
Potential Application of MicroRNAs and Some Other Molecular Biomarkers in Alzheimer’s Disease

 , and
, and
Abstract
1. Introduction
2. Molecular Mechanisms of Alzheimer’s Disease Pathogenesis
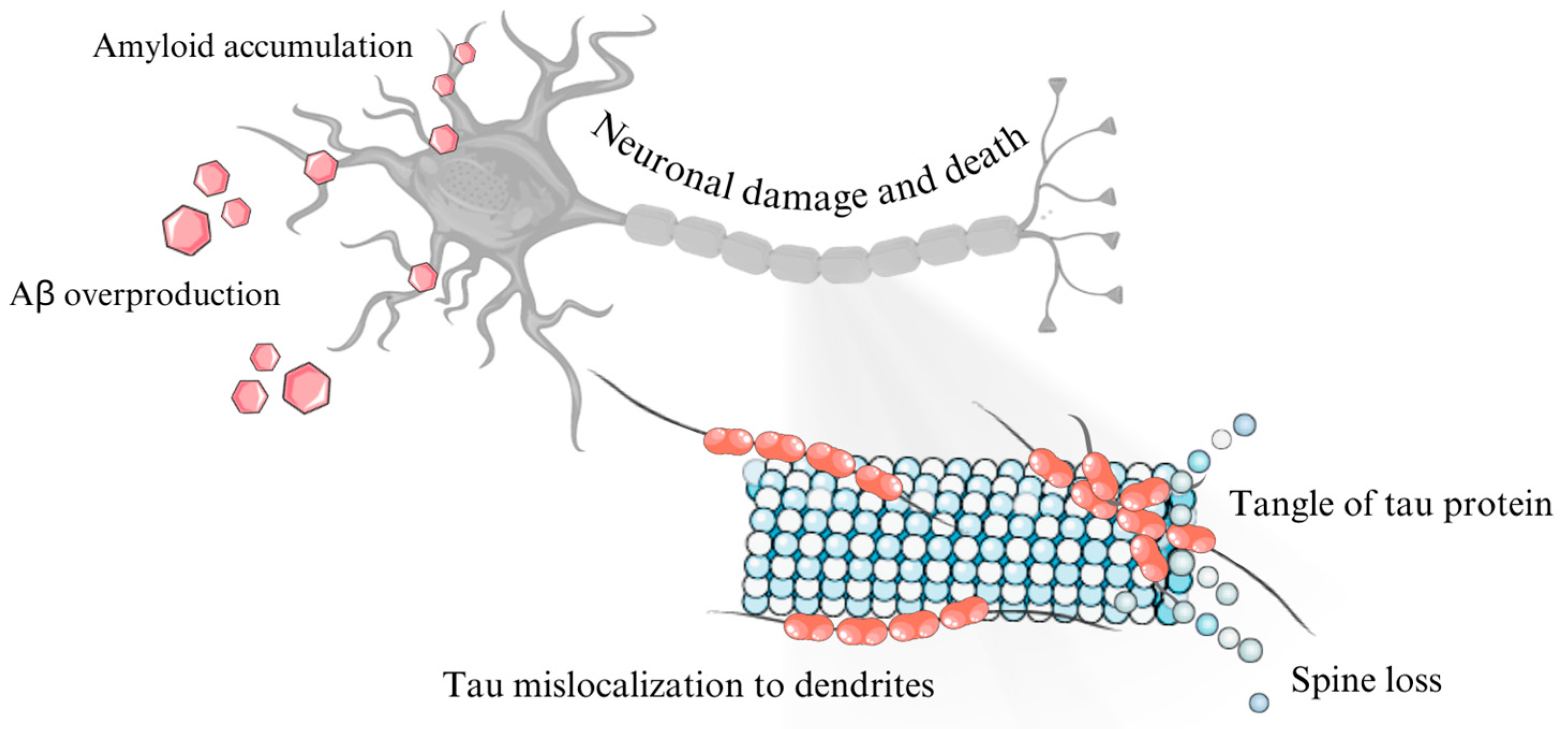
2.1. Role of β-Amyloid Precursor Protein (APP) and β-Amyloid
2.2. Tau Protein in Alzheimer’s Disease
2.3. Cellular Prion Protein in the Development of Alzheimer’s Disease
2.4. Role of Neurofibrillary Tangles in Alzheimer’s Disease
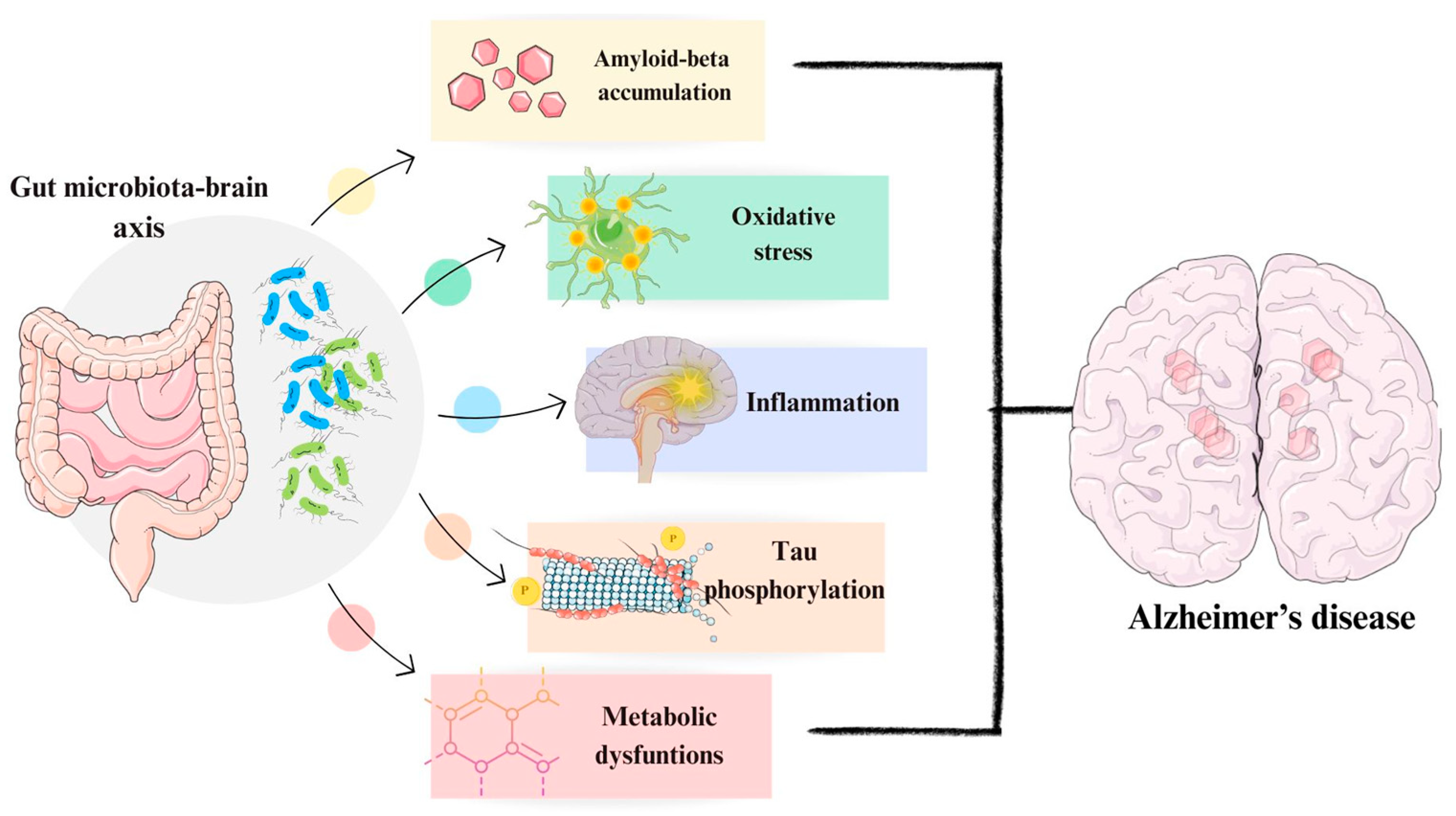
2.5. Role of Microbiome in Alzheimer’s Disease
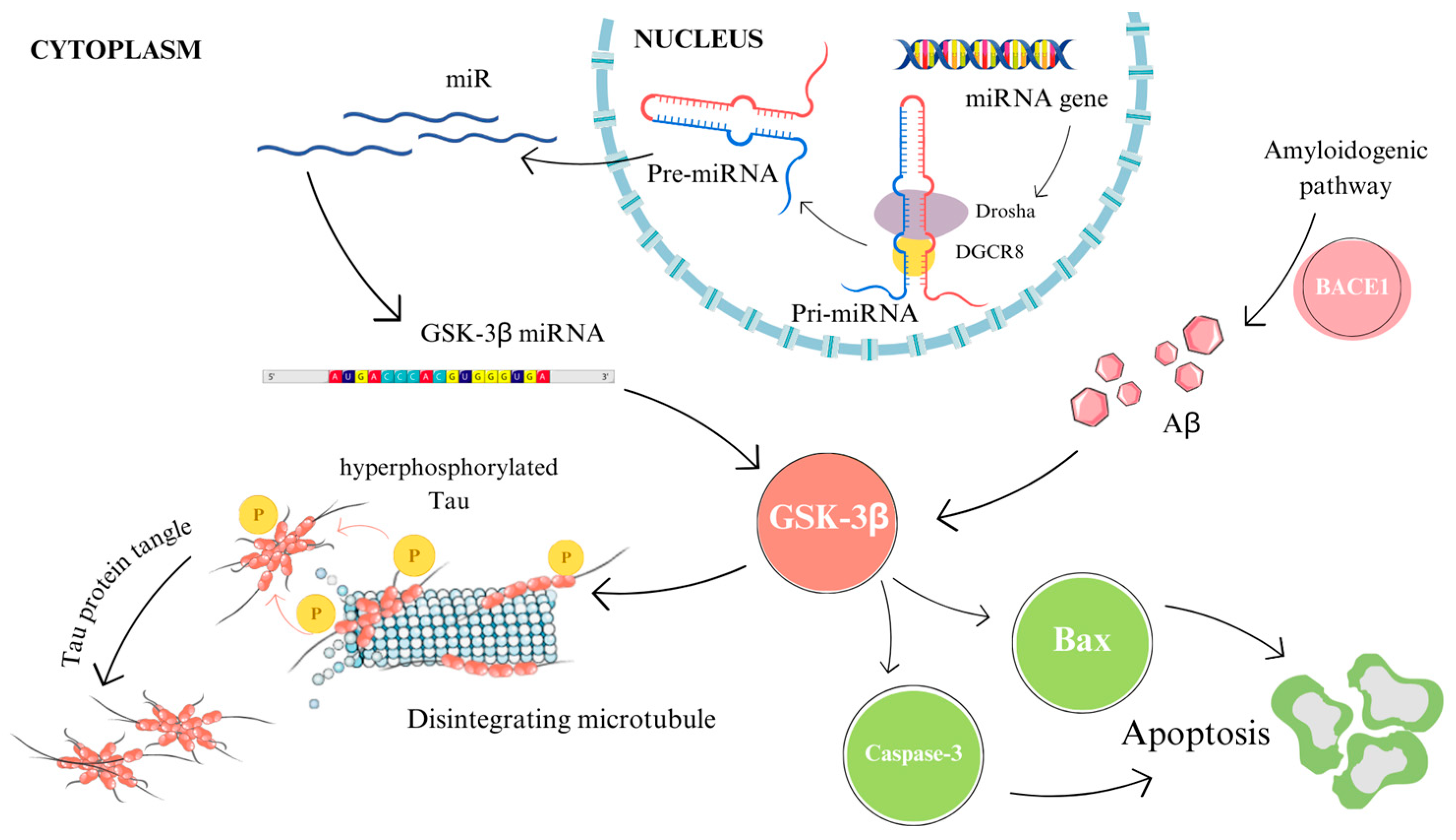
3. Association of MicroRNAs with Alzheimer’s Disease
3.1. Influence of MicroRNAs on β-Amyloid Formation
3.2. Relating MicroRNAs with Tau Protein
3.3. Involvement of MicroRNAs in Neurogenesis and Synaptic Plasticity
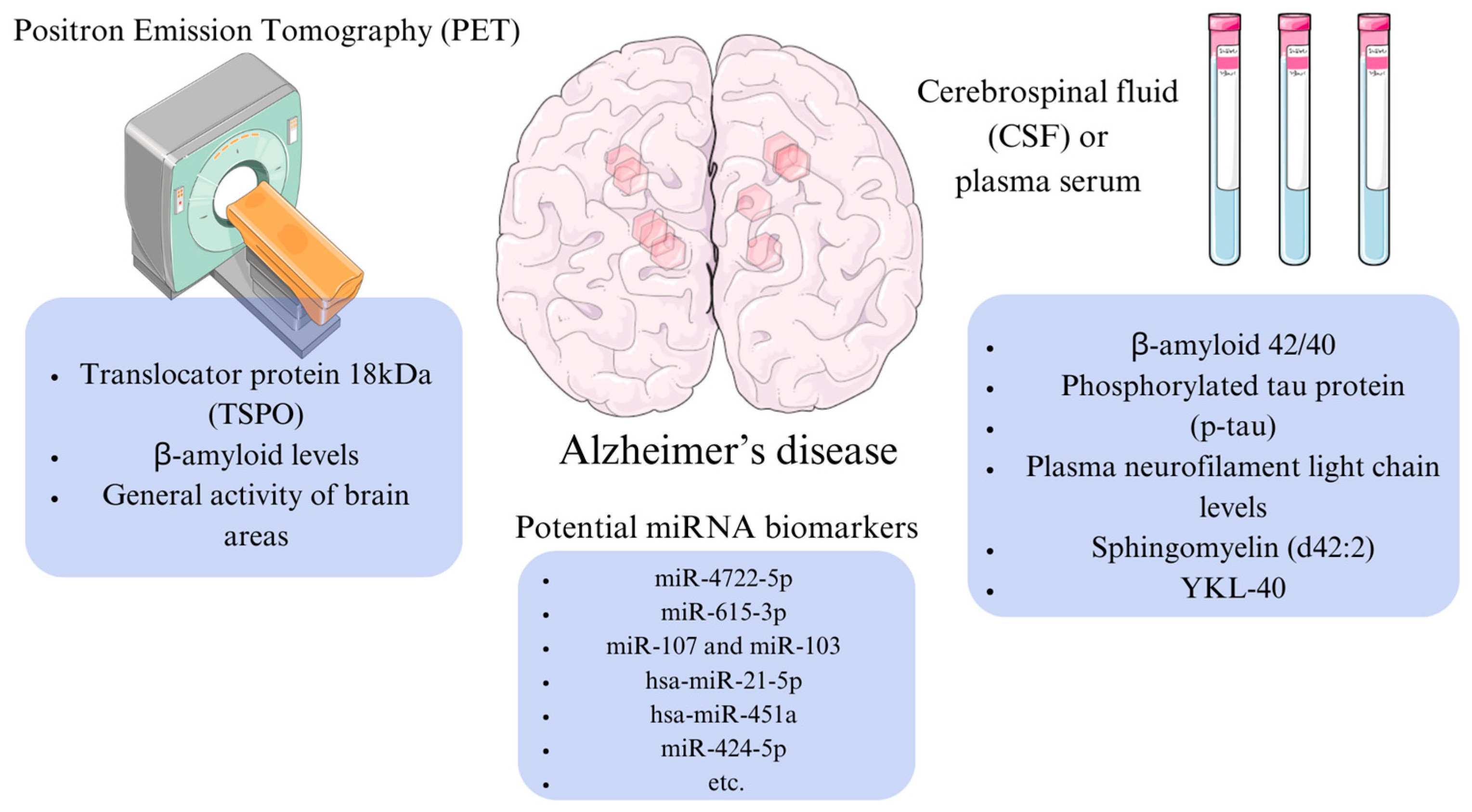
4. Potential Molecular Markers of Alzheimer’s Disease
5. Application of microRNAs as Biomarkers for Alzheimer’s Disease Diagnostics
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Klyucherev, T.O.; Olszewski, P.; Shalimova, A.A.; Chubarev, V.N.; Tarasov, V.V.; Attwood, M.M.; Syvänen, S.; Schiöth, H.B. Advances in the development of new biomarkers for Alzheimer’s disease. Transl. Neurodegener. 2022, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Rostagno, A.A. Pathogenesis of Alzheimer’s disease. Int. J. Mol. Sci. 2022, 24, 107. [Google Scholar] [CrossRef] [PubMed]
- John, A.; Reddy, P.H. Synaptic basis of Alzheimer’s disease: Focus on synaptic amyloid beta, p-tau and nitochondria. Ageing Res. Rev. 2021, 65, 101208. [Google Scholar] [CrossRef] [PubMed]
- Nikolac Perkovic, M.; Videtic Paska, A.; Konjevod, M.; Kouter, K.; Svob Strac, D.; Nedic Erjavec, G.; Pivac, N. Epigenetics of Alzheimer’s disease. Biomolecules 2021, 11, 195. [Google Scholar] [CrossRef] [PubMed]
- Bertram, L.; Tanzi, R.E. Genomic mechanisms in Alzheimer’s disease. Brain Pathol. 2020, 30, 966–977. [Google Scholar] [CrossRef]
- Andrade-Guerrero, J.; Santiago-Balmaseda, A.; Jeronimo-Aguilar, P.; Vargas-Rodríguez, I.; Cadena-Suárez, A.R.; Sánchez-Garibay, C.; Pozo-Molina, G.; Méndez-Catalá, C.F.; Cardenas-Aguayo, M.-C.; Diaz-Cintra, S.; et al. Alzheimer’s disease: An updated overview of its genetics. Int. J. Mol. Sci. 2023, 24, 3754. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.V.F.; Loures, C.d.M.G.; Alves, L.C.V.; de Souza, L.C.; Borges, K.B.G.; Carvalho, M.d.G. Alzheimer’s disease: Risk factors and potentially protective measures. J. Biomed. Sci. 2019, 26, 33. [Google Scholar] [CrossRef]
- Parashat, T.; Sumathi, V. Identification of Alzheimer’s disease by imaging: A comprehensive review. Int. J. Environ. Res. Public Health 2023, 20, 1273. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Barve, K.H.; Kumar, M.S. Recent advancements in pathogenesis, diagnostics and treatment of Alzheimer’s disease. Curr. Neuropharmacol. 2020, 18, 1106–1125. [Google Scholar] [CrossRef] [PubMed]
- Atri, A. The Alzheimer’s disease clinical spectrum: Diagnosis and management. Med. Clin. N. Am. 2019, 103, 263–293. [Google Scholar] [CrossRef]
- Porsteinsson, A.P.; Isaacson, R.S.; Knox, S.; Sabbagh, M.N.; Rubino, I. Diagnosis of early Alzheimer’s disease: Clinical practice in 2021. J. Prev. Alzheimers Dis. 2021, 8, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Graff-Radford, J.; Yong, K.X.X.; Apostolova, L.G.; Bouwman, F.H.; Carrillo, M.; Dickerson, B.C.; Rabinovici, G.D.; Schott, J.M.; Jones, D.T.; Murray, M.E. New insights into atypical Alzheimer’s disease in the era of biomarkers. Lancet Neurol. 2021, 20, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Knapskog, A.-B.; Engedal, K.; Selbæk, G.; Øksengård, A.-R. Alzheimer’s disease—Diagnosis and treatment. Tidsskr Nor Legeforen 2021, 141, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Balázs, N.; Bereczki, D.; Kovács, T. Cholinesterase inhibitors and memantine for the treatment of Alzheimer and non-Alzheimer dementias. Ideggyogyaszati Szle 2021, 74, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Athar, T.; Al Balushi, K.; Khan, S.A. Recent advances on drug development and emerging therapeutic agents for Alzheimer’s disease. Mol. Biol. Rep. 2021, 48, 5629–5645. [Google Scholar] [CrossRef] [PubMed]
- Passeri, E.; Elkhoury, K.; Morsink, M.; Broersen, K.; Linder, M.; Tamayol, A.; Malaplate, C.; Yen, F.T.; Arab-Tehrany, E. Alzheimer’s disease: Treatment strategies and their limitations. Int. J. Mol. Sci. 2022, 23, 13954. [Google Scholar] [CrossRef]
- Lloret, A.; Esteve, D.; Lloret, M.-A.; Cervera-Ferri, A.; Lopez, B.; Nepomuceno, M.; Monllor, P. When does Alzheimer′s disease really start? The role of biomarkers. Int. J. Mol. Sci. 2019, 20, 5536. [Google Scholar] [CrossRef]
- Hansson, O.; Edelmayer, R.M.; Boxer, A.L.; Carrillo, M.C.; Mielke, M.M.; Rabinovici, G.D.; Salloway, S.; Sperling, R.; Zetterberg, H.; Teunissen, C.E. The Alzheimer’s association appropriate use recommendations for blood biomarkers in Alzheimer’s disease. Alzheimers Dement. 2022, 18, 2669–2686. [Google Scholar] [CrossRef]
- Thijssen, E.H.; La Joie, R.; Strom, A.; Fonseca, C.; Iaccarino, L.; Wolf, A.; Spina, S.; Allen, I.E.; Cobigo, Y.; Heuer, H.; et al. Plasma phosphorylated tau 217 and phosphorylated tau 181 as biomarkers in Alzheimer’s disease and frontotemporal lobar degeneration: A retrospective diagnostic performance study. Lancet Neurol. 2021, 20, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Saunders, T.; Gunn, C.; Blennow, K.; Kvartsberg, H.; Zetterberg, H.; Shenkin, S.D.; Cox, S.R.; Deary, I.J.; Smith, C.; King, D.; et al. Neurogranin in Alzheimer’s disease and ageing: A human post-mortem study. Neurobiol. Dis. 2023, 177, 105991. [Google Scholar] [CrossRef]
- Chang, C.-H.; Lin, C.-H.; Lane, H.-Y. Machine learning and novel biomarkers for the diagnosis of Alzheimer’s disease. Int. J. Mol. Sci. 2021, 22, 2761. [Google Scholar] [CrossRef] [PubMed]
- Headley, A.; De Leon-Benedetti, A.; Dong, C.; Levin, B.; Loewenstein, D.; Camargo, C.; Rundek, T.; Zetterberg, H.; Blennow, K.; Wright, C.B.; et al. Alzheimer’s Disease Neuroimaging Initiative. Neurogranin as a predictor of memory and executive function decline in MCI patients. Neurology 2018, 90, e887–e895. [Google Scholar] [CrossRef] [PubMed]
- Stevenson-Hoare, J.; Heslegrave, A.; Leonenko, G.; Fathalla, D.; Bellou, E.; Luckcuck, L.; Marshall, R.; Sims, R.; Morgan, B.P.; Hardy, J.; et al. Plasma biomarkers and genetics in the diagnosis and prediction of Alzheimer’s disease. Brain 2022, 146, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Leuzy, A.; Mattsson-Carlgren, N.; Palmqvist, S.; Janelidze, S.; Dage, J.L.; Hansson, O. Blood-based biomarkers for Alzheimer’s disease. EMBO Mol. Med. 2022, 14, e14408. [Google Scholar] [CrossRef]
- Pereira, J.B.; Westman, E.; Hansson, O. Alzheimer’s Disease Neuroimaging Initiative. Association between cerebrospinal fluid and plasma neurodegeneration biomarkers with brain atrophy in Alzheimer’s disease. Neurobiol. Aging 2017, 58, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Preische, O.; Schultz, S.A.; Apel, A.; Kuhle, J.; Kaeser, S.A.; Barro, C.; Gräber, S.; Kuder-Buletta, E.; LaFougere, C.; Laske, C.; et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer’s disease. Nat. Med. 2019, 25, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.Y.; Chang, D.C.; Lin, S.L. The microRNA (miRNA): Overview of the RNA genes that modulate gene function. Mol. Biotechnol. 2008, 38, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Thul, P.J.; Lindskog, C. The human protein atlas: A spatial map of the human proteome. Protein Sci. 2018, 27, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Balzano, F.; Deiana, M.; Dei Giudici, S.; Oggiano, A.; Baralla, A.; Pasella, S.; Mannu, A.; Pescatori, M.; Porcu, B.; Fanciulli, G.; et al. miRNA stability in frozen plasma samples. Molecules 2015, 20, 19030–19040. [Google Scholar] [CrossRef]
- Sethi, P.; Lukiw, W.J. Micro-RNA abundance and stability in human brain: Specific alterations in Alzheimer’s disease temporal lobe neocortex. Neurosci. Lett. 2009, 459, 100–104. [Google Scholar] [CrossRef]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Takayama, K.; Hashimoto, M.; Inoue, S. Transcriptional and post-transcriptional regulations of amyloid-β precursor protein (APP) mRNA. Front. Aging 2021, 2, 721579. [Google Scholar] [CrossRef] [PubMed]
- Delport, A.; Hewer, R. The amyloid precursor protein: A converging point in Alzheimer’s disease. Mol. Neurobiol. 2022, 59, 4501–4516. [Google Scholar] [CrossRef] [PubMed]
- Dunot, J.; Ribera, A.; Pousinha, P.A.; Marie, H. Spatiotemporal insights of APP function. Curr. Opin. Neurobiol. 2023, 82, 102754. [Google Scholar] [CrossRef] [PubMed]
- Sehar, U.; Rawat, P.; Reddy, A.P.; Kopel, J.; Reddy, P.H. Amyloid beta in aging and Alzheimer’s disease. Int. J. Mol. Sci. 2022, 23, 12924. [Google Scholar] [CrossRef] [PubMed]
- Yarns, B.C.; Holiday, K.A.; Carlson, D.M.; Cosgrove, C.K.; Melrose, R.J. Pathophysiology of Alzheimer’s disease. Psychiatr. Clin. N. Am. 2022, 45, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Tabeshmehr, P.; Eftekharpour, E. Tau; one protein, so many diseases. Biology 2023, 12, 244. [Google Scholar] [CrossRef] [PubMed]
- Rawat, P.; Sehar, U.; Bisht, J.; Selman, A.; Culberson, J.; Reddy, P.H. Phosphorylated tau in Alzheimer’s disease and other tauopathies. Int. J. Mol. Sci. 2022, 23, 12841. [Google Scholar] [CrossRef]
- Corsi, A.; Bombieri, C.; Valenti, M.T.; Romanelli, M.G. Tau isoforms: Gaining insight into MAPT alternative splicing. Int. J. Mol. Sci. 2022, 23, 15383. [Google Scholar] [CrossRef]
- Antón-Fernández, A.; Vallés-Saiz, L.; Avila, J.; Hernández, F. Neuronal nuclear tau and neurodegeneration. Neuroscience 2023, 518, 178–184. [Google Scholar] [CrossRef]
- Lester, E.; Van Alstyne, M.; McCann, K.L.; Reddy, S.; Cheng, L.Y.; Kuo, J.; Pratt, J.; Parker, R. Cytosolic condensates rich in polyserine define subcellular sites of tau aggregation. Proc. Natl. Acad. Sci. USA 2023, 120, e2217759120. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, S.; Biernat, J.; Mandelkow, E. A current view on tau protein phosphorylation in Alzheimer’s disease. Curr. Opin. Neurobiol. 2021, 69, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Jackson, N.A.; Guerrero-Muñoz, M.J.; Castillo-Carranza, D.L. The prion-like transmission of tau oligomers via exosomes. Front. Aging Neurosci. 2022, 14, 974414. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, K.J.; Correll, C.M. Prion disease. Semin. Neurol. 2019, 39, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Panes, J.D.; Saavedra, P.; Pineda, B.; Escobar, K.; Cuevas, M.E.; Moraga-Cid, G.; Fuentealba, J.; Rivas, C.I.; Rezaei, H.; Muñoz-Montesino, C. PrPC as a transducer of physiological and pathological signals. Front. Mol. Neurosci. 2021, 14, 762918. [Google Scholar] [CrossRef] [PubMed]
- Legname, G.; Scialò, C. On the role of the cellular prion protein in the uptake and signaling of pathological aggregates in neurodegenerative diseases. Prion 2020, 14, 257–270. [Google Scholar] [CrossRef]
- Beraldo, F.H.; Ostapchenko, V.G.; Caetano, F.A.; Guimaraes, A.L.S.; Ferretti, G.D.S.; Daude, N.; Bertram, L.; Nogueira, K.O.P.C.; Silva, J.L.; Westaway, D.; et al. Regulation of amyloid β oligomer binding to neurons and neurotoxicity by the prion protein-mGluR5 complex. J. Biol. Chem. 2016, 291, 21945–21955. [Google Scholar] [CrossRef] [PubMed]
- Balducci, C.; Beeg, M.; Stravalaci, M.; Bastone, A.; Sclip, A.; Biasini, E.; Tapella, L.; Colombo, L.; Manzoni, C.; Borsello, T.; et al. Synthetic amyloid-β oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. USA 2010, 107, 2295–2300. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M. Structure of NFT: Biochemical pproach. Adv. Exp. Med. Biol. 2019, 1184, 23–34. [Google Scholar]
- Trejo-Lopez, J.A.; Yachnis, A.T.; Prokop, S. Neuropathology of Alzheimer’s disease. Neurotherapeutics 2022, 19, 173–185. [Google Scholar] [CrossRef]
- Nguyen, N.M.; Cho, J.; Lee, C. Gut microbiota and Alzheimer’s disease: How to study and apply their relationship. Int. J. Mol. Sci. 2023, 24, 4047. [Google Scholar] [CrossRef] [PubMed]
- Dissanayaka, D.M.S.; Jayasena, V.; Rainey-Smith, S.R.; Martins, R.N.; Fernando, W.M.A.D.B. The role of diet and gut microbiota in Alzheimer’s disease. Nutrients 2024, 16, 412. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, F.; Cechova, K.; Amlerova, J.; Hort, J. Antibiotics, gut microbiota, and Alzheimer’s disease. J. Neuroinflammation 2019, 16, 108. [Google Scholar] [CrossRef] [PubMed]
- Tarawneh, R.; Penhos, E. The gut microbiome and Alzheimer’s disease: Complex and bidirectional interactions. Neurosci. Biobehav. Rev. 2022, 141, 104814. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Sisodia, S.S.; Vassar, R.J. The gut microbiome in Alzheimer’s disease: What we know and what remains to be explored. Mol. Neurodegener. 2023, 18, 9. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, A.L.; Choi, J.; Ryou, J.; Newcomer, E.P.; Thompson, R.; Bollinger, R.M.; Hall-Moore, C.; Ndao, I.M.; Sax, L.; Benzinger, T.L.S.; et al. Gut microbiome composition may be an indicator of preclinical Alzheimer’s disease. Sci. Transl. Med. 2023, 15, eabo2984. [Google Scholar] [CrossRef] [PubMed]
- Swarbrick, S.; Wragg, N.; Ghosh, S.; Stolzing, A. Systematic review of miRNA as biomarkers in Alzheimer’s disease. Mol. Neurobiol. 2019, 56, 6156–6167. [Google Scholar] [CrossRef] [PubMed]
- Groot, M.; Lee, H. Sorting mechanisms for microRNAs into extracellular vesicles and their associated diseases. Cells 2020, 9, 1044. [Google Scholar] [CrossRef]
- Li, X.; Chen, S.-C.; Ip, J.P.K. Diverse and composite roles of miRNA in non-neuronal cells and neuronal synapses in Alzheimer’s disease. Biomolecules 2022, 12, 1505. [Google Scholar] [CrossRef]
- Koh, H.S.; Lee, S.; Lee, H.J.; Min, J.-W.; Iwatsubo, T.; Teunissen, C.E.; Cho, H.-J.; Ryu, J.-H. Targeting microRNA-485-3p blocks Alzheimer’s disease progression. Int. J. Mol. Sci. 2021, 22, 13136. [Google Scholar] [CrossRef]
- Chen, M.-L.; Hong, C.-G.; Yue, T.; Li, H.-M.; Duan, R.; Hu, W.-B.; Cao, J.; Wang, Z.-X.; Chen, C.-Y.; Hu, X.-K.; et al. Inhibition of miR-331-3p and miR-9-5p ameliorates Alzheimer’s disease by enhancing autophagy. Theranostics 2021, 11, 2395–2409. [Google Scholar] [CrossRef]
- Jiang, Y.; Bian, W.; Chen, J.; Cao, X.; Dong, C.; Xiao, Y.; Xu, B.; Sun, X. miRNA-137-5p improves spatial memory and cognition in Alzheimer’s mice by targeting ubiquitin-specific peptidase 30. Anim. Models Exp. Med. 2023, 6, 526–536. [Google Scholar] [CrossRef]
- Zheng, K.; Hu, F.; Zhou, Y.; Zhang, J.; Zheng, J.; Lai, C.; Xiong, W.; Cui, K.; Hu, Y.-Z.; Han, Z.-T.; et al. miR-135a-5p mediates memory and synaptic impairments via the Rock2/Adducin1 signaling pathway in a mouse model of Alzheimer’s disease. Nat. Commun. 2021, 12, 1903. [Google Scholar] [CrossRef]
- Wang, R.; Chopra, N.; Nho, K.; Maloney, B.; Obukhov, A.G.; Nelson, P.T.; Counts, S.E.; Lahiri, D.K. Human microRNA (miR-20b-5p) modulates Alzheimer’s disease pathways and neuronal function, and a specific polymorphism close to the MIR20B gene influences Alzheimer’s biomarkers. Mol. Psychiatry 2022, 27, 1256–1273. [Google Scholar] [CrossRef]
- Nagaraj, S.; Want, A.; Laskowska-Kaszub, K.; Fesiuk, A.; Vaz, S.; Logarinho, E.; Wojda, U. Candidate Alzheimer’s disease biomarker miR-483-5p lowers TAU phosphorylation by direct ERK1/2 repression. Int. J. Mol. Sci. 2021, 22, 3653. [Google Scholar] [CrossRef]
- Walgrave, H.; Balusu, S.; Snoeck, S.; Vanden Eynden, E.; Craessaerts, K.; Thrupp, N.; Wolfs, L.; Horré, K.; Fourne, Y.; Ronisz, A.; et al. Restoring miR-132 expression rescues adult hippocampal neurogenesis and memory deficits in Alzheimer’s disease. Cell Stem Cell 2021, 28, 1805–1821. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, Y.; Yu, M. MicroRNA-4722-5p and microRNA-615-3p serve as potential biomarkers for Alzheimer’s disease. Exp. Ther. Med. 2022, 23, 241. [Google Scholar] [CrossRef]
- Dhiman, K.; Blennow, K.; Zetterberg, H.; Martins, R.N.; Gupta, V.B. Cerebrospinal fluid biomarkers for understanding multiple aspects of Alzheimer’s disease pathogenesis. Cell Mol. Life Sci. 2019, 76, 1833–1863. [Google Scholar] [CrossRef]
- Maltais, M.; De Souto Barreto, P.; Hooper, C.; Payoux, P.; Rolland, Y.; Vellas, B.; MAPT/DSA Study Group. Association between brain β-amyloid and frailty in older adults. J. Gerontol. Ser. A 2019, 74, 1747–1752. [Google Scholar] [CrossRef]
- Lopes Alves, I.; Collij, L.E.; Altomare, D.; Frisoni, G.B.; Saint-Aubert, L.; Payoux, P.; Kivipelto, M.; Jessen, F.; Drzezga, A.; Leeuwis, A.; et al. Quantitative amyloid PET in Alzheimer’s disease: The AMYPAD prognostic and natural history study. Alzheimers Dement. 2020, 16, 750–758. [Google Scholar] [CrossRef]
- Pereira, J.B.; Janelidze, S.; Stomrud, E.; Palmqvist, S.; van Westen, D.; Dage, J.L.; Mattsson-Carlgren, N.; Hansson, O. Plasma markers predict changes in amyloid, tau, atrophy and cognition in non-demented subjects. Brain 2021, 144, 2826–2836. [Google Scholar] [CrossRef]
- Lu, W.-H.; Giudici, K.V.; Rolland, Y.; Guyonnet, S.; Li, Y.; Bateman, R.J.; de Souto Barreto, P.; Vellas, B. Prospective associations between plasma amyloid-beta 42/40 and frailty in community-dwelling older adults. J. Prev. Alzheimers Dis. 2021, 8, 41–47. [Google Scholar] [CrossRef]
- Peña-Bautista, C.; Roca, M.; Hervás, D.; Cuevas, A.; López-Cuevas, R.; Vento, M.; Baquero, M.; García-Blanco, A.; Cháfer-Pericás, C. Plasma metabolomics in early Alzheimer’s disease patients diagnosed with amyloid biomarker. J. Proteomics 2019, 200, 144–152. [Google Scholar] [CrossRef]
- Bergland, A.K.; Proitsi, P.; Kirsebom, B.-E.; Soennesyn, H.; Hye, A.; Larsen, A.I.; Xu, J.; Legido-Quigley, C.; Rajendran, L.; Fladby, T.; et al. Exploration of plasma lipids in mild cognitive impairment due to Alzheimer’s disease. J. Alzheimers Dis. 2020, 77, 1117–1127. [Google Scholar] [CrossRef]
- Ashton, N.J.; Moseby-Knappe, M.; Benedet, A.L.; Grötschel, L.; Lantero-Rodriguez, J.; Karikari, T.K.; Hassager, C.; Wise, M.P.; Stammet, P.; Kjaergaard, J.; et al. Alzheimer disease blood biomarkers in patients with out-of-hospital cardiac Arrest. JAMA Neurol. 2023, 80, 388–396. [Google Scholar] [CrossRef]
- Gill, J.; Mustapic, M.; Diaz-Arrastia, R.; Lange, R.; Gulyani, S.; Diehl, T.; Motamedi, V.; Osier, N.; Stern, R.A.; Kapogiannis, D. Higher exosomal tau, amyloid-beta 42 and IL-10 are associated with mild TBIs and chronic symptoms in military personnel. Brain Inj. 2018, 32, 1277–1284. [Google Scholar] [CrossRef]
- Brett, B.L.; Gardner, R.C.; Godbout, J.; Dams-O’Connor, K.; Keene, C.D. Traumatic brain injury and risk of neurodegenerative disorder. Biol. Psychiatry 2022, 91, 498–507. [Google Scholar] [CrossRef]
- Prins, S.; de Kam, M.L.; Teunissen, C.E.; Groeneveld, G.J. Inflammatory plasma biomarkers in subjects with preclinical Alzheimer’s disease. Alzheimers Res. Ther. 2022, 14, 106. [Google Scholar] [CrossRef]
- Chaney, A.; Williams, S.R.; Boutin, H. In vivo molecular imaging of neuroinflammation in Alzheimer’s disease. J. Neurochem. 2019, 149, 438–451. [Google Scholar] [CrossRef]
- El Idrissi, F.; Gressier, B.; Devos, D.; Belarbi, K. A Computational exploration of the molecular network associated to neuroinflammation in Alzheimer’s disease. Front. Pharmacol. 2021, 12, 630003. [Google Scholar] [CrossRef]
- Carlini, V.; Verduci, I.; Cianci, F.; Cannavale, G.; Fenoglio, C.; Galimberti, D.; Mazzanti, M. CLIC1 Protein Accumulates in Circulating Monocyte Membrane during Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 1484. [Google Scholar] [CrossRef]
- Kenny, A.; Jiménez-Mateos, E.M.; Zea-Sevilla, M.A.; Rábano, A.; Gili-Manzanaro, P.; Prehn, J.H.M.; Henshall, D.C.; Ávila, J.; Engel, T.; Hernández, F. Proteins and microRNAs are differentially expressed in tear fluid from patients with Alzheimer’s disease. Sci. Rep. 2019, 9, 15437. [Google Scholar] [CrossRef]
- Fotuhi, S.N.; Khalaj-Kondori, M.; Hoseinpour Feizi, M.A.; Talebi, M. Long non-coding RNA BACE1-AS may serve as an Alzheimer’s disease blood-based biomarker. J. Mol. Neurosci. 2019, 69, 351–359. [Google Scholar] [CrossRef]
- Nie, C.; Sun, Y.; Zhen, H.; Guo, M.; Ye, J.; Liu, Z.; Yang, Y.; Zhang, X. Differential expression of plasma exo-miRNA in neurodegenerative diseases by next-generation sequencing. Front. Neurosci. 2020, 14, 438. [Google Scholar] [CrossRef]
- Visconte, C.; Fenoglio, C.; Serpente, M.; Muti, P.; Sacconi, A.; Rigoni, M.; Arighi, A.; Borracci, V.; Arcaro, M.; Arosio, B.; et al. Altered extracellular vesicle miRNA profile in prodromal Alzheimer’s disease. Int. J. Mol. Sci. 2023, 24, 14749. [Google Scholar] [CrossRef]
- Liu, W.-L.; Lin, H.-W.; Lin, M.-R.; Yu, Y.; Liu, H.-H.; Dai, Y.-L.; Chen, L.-W.; Jia, W.-W.; He, X.-J.; Li, X.-L.; et al. Emerging blood exosome-based biomarkers for preclinical and clinical Alzheimer’s disease: A meta-analysis and systematic review. Neural Regen Res 2022, 17, 2381–2390. [Google Scholar]
- Kouhnavardi, S.; Cabatic, M.; Mañas-Padilla, M.C.; Malabanan, M.A.; Smani, T.; Cicvaric, A.; Muñoz Aranzalez, E.A.; Koenig, X.; Urban, E.; Lubec, G.; et al. miRNA-132/212 deficiency disrupts selective corticosterone modulation of dorsal vs. ventral hippocampal metaplasticity. Int. J. Mol. Sci. 2023, 24, 9565. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, X.; Pei, H. miRNA-384-3p alleviates sevoflurane-induced nerve injury by inhibiting Aak1 kinase in neonatal rats. Brain Behav. 2022, 12, e2556. [Google Scholar] [CrossRef]
- Liu, C.; Tong, Z.; Tan, J.; Xin, Z.; Wang, Z.; Tian, L. MicroRNA-21-5p targeting PDCD4 suppresses apoptosis via regulating the PI3K/AKT/FOXO1 signaling pathway in tongue squamous cell carcinoma. Exp. Ther. Med. 2019, 18, 3543–3551. [Google Scholar] [CrossRef]
- Duan, X.; Zheng, Q.; Liang, L.; Zhou, L. Serum exosomal miRNA-125b and miRNA-451a are potential diagnostic biomarker for Alzheimer’s diseases. Degener. Neurol. Neuromuscul. Dis. 2024, 14, 21–31. [Google Scholar] [CrossRef]
- Wang, J.; Chen, C.; Zhang, Y. An investigation of microRNA-103 and microRNA-107 as potential blood-based biomarkers for disease risk and progression of Alzheimer’s disease. J. Clin. Lab. Anal. 2020, 34, e23006. [Google Scholar] [CrossRef]
- Souza, V.C.; Morais, G.S.; Henriques, A.D.; Machado-Silva, W.; Perez, D.I.V.; Brito, C.J.; Camargos, E.F.; Moraes, C.F.; Nóbrega, O.T. Whole-blood levels of microRNA-9 are decreased in patients with late-onset Alzheimer disease. Am. J. Alzheimers Dis. Other Demen. 2020, 35, 1533317520911573. [Google Scholar] [CrossRef]
- Li, Q.; Wang, Y.; Peng, W.; Jia, Y.; Tang, J.; Li, W.; Zhang, J.H.; Yang, J. MicroRNA-101a regulates autophagy phenomenon via the MAPK pathway to modulate Alzheimer’s-associated pathogenesis. Cell Transplant. 2019, 28, 1076–1084. [Google Scholar] [CrossRef]
- Li, S.; Poon, C.H.; Zhang, Z.; Yue, M.; Chen, R.; Zhang, Y.; Hossain, M.F.; Pan, Y.; Zhao, J.; Rong, L.; et al. MicroRNA-128 suppresses tau phosphorylation and reduces amyloid-beta accumulation by inhibiting the expression of GSK3β, APPBP2, and mTOR in Alzheimer’s disease. CNS Neurosci. Ther. 2023, 29, 1848–1864. [Google Scholar] [CrossRef]
- Kim, S.J.; Russell, A.E.; Wang, W.; Gemoets, D.E.; Sarkar, S.N.; Simpkins, J.W.; Brown, C.M. miR-146a dysregulates energy metabolism during neuroinflammation. J. Neuroimmune Pharmacol. 2022, 17, 228–241. [Google Scholar] [CrossRef]
- Cao, Y.; Tan, X.; Lu, Q.; Huang, K.; Tang, X.; He, Z. MiR-29c-3p may promote the progression of Alzheimer’s disease through BACE1. J. Healthc. Eng. 2021, 2021, 2031407. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, M.; Hong, Z.; Kang, J.; Pan, H.; Yan, C. MiR-130a-3p has protective effects in Alzheimer’s disease via targeting DAPK1. Am. J. Alzheimers Dis. Other Dement. 2021, 36, 15333175211020572. [Google Scholar] [CrossRef]
- Feng, H.; Hu, P.; Chen, Y.; Sun, H.; Cai, J.; He, X.; Cao, Q.; Yin, M.; Zhang, Y.; Li, Q.; et al. Decreased miR-451a in cerebrospinal fluid, a marker for both cognitive impairment and depressive symptoms in Alzheimer’s disease. Theranostics 2023, 13, 3021–3040. [Google Scholar] [CrossRef]
- Fu, L.; Jiang, G.; Weng, H.; Dick, G.M.; Chang, Y.; Kassab, G.S. Cerebrovascular miRNAs correlate with the clearance of Aβ through perivascular route in younger 3xTg-AD mice. Brain Pathol. 2019, 30, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Ortiz, C.J.; Prieto, G.A.; Martini, A.C.; Forner, S.; Trujillo-Estrada, L.; LaFerla, F.M.; Baglietto-Vargas, D.; Cotman, C.W.; Kitazawa, M. miR-181a negatively modulates synaptic plasticity in hippocampal cultures and its inhibition rescues memory deficits in a mouse model of Alzheimer’s disease. Aging Cell 2020, 19, e13118. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Xian, X.; Fang, M.; Fan, S.; Li, W. Loss of miR-369 promotes tau phosphorylation by targeting the Fyn and serine/threonine-protein kinase 2 signaling pathways in Alzheimer’s disease mice. Front. Aging Neurosci. 2020, 11, 365. [Google Scholar] [CrossRef] [PubMed]
- Hosseinian, S.; Arefian, E.; Rakhsh-Khorshid, H.; Eivani, M.; Rezayof, A.; Pezeshk, H.; Marashi, S.-A. A meta-analysis of gene expression data highlights synaptic dysfunction in the hippocampus of brains with Alzheimer’s disease. Sci. Rep. 2020, 10, 8384. [Google Scholar] [CrossRef]
- Estfanous, S.; Daily, K.P.; Eltobgy, M.; Deems, N.P.; Anne, M.N.K.; Krause, K.; Badr, A.; Hamilton, K.; Carafice, C.; Hegazi, A.; et al. Elevated expression of miR-17 in microglia of Alzheimer’s disease patients abrogates autophagy-mediated amyloid-β degradation. Front. Immunol. 2021, 12, 705581. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Zhang, K.; Zhou, H.; Jiang, L.; Xie, B.; Wang, R.; Xia, W.; Yin, Y.; Gao, Z.; Cui, D.; et al. Increased miR-34c mediates synaptic deficits by targeting synaptotagmin 1 through ROS-JNK-p53 pathway in Alzheimer’s disease. Aging Cell 2020, 19, e13125. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Chen, Q.; Zhang, X. MicroRNA-455–5p/CPEB1 pathway mediates Aβ-related learning and memory deficits in a mouse model of Alzheimer’s disease. Brain Res. Bull. 2021, 177, 282–294. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-G.; Zhao, Y.; Lu, Y.; Wang, P.-C. ABCA1-labeled exosomes in serum contain higher microRNA-193b levels in Alzheimer’s disease. BioMed Res. Int. 2021, 2021, 5450397. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Meng, S.; Di, W.; Xia, M.; Dong, L.; Zhao, Y.; Ling, S.; He, J.; Xue, X.; Chen, X.; et al. Amyloid-β protein and microRNA-384 in NCAM-labeled exosomes from peripheral blood are potential diagnostic markers for Alzheimer’s disease. CNS Neurosci. Ther. 2022, 28, 1093–1107. [Google Scholar] [CrossRef]
- Siedlecki-Wullich, D.; Miñano-Molina, A.J.; Rodríguez-Álvarez, J. microRNAs as early biomarkers of Alzheimer’s disease: A synaptic perspective. Cells 2021, 10, 113. [Google Scholar] [CrossRef]
- Jaber, V.R.; Zhao, Y.; Sharfman, N.M.; Li, W.; Lukiw, W.J. Addressing Alzheimer’s disease (AD) neuropathology using anti-microRNA (AM) strategies. Mol. Neurobiol. 2019, 56, 8101–8108. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | Function | Biological Material | Ref. |
|---|---|---|---|
| miRNA-101a | Negative modulation of autophagy | Human blood plasma, APPSwe/PS1ΔEP transgenic mice, Human SH-SY5U immature neuroblastoma cells | [93] |
| miRNA-128 | Inhibition of tau protein phosphorylation and β-amyloid accumulation | Human cerebral cortex tissues from patients with AD and control patients, mouse Neuro-2a neuroblast cells, human SH-SY5Y immature neuroblastoma cells, transformed human embryonic kidney cells, HEK-293T | [94] |
| miRNA-146a | Pro-inflammatory function | Rat brain tissue (culture of glial and cortical neurons), Mouse hippocampal neuronal cell line, HT-22 cells, mouse cerebellar microglia, mouse vascular endothelial cells, mouse C8-B4 cells, mouse bEnd cells.3; Human brain tissue of the frontal cortex, temporal cortex, and cerebellum of patients with AD and controls | [95] |
| miRNA-29c-3p | Negative regulation of BACE1, a key enzyme that degrades amyloid precursor protein | SPF C57BL/6J mice, PC12 cell line | [96] |
| miRNA-130a | Protective function resulting from targeting DAPK1, a serine/threonine kinase gene associated with neuronal apoptosis | SH-SY5Y cell line, AD APPSwe/PS1dE9 mice, wild-type mice | [97] |
| miRNA-451a | Protective function, inhibition of BACE1, inhibition of inflammatory processes by negative regulation of NLRP3 | Human cerebrospinal fluid, APP/PS1 transgenic mice, wild-type mice, mouse line Neuro-2a, human line HEK293, | [98] |
| miRNA-485-3p | Accumulation of β-amyloid, pathologies of tau protein, development of the inflammatory process of the nervous system | Human frontal cortex, medial cortex, and CSF samples from AD and control patients, human plasma samples, B6SJLF1/J mice, 5XFAD transgenic mice, C57BL/6 mice | [60] |
| miRNA-126, miRNA-135 | Protective function related to the removal of β-amyloid | Mice 3xTG-AD, B6129SF2 control strain | [99] |
| miRNA-181a | Toxicity associated with β-amyloid, a negative regulator of synaptic plasticity | Brain tissue and cell cultures, C57BL/J, 3xTg-AD mice, 129/C57BL6 hybrid control mice | [100] |
| miRNA-31 | Protective function associated with targeting APP and BACE1 genes | Mice 3xTG-AD, mouse line HT-22, human line HEK293, human line SH-SY5Y | [101] |
| miRNA-369 | Phosphorylation of tau protein | miRNA-369 KO 3xTG-AD mice, C57/B6 mice, 3xTG-AD mice, 293T cells | [102] |
| miRNA-129 | Modulation of MAPK1 and ERK genes | Wistar rats | [103] |
| miRNA-34c | Modulation of the SYT1 gene | HT-22 cell line, line 293a | [104] |
| miRNA-455-5p | CPEB gene regulation | APP/PS1 mice, wild-type control mice | [105] |
| miRNA-193b | The impact of the decrease in APP | Blood from patients with Alzheimer’s dementia, mild cognitive impairment, and subjective cognitive decline, patients as controls, APP/PS1 double transgenic mice, wild-type mice, mouse HT-22 line | [106] |
| miRNA-17 | Downregulation of autophagy-related genes | Brain tissues of the human temporal lobe of patients with AD and controls, C57BL/6 wild-type mice, 5xFAD mice, Atg 5−/− mice | [103] |
| miRNA-384 | Decreased expression of genes encoding APP and β-secretase | Blood and CSF of patients with dementia due to AD, with mild cognitive impairment and subjective cognitive decline, control patients | [107] |
| miRNA-26b | Increase in β-amyloid production mediated by IGF1 regulation | n/d | [108] |
| miRNA-125b | Targeting 15-lipoxygenase, synapsin, complement factor H, tetraspanin 7 and tetraspanin 12 | n/d | [109] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paprzycka, O.; Wieczorek, J.; Nowak, I.; Madej, M.; Strzalka-Mrozik, B. Potential Application of MicroRNAs and Some Other Molecular Biomarkers in Alzheimer’s Disease. Curr. Issues Mol. Biol. 2024, 46, 5066-5084. https://doi.org/10.3390/cimb46060304
Paprzycka O, Wieczorek J, Nowak I, Madej M, Strzalka-Mrozik B. Potential Application of MicroRNAs and Some Other Molecular Biomarkers in Alzheimer’s Disease. Current Issues in Molecular Biology. 2024; 46(6):5066-5084. https://doi.org/10.3390/cimb46060304
Chicago/Turabian StylePaprzycka, Olga, Jan Wieczorek, Ilona Nowak, Marcel Madej, and Barbara Strzalka-Mrozik. 2024. "Potential Application of MicroRNAs and Some Other Molecular Biomarkers in Alzheimer’s Disease" Current Issues in Molecular Biology 46, no. 6: 5066-5084. https://doi.org/10.3390/cimb46060304
APA StylePaprzycka, O., Wieczorek, J., Nowak, I., Madej, M., & Strzalka-Mrozik, B. (2024). Potential Application of MicroRNAs and Some Other Molecular Biomarkers in Alzheimer’s Disease. Current Issues in Molecular Biology, 46(6), 5066-5084. https://doi.org/10.3390/cimb46060304