; Kowal, J.; Yakovleva, T.; Usynin, I.; Ekström, T.J.; Porreca, F.; Bakalkin, G. Control of chronic pain by the ubiquitin proteasome system in the spinal cord. J. Neurosci. 2007, 27, 8226–8237. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.S.; Li, J.; Erlandsson-Harris, H.; Stark, A.; Bakalkin, G.; Ahmed, M. Suppression of pain and joint destruction by inhibition of the proteasome system in experimental osteoarthritis. Pain 2012, 153, 18–26. [Google Scholar] [CrossRef]
- Mika, J.; Zychowska, M.; Popiolek-Barczyk, K.; Rojewska, E.; Przewlocka, B. Importance of glial activation in neuropathic pain. Eur. J. Pharmacol. 2013, 716, 106–119. [Google Scholar] [CrossRef]
- Vallejo, R.; Tilley, D.M.; Vogel, L.; Benyamin, R. The role of glia and the immune system in the development and maintenance of neuropathic pain. Pain Pract. 2010, 10, 167–184. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, H.; Ying, Z.; Gao, Q. Ibudilast enhances the clearance of SOD1 and TDP-43 aggregates through TFEB-mediated autophagy and lysosomal biogenesis: The new molecular mechanism of ibudilast and its implication for neuroprotective therapy. Biochem. Biophys. Res. Commun. 2020, 526, 231–238. [Google Scholar] [CrossRef]
- Ruiz, A.; Zuazo, J.; Ortiz-Sanz, C.; Luchena, C.; Matute, C.; Alberdi, E. Sephin1 protects neurons against excitotoxicity independently of the integrated stress response. Int. J. Mol. Sci. 2020, 21, 6088. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Potapova, E.V.; Dremin, V.V.; Dunaev, A.V. Interaction of Oxidative Stress and Misfolded Proteins in the Mechanism of Neurodegeneration. Life 2020, 10, 101. [Google Scholar] [CrossRef]
- Atkuri, K.R.; Mantovani, J.J.; Herzenberg, L.A.; Herzenberg, L.A. N-Acetylcysteine—A Safe Antidote for Cysteine/Glutathione Deficiency. Curr. Opin. Pharmacol. 2007, 7, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Gleixner, A.M.; Hutchison, D.F.; Sannino, S.; Bhatia, T.N.; Leak, L.C.; Flaherty, P.T.; Wipf, P.; Brodsky, J.L.; Leak, R.K. N-Acetyl-l-Cysteine Protects Astrocytes against Proteotoxicity without Recourse to Glutathione. Mol. Pharmacol. 2017, 92, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Mehringer, J.; Navarro, J.A.; Touraud, D.; Schneuwly, S.; Kunz, W. Phosphorylated Resveratrol as a Protein Aggregation Suppressor In Vitro and In Vivo. RSC Chem. Biol. 2022, 3, 250–260. [Google Scholar] [CrossRef]
- Sharma, S.; Gojal, H.; Joshi, S.; Nehru, B.; Saini, A. Molecular Interactions of Resveratrol with Aβ42 Peptide and Fibril during In Vitro Aβ42 Aggregation. Adv. Redox Res. 2023, 7, 100060. [Google Scholar] [CrossRef]
- Ahmad, B.; Borana, M.S.; Chaudhary, A.P. Understanding Curcumin-Induced Modulation of Protein Aggregation. Int. J. Biol. Macromol. 2017, 100, 89–96. [Google Scholar] [CrossRef]
- Mukherjee, S.; Mishra, A.K.; Peer, G.D.G.; Bagabir, S.A.; Haque, S.; Pandey, R.P.; Raj, V.S.; Jain, N.; Pandey, A.; Kar, S.K. The Interplay of the Unfolded Protein Response in Neurodegenerative Diseases: A Therapeutic Role of Curcumin. Front. Aging Neurosci. 2021, 13, 767493. [Google Scholar] [CrossRef]
- Fink, B.; Coppey, L.; Davidson, E.; Shevalye, H.; Obrosov, A.; Chheda, P.R.; Kerns, R.; Sivitz, W.; Yorek, M. Effect of Mitoquinone (Mito-Q) on Neuropathic Endpoints in an Obese and Type 2 Diabetic Rat Model. Free Radic. Res. 2020, 54, 311–318. [Google Scholar] [CrossRef]
- McManus, M.J.; Murphy, M.P.; Franklin, J.L. The Mitochondria-Targeted Antioxidant MitoQ Prevents Loss of Spatial Memory Retention and Early Neuropathology in a Transgenic Mouse Model of Alzheimer’s Disease. J. Neurosci. 2011, 31, 15703–15715. [Google Scholar] [CrossRef]
- Simioni, C.; Zauli, G.; Martelli, A.M.; Vitale, M.; Sacchetti, G.; Gonelli, A.; Neri, L.M. Oxidative Stress: Role of Physical Exercise and Antioxidant Nutraceuticals in Adulthood and Aging. Oncotarget 2018, 9, 17181–17198. [Google Scholar] [CrossRef]
- Ulbricht, A.; Gehlert, S.; Leciejewski, B.; Schiffer, T.; Bloch, W.; Höhfeld, J. Induction and Adaptation of Chaperone-Assisted Selective Autophagy CASA in Response to Resistance Exercise in Human Skeletal Muscle. Autophagy 2015, 11, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Freyssin, A.; Page, G.; Fauconneau, B.; Rioux Bilan, A. Natural Polyphenols Effects on Protein Aggregates in Alzheimer’s and Parkinson’s Prion-Like Diseases. Neural Regen. Res. 2018, 13, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Alomar, S.Y.; Gheit, R.E.A.E.; Enan, T.; El-Bayoumi, K.S.; Shoaeir, M.Z.; Elkazaz, A.Y.; Al Thagfan, S.S.; Zaitone, S.A.; El-Sayed, R.M. Novel Mechanism for Memantine in Attenuating Diabetic Neuropathic Pain in Mice via Downregulating the Spinal HMGB1/TRL4/NF-kB Inflammatory Axis. Pharmaceuticals 2021, 14, 307. [Google Scholar] [CrossRef]
- Bidari, A.; Ghavidel-Parsa, B.; Gharibpoor, F. Comparison of the Serum Brain-Derived Neurotrophic Factor (BDNF) between Fibromyalgia and Nociceptive Pain Groups; and Effect of Duloxetine on the BDNF Level. BMC Musculoskelet. Disord. 2022, 23, 411. [Google Scholar] [CrossRef] [PubMed]
- Vuic, B.; Milos, T.; Tudor, L.; Konjevod, M.; Nikolac Perkovic, M.; Jazvinscak Jembrek, M.; Nedic Erjavec, G.; Svob Strac, D. Cannabinoid CB2 Receptors in Neurodegenerative Proteinopathies: New Insights and Therapeutic Potential. Biomedicines 2022, 10, 3000. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Author | Year | Drug/Target | Mechanism of Action | Preclinical/Clinical Evidence | Challenges and Future Directions |
|---|---|---|---|---|---|
| Jiang et al. [193] | 2017 | N-acetylcysteine | Restores glutathione, modulates HSPs, protects against proteasome inhibitor toxicity | Preclinical (neuroblastoma cell model) | Requires clinical trials in pain conditions |
| Gao et al. [194] | 2019 | Aspirin | Inhibits proteasomal degradation, promotes aggregate clearance via K63 ubiquitination | Preclinical (PD mouse model) | Repurposing needs validation; risk of systemic effects |
| Kan et al. [37] | 2024 | PKCε inhibitor | Reduces ER stress and autophagy activation | Preclinical (diabetic neuropathy model) | Needs human translation |
| Krukowski et al. [195] | 2017 | PFM-µ (Hsp70/Hsp90 inhibitor) | Inhibits chaperone activity and p53, prevents mitochondrial p53 aggregation | Preclinical (chemotherapy-induced neuropathy models) | No clinical trials |
| Stine et al. [196] | 2019 | Hsp90 inhibitors | Block opioid-induced analgesia via TLR4 modulation | Preclinical (mouse models of bone pain) | Mixed effects; may impair pain relief from opioids |
| Tikka et al. [197] | 2001 | Minocycline | Inhibits microglial activation and inflammatory cytokine production | Preclinical (primary neuronal cultures model) | Requires exploration in chronic pain with misfolding |
| Lisi et al. [198] | 2012 | Rapamycin (mTOR inhibitor) | Enhances autophagy, reduces inflammation | Preclinical (autoimmune encephalitis model) | Immunosuppression risk; unclear dosing for pain relief |
| Kraft et al. [199] | 2021 | Trehalose | Activates TFEB to enhance autophagy, clears protein aggregates | Preclinical (mice spared nerve injury model) | Sedation reported; require further safety and efficacy studies |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brezic, N.; Gligorevic, S.; Sic, A.; Knezevic, N.N. Protein Misfolding and Aggregation as a Mechanistic Link Between Chronic Pain and Neurodegenerative Diseases. Curr. Issues Mol. Biol. 2025, 47, 259. https://doi.org/10.3390/cimb47040259
Brezic N, Gligorevic S, Sic A, Knezevic NN. Protein Misfolding and Aggregation as a Mechanistic Link Between Chronic Pain and Neurodegenerative Diseases. Current Issues in Molecular Biology. 2025; 47(4):259. https://doi.org/10.3390/cimb47040259
Chicago/Turabian StyleBrezic, Nebojsa, Strahinja Gligorevic, Aleksandar Sic, and Nebojsa Nick Knezevic. 2025. "Protein Misfolding and Aggregation as a Mechanistic Link Between Chronic Pain and Neurodegenerative Diseases" Current Issues in Molecular Biology 47, no. 4: 259. https://doi.org/10.3390/cimb47040259
APA StyleBrezic, N., Gligorevic, S., Sic, A., & Knezevic, N. N. (2025). Protein Misfolding and Aggregation as a Mechanistic Link Between Chronic Pain and Neurodegenerative Diseases. Current Issues in Molecular Biology, 47(4), 259. https://doi.org/10.3390/cimb47040259