
Effects of Blood-Derived Products on Cellular Senescence and Inflammatory Response: A Study on Skin Rejuvenation

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Media
2.2. Preparation of Blood Products
2.3. Growth Curves
2.4. Procedure for SA-ß-gal Staining
2.5. Immuno Fluorescence
2.6. Western Blot Analysis
2.7. XTT Assay
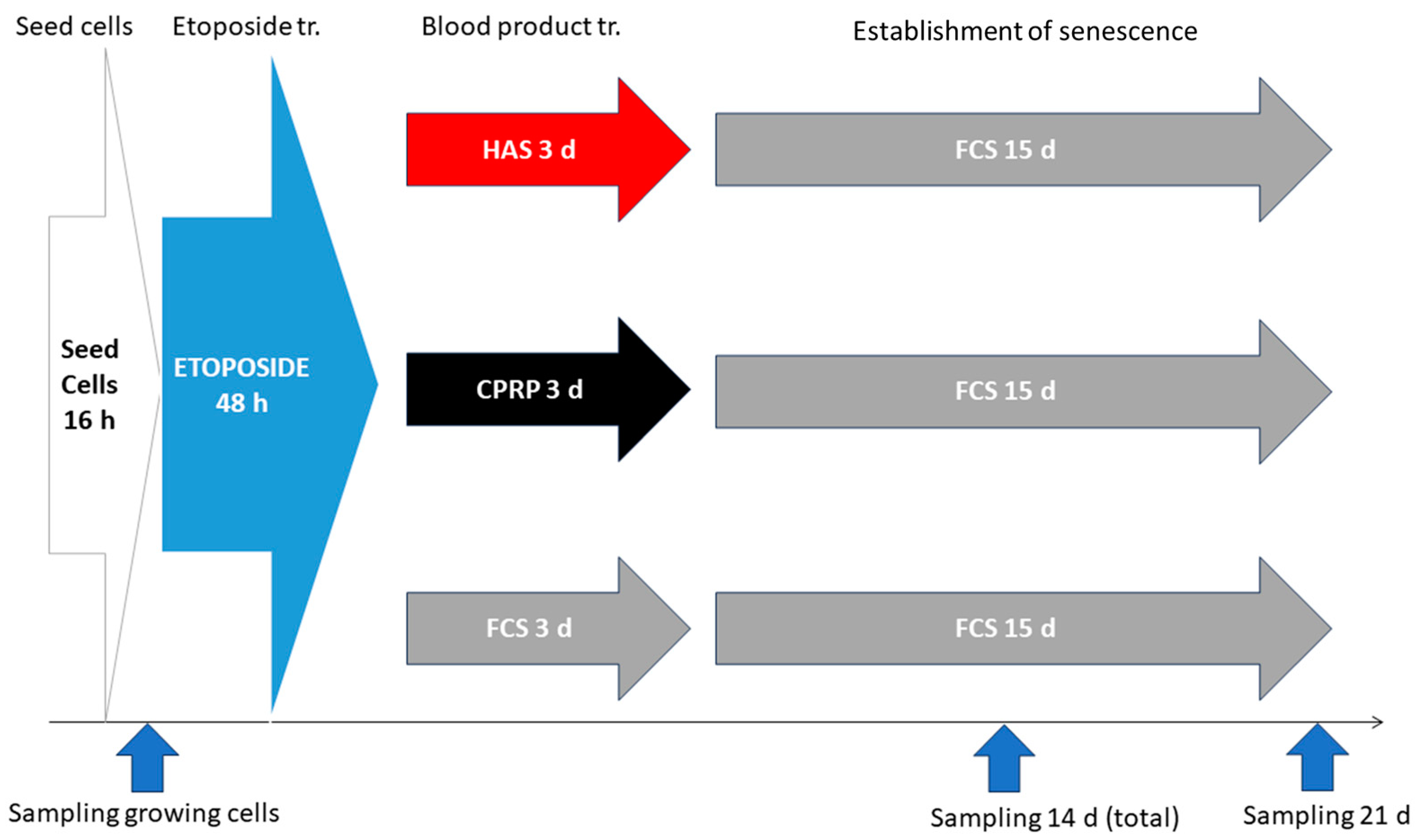
2.8. BP-Treatment for Early Treatemnt Group
2.8.1. Cell Seeding
2.8.2. Etoposide Treatment
2.8.3. BP-Treatment
2.8.4. Establishment of Cellular Senescence
2.8.5. Harvesting of Cells
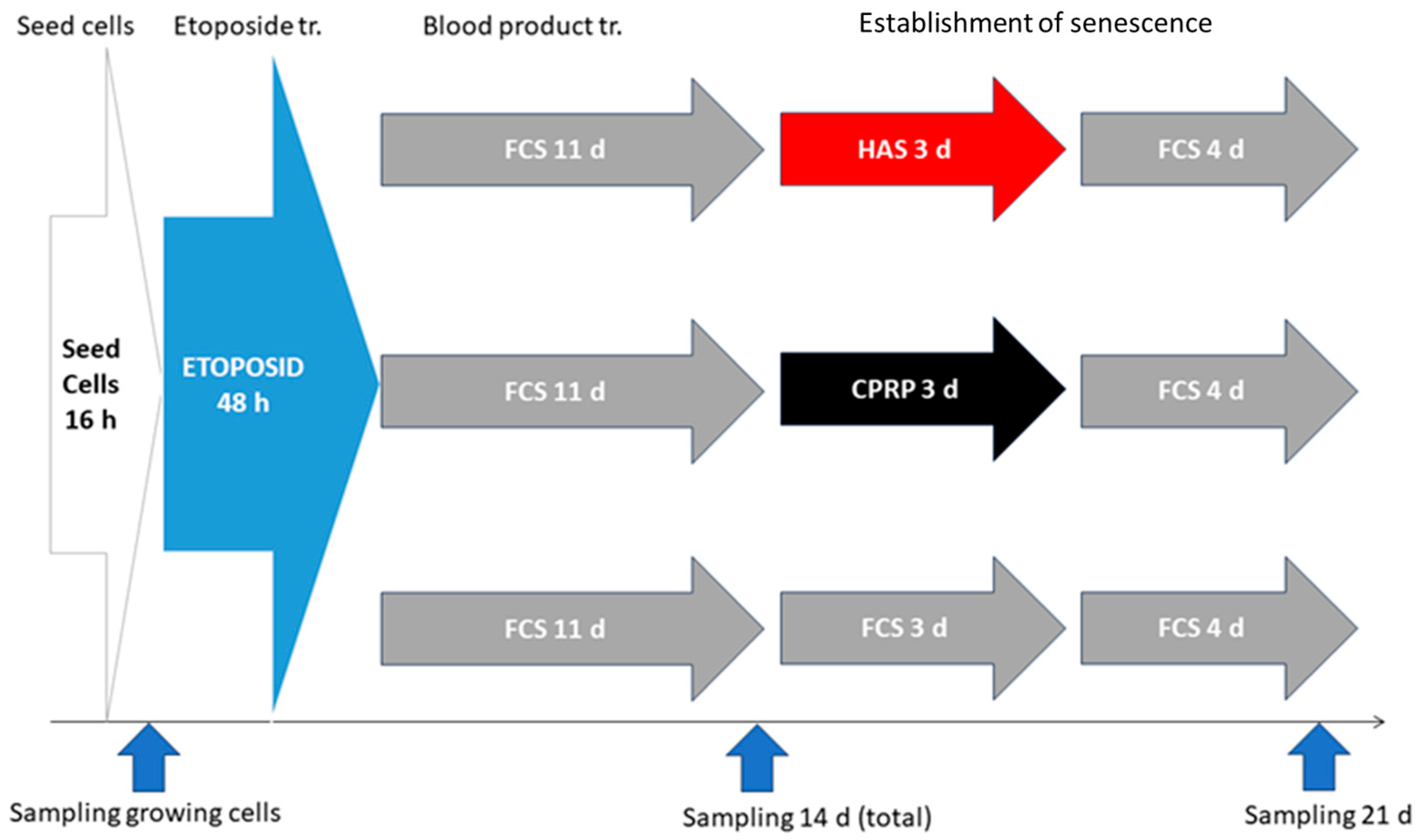
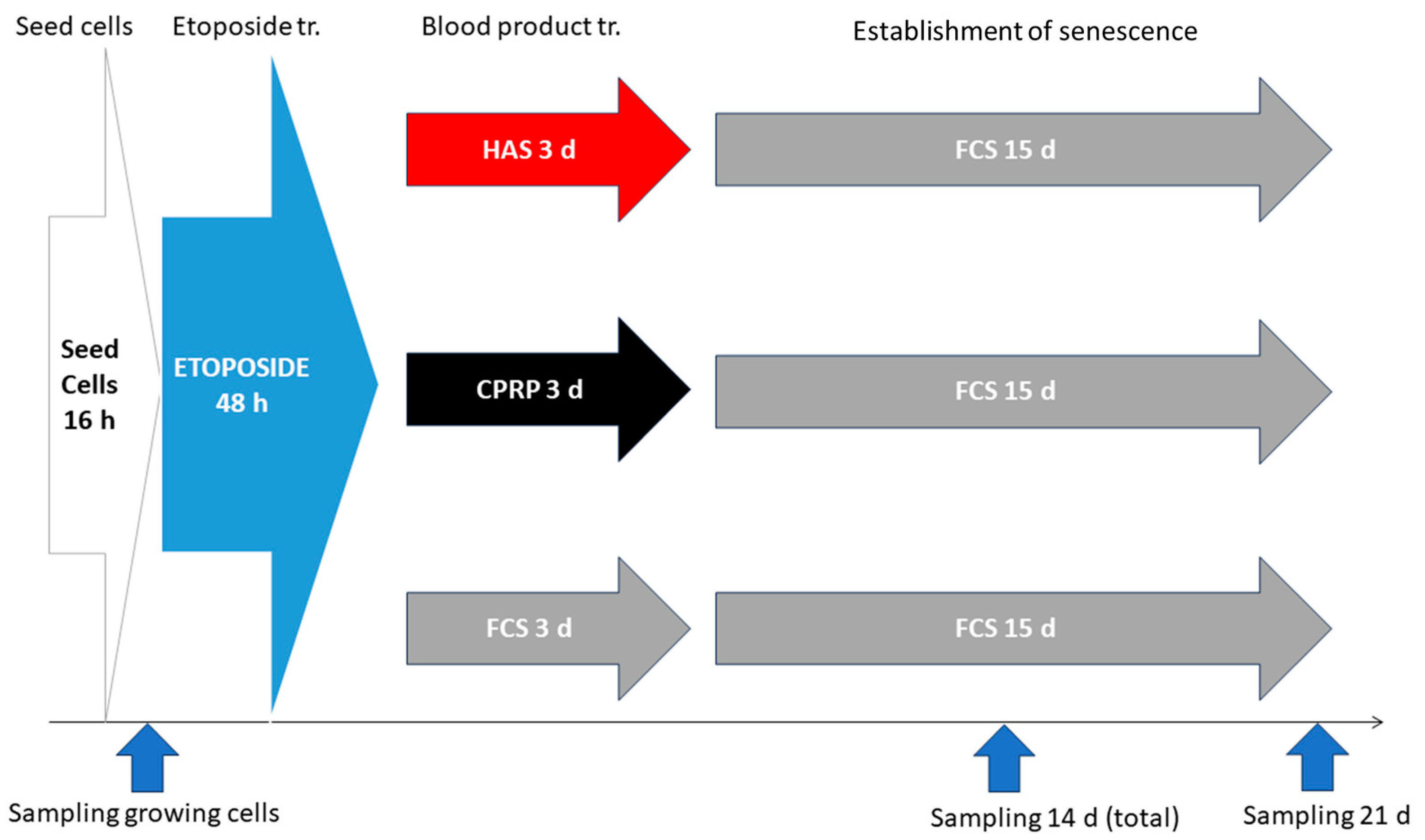
2.9. BP-Treatment for Late Exposure Group
2.9.1. Seeding of Cells and Etoposide Treatment
2.9.2. Establishment of Cellular Senescence
2.9.3. BP-Treatment
2.9.4. Final Incubation and Harvest
2.10. RNA Extraction and qPCR
2.11. Luminex Assay
2.12. Statistics
3. Results
3.1. Definition of Reagents
Blood-Derived Products
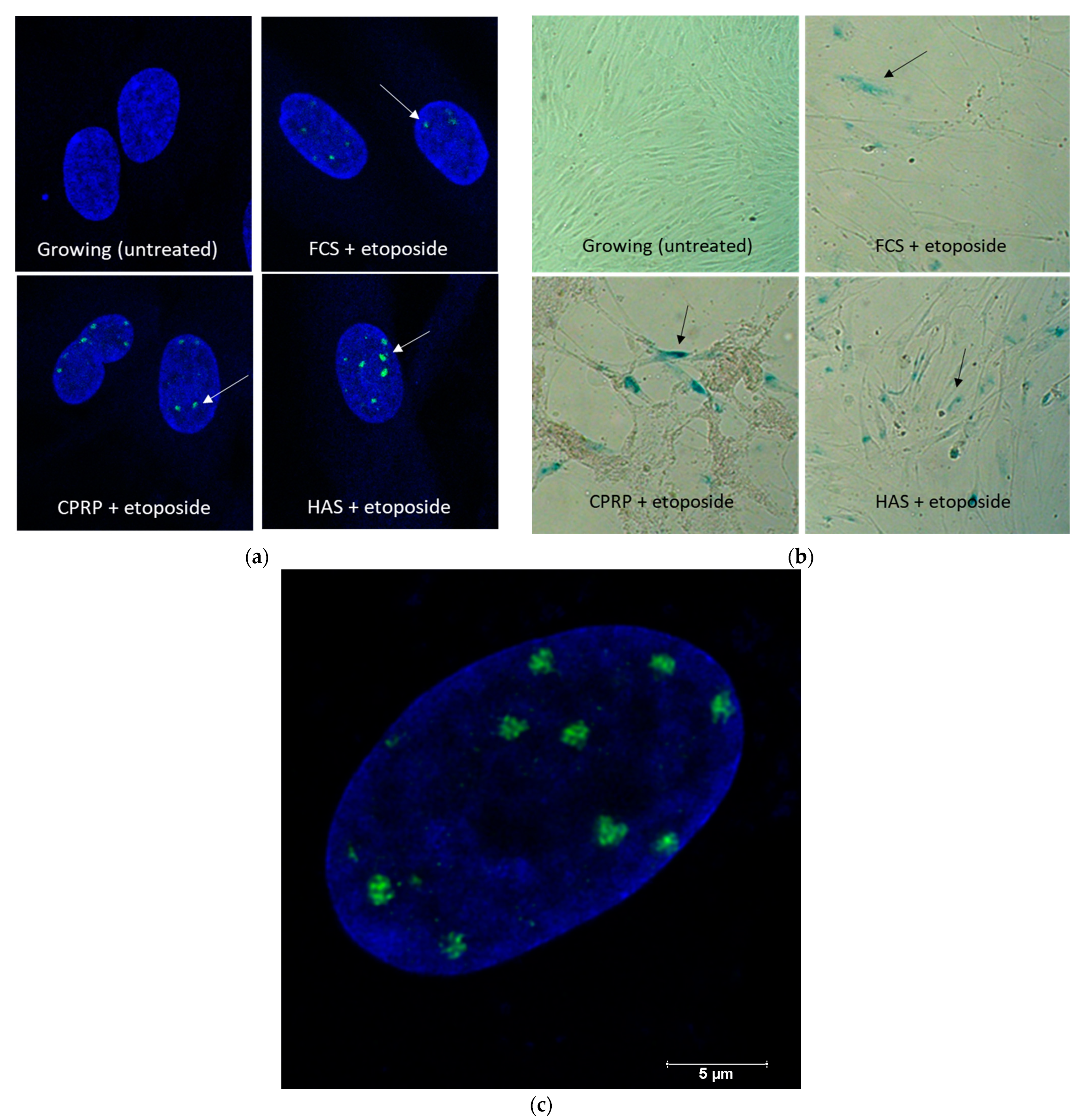
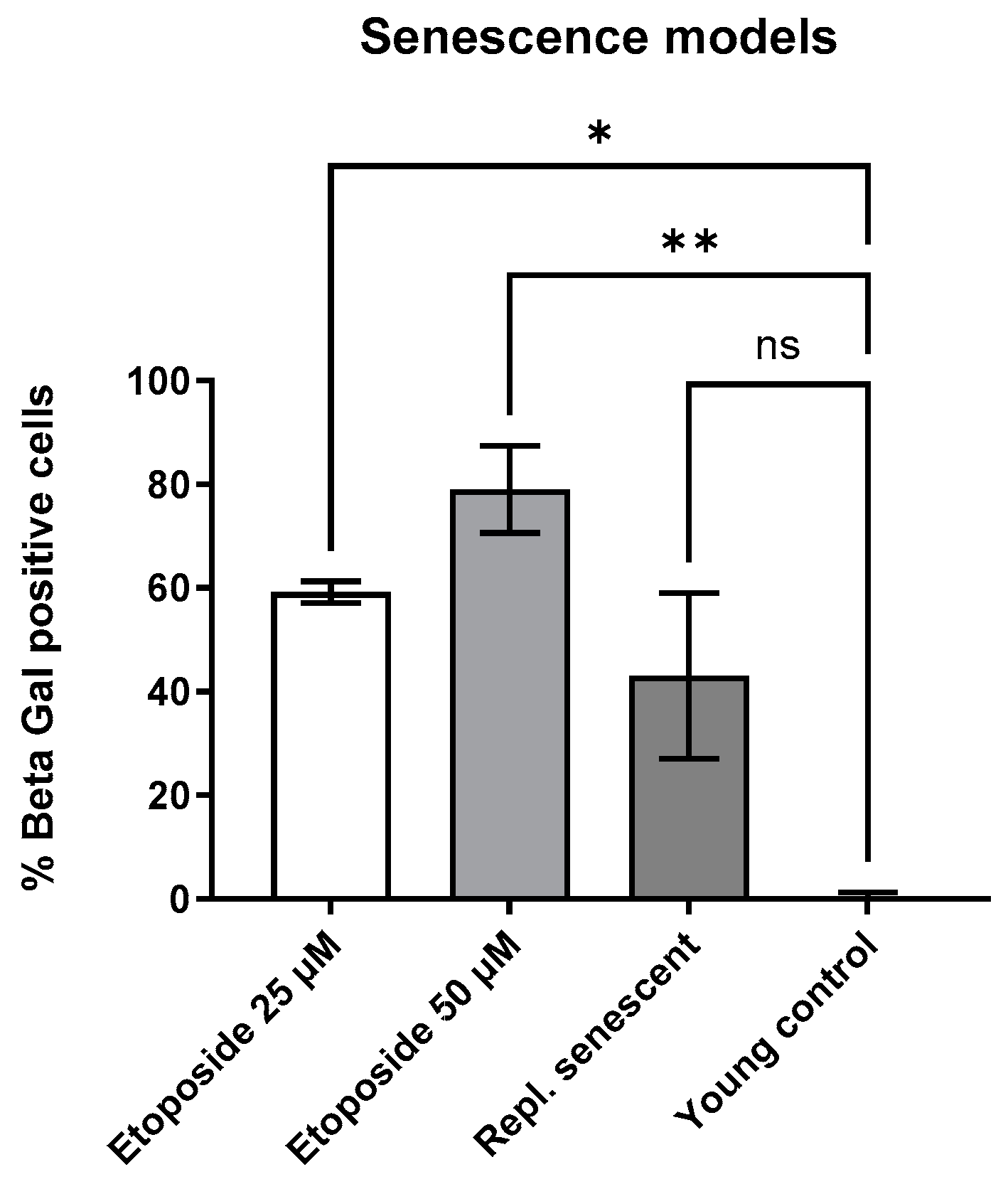
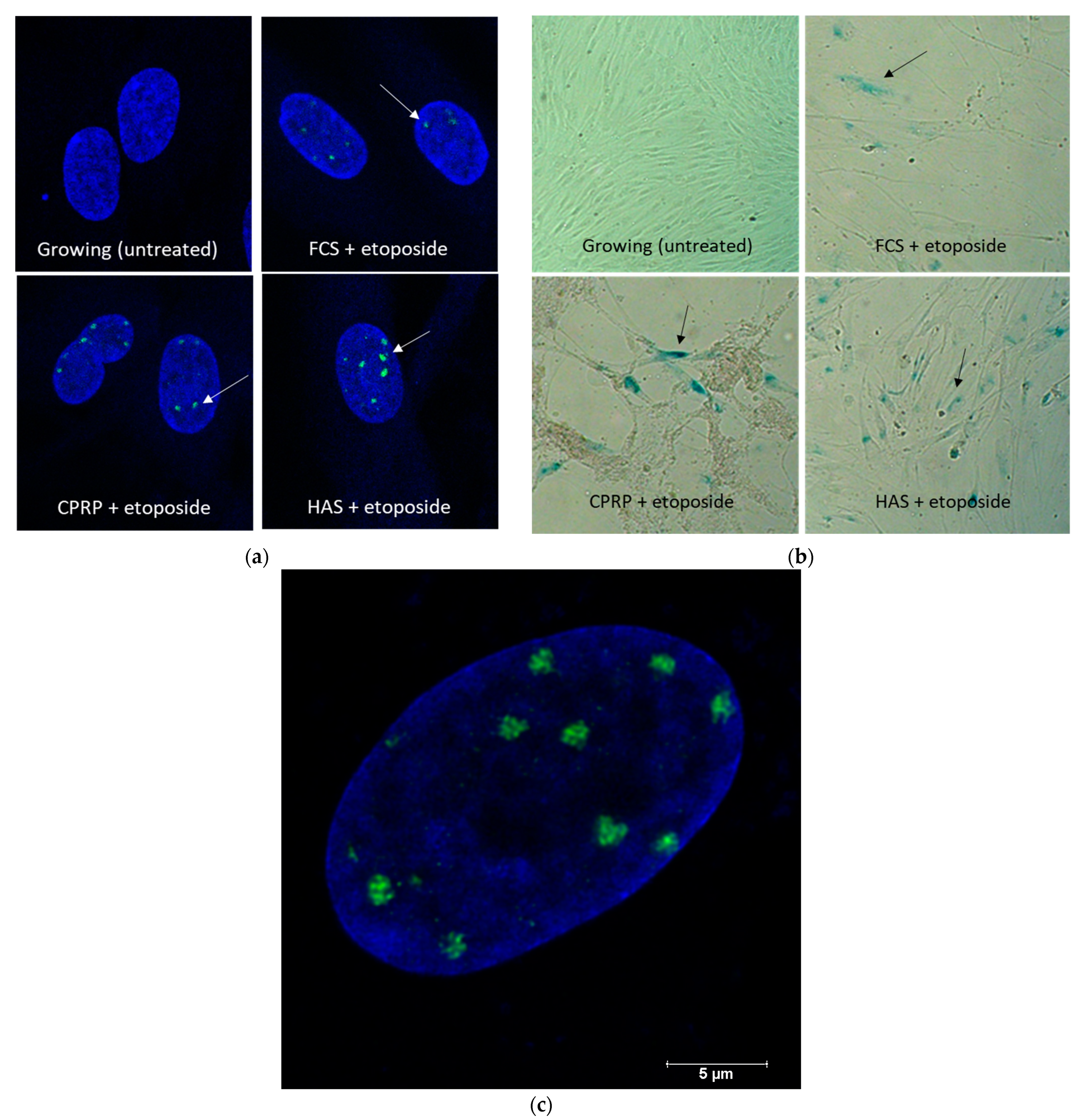
3.2. Induction of Senescent State
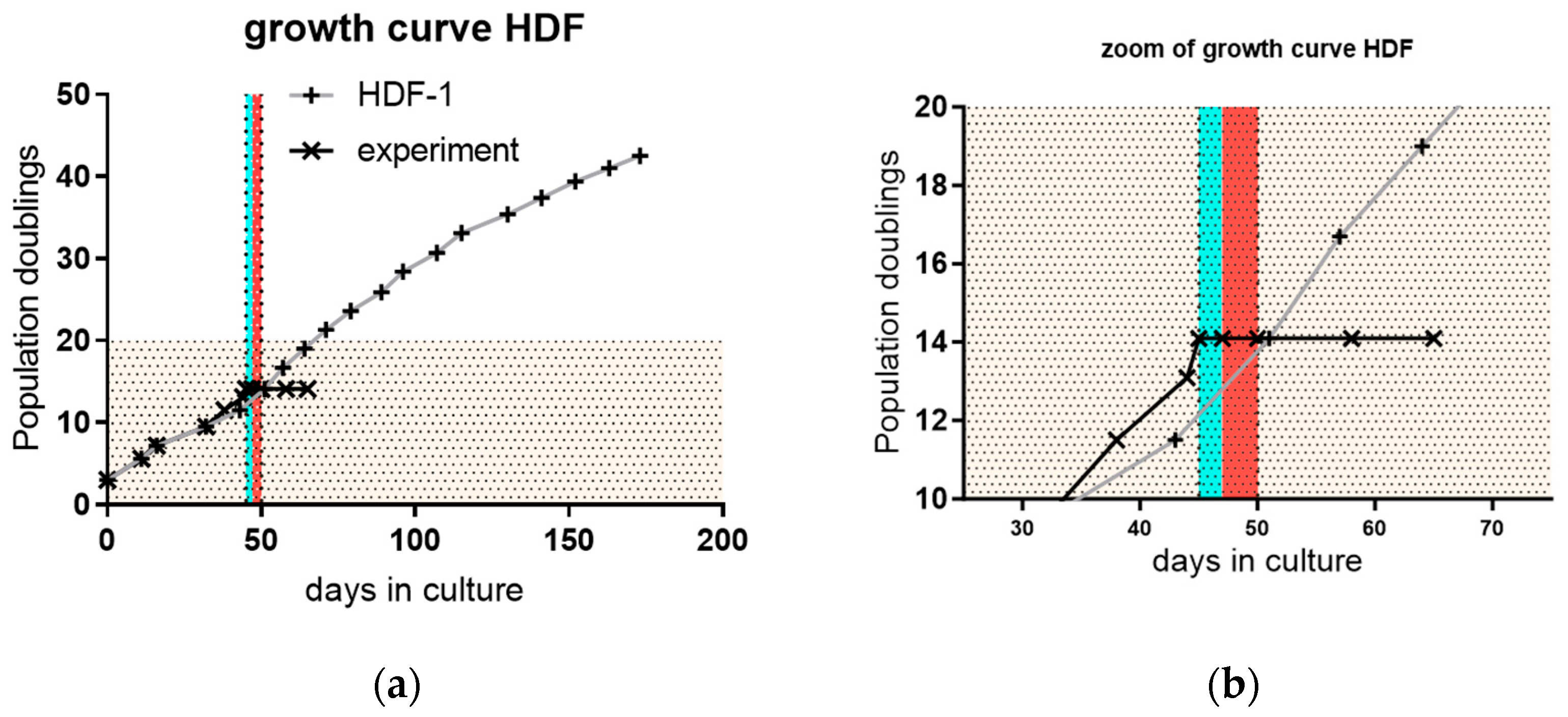
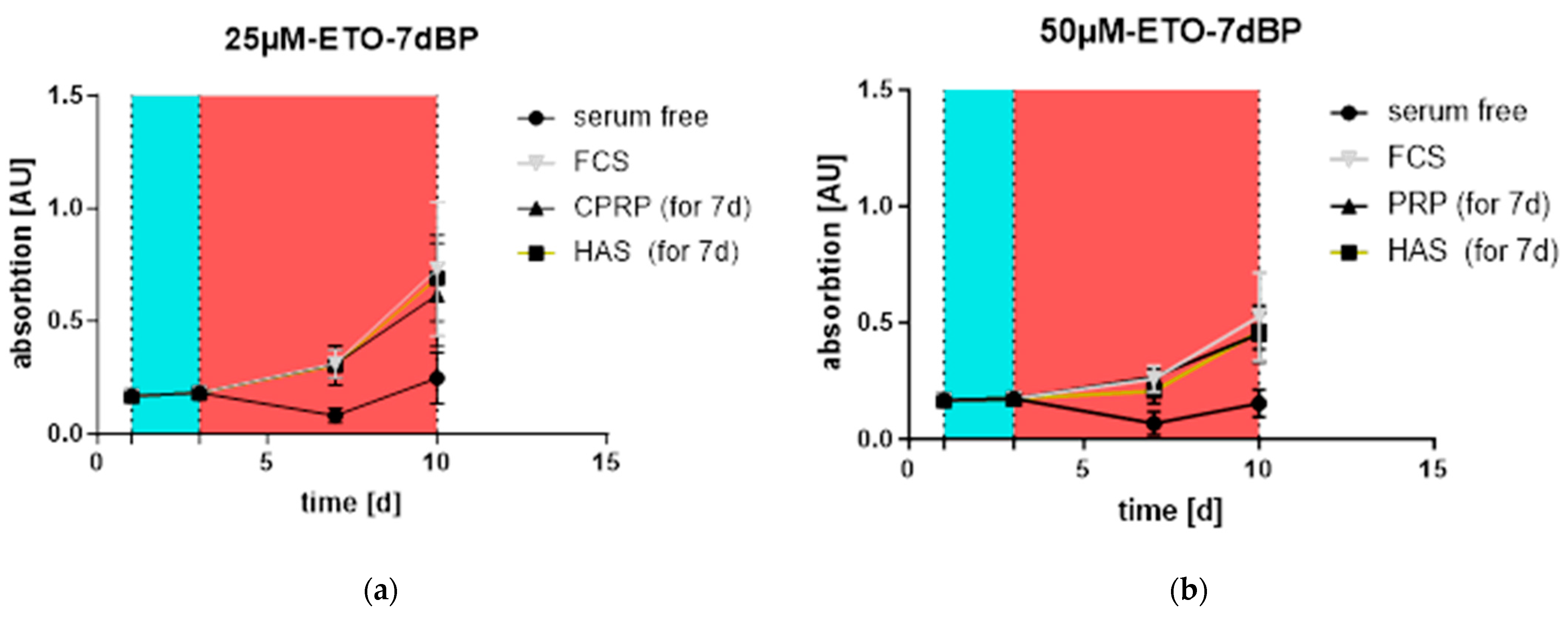
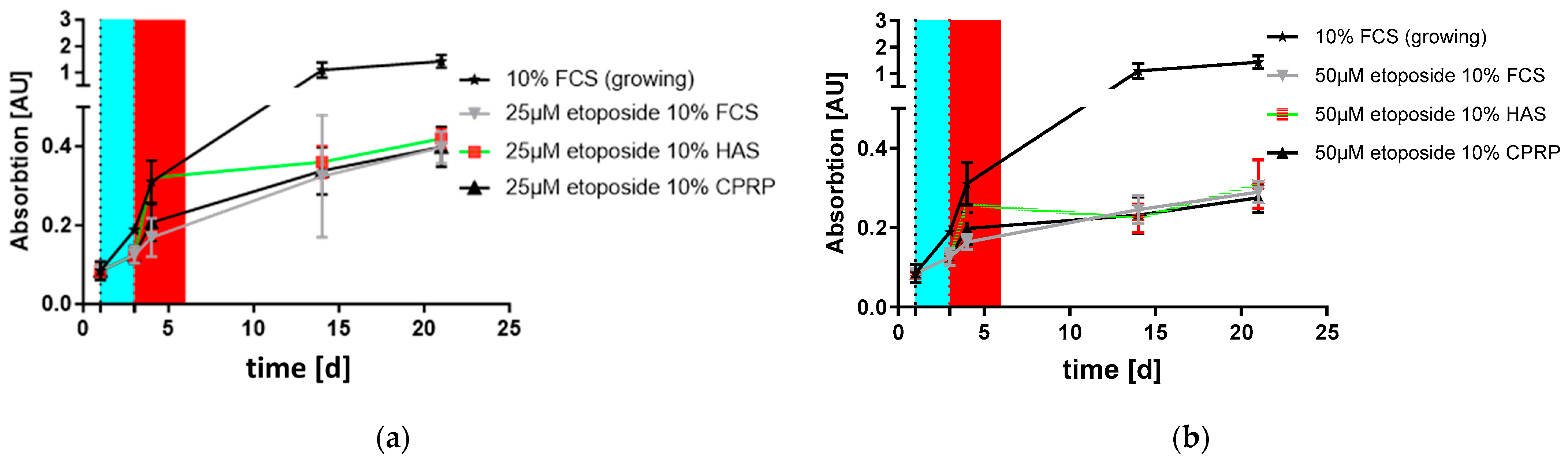
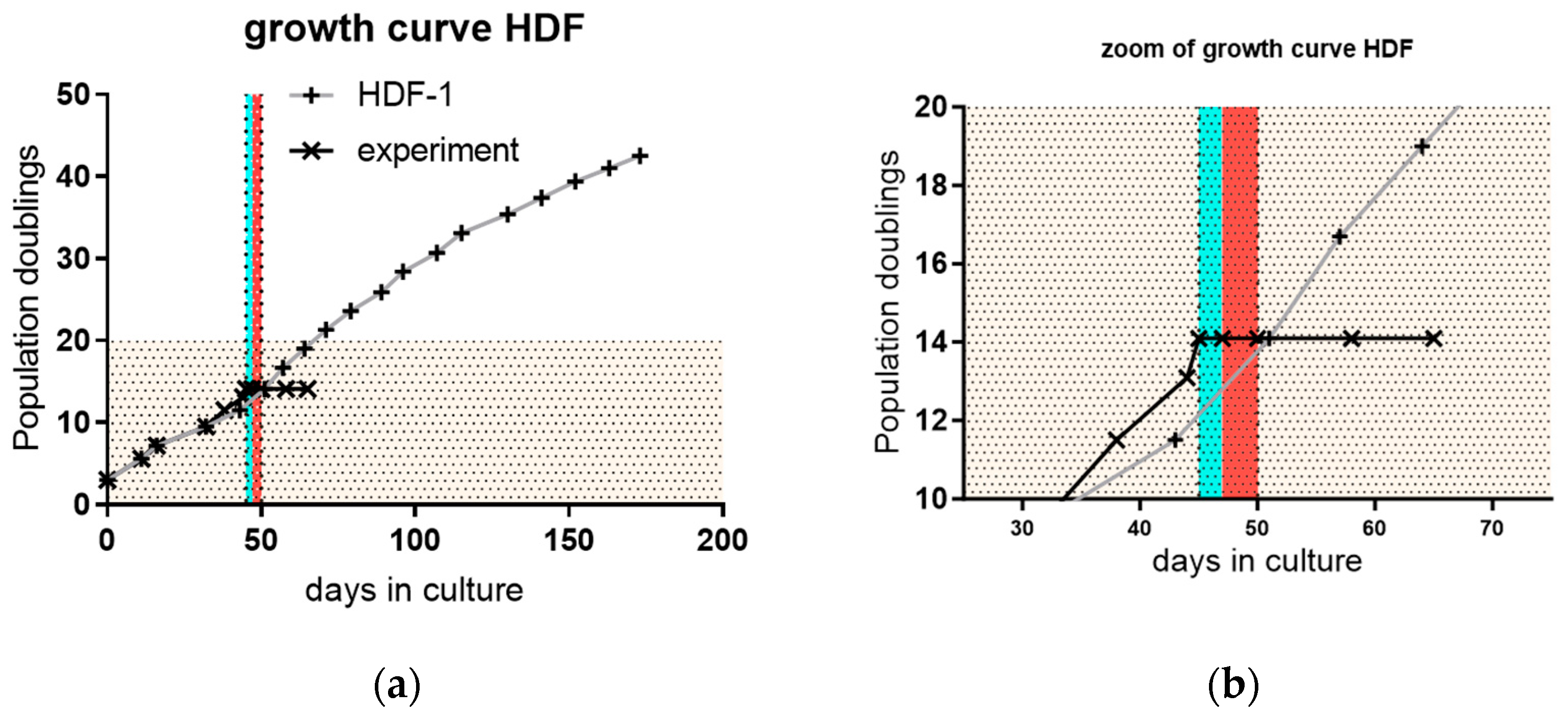
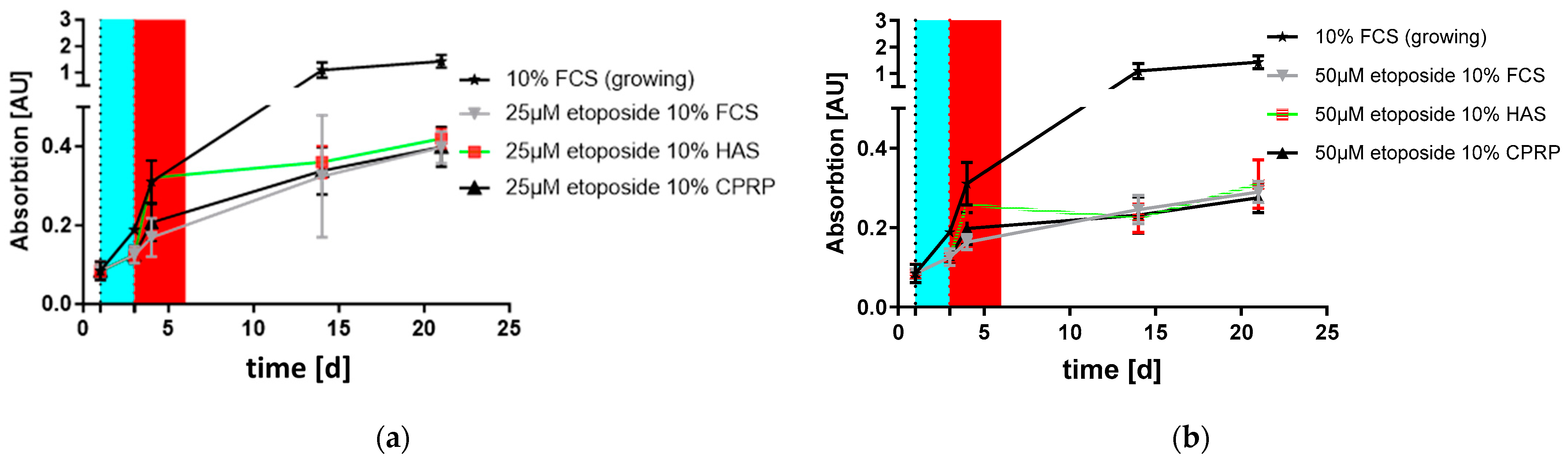
3.3. Proliferation and Outgrowth of HDF Cells
3.4. Investigations in Cellular Senescence and SASP-Factors IL-6 and IL-8
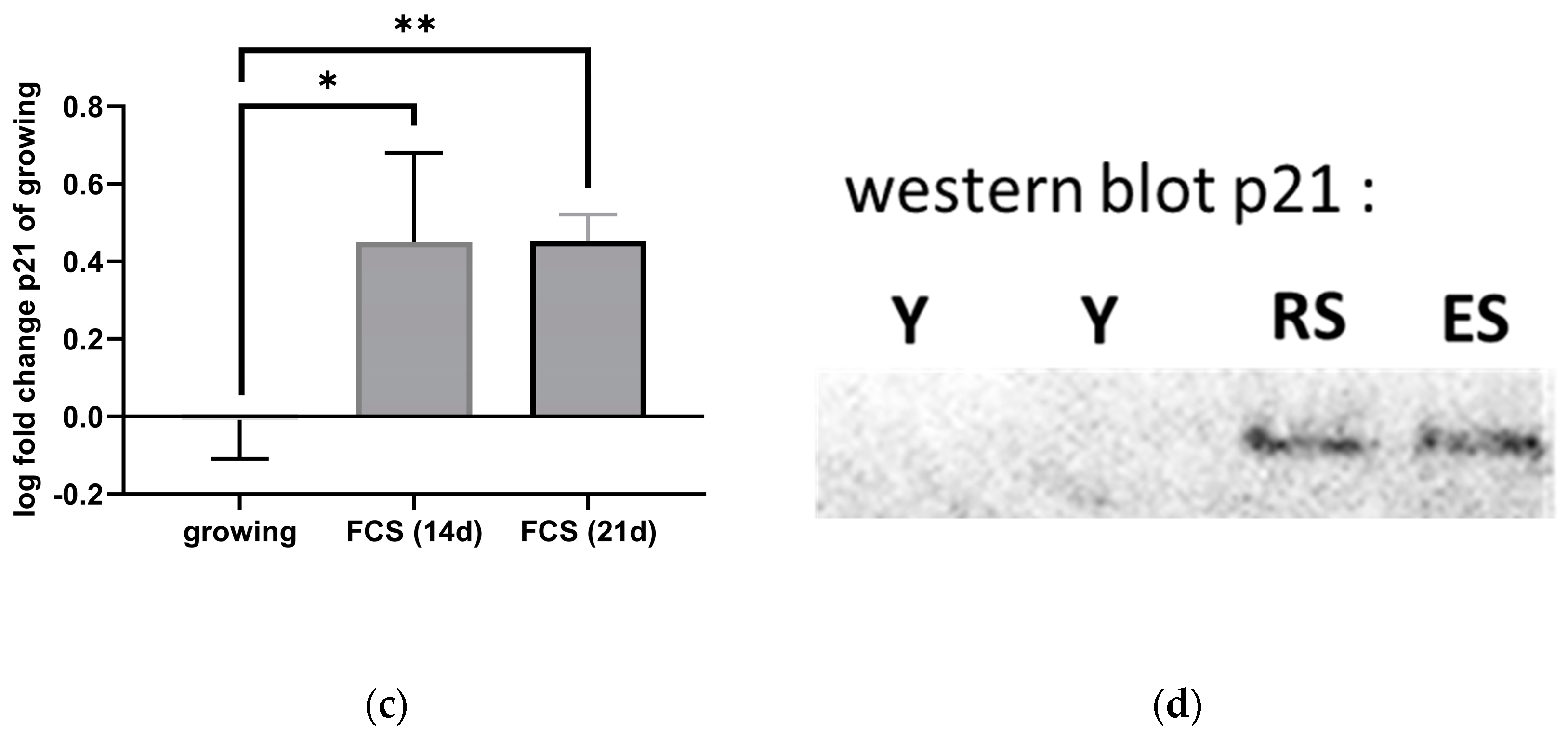
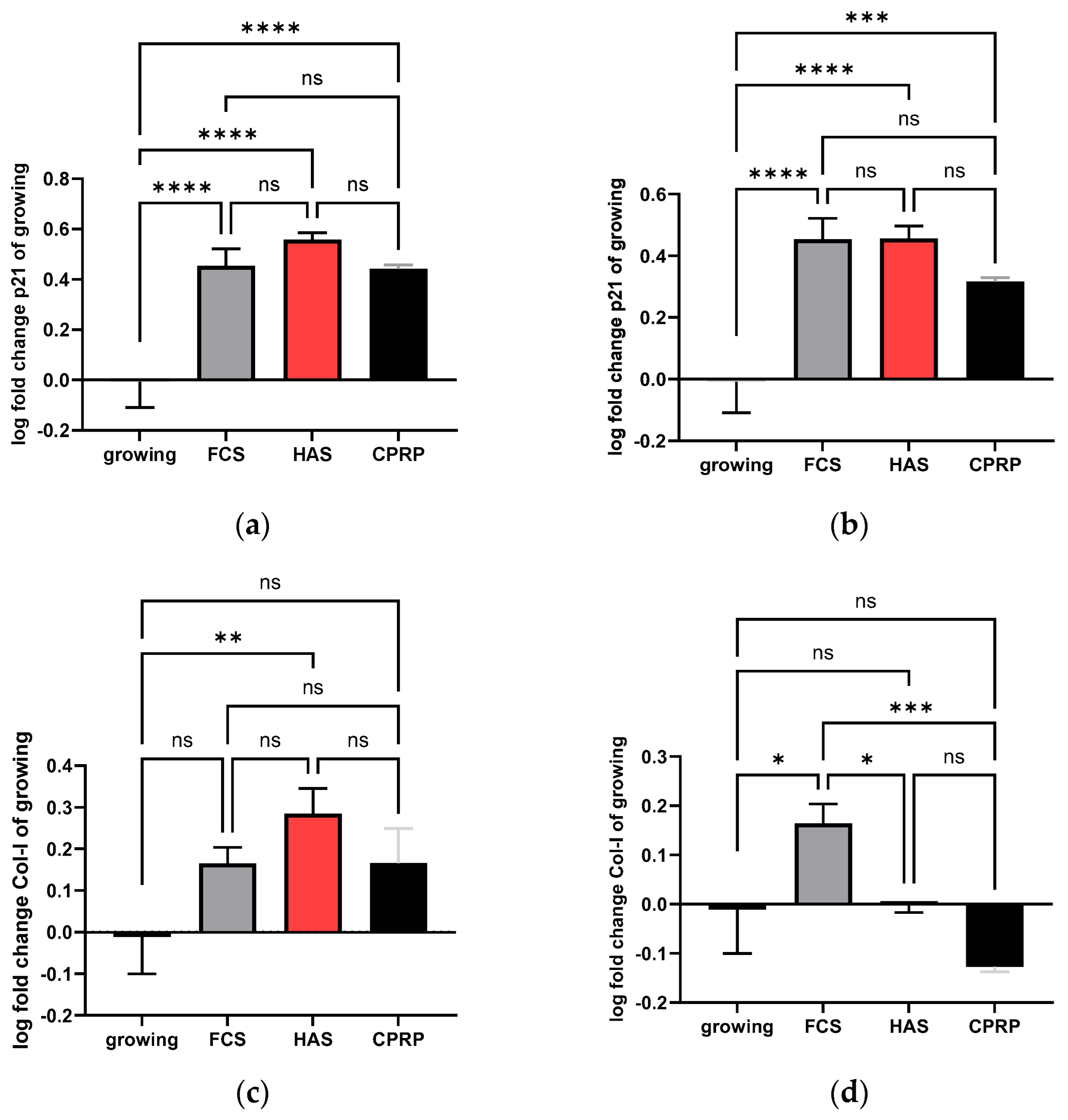
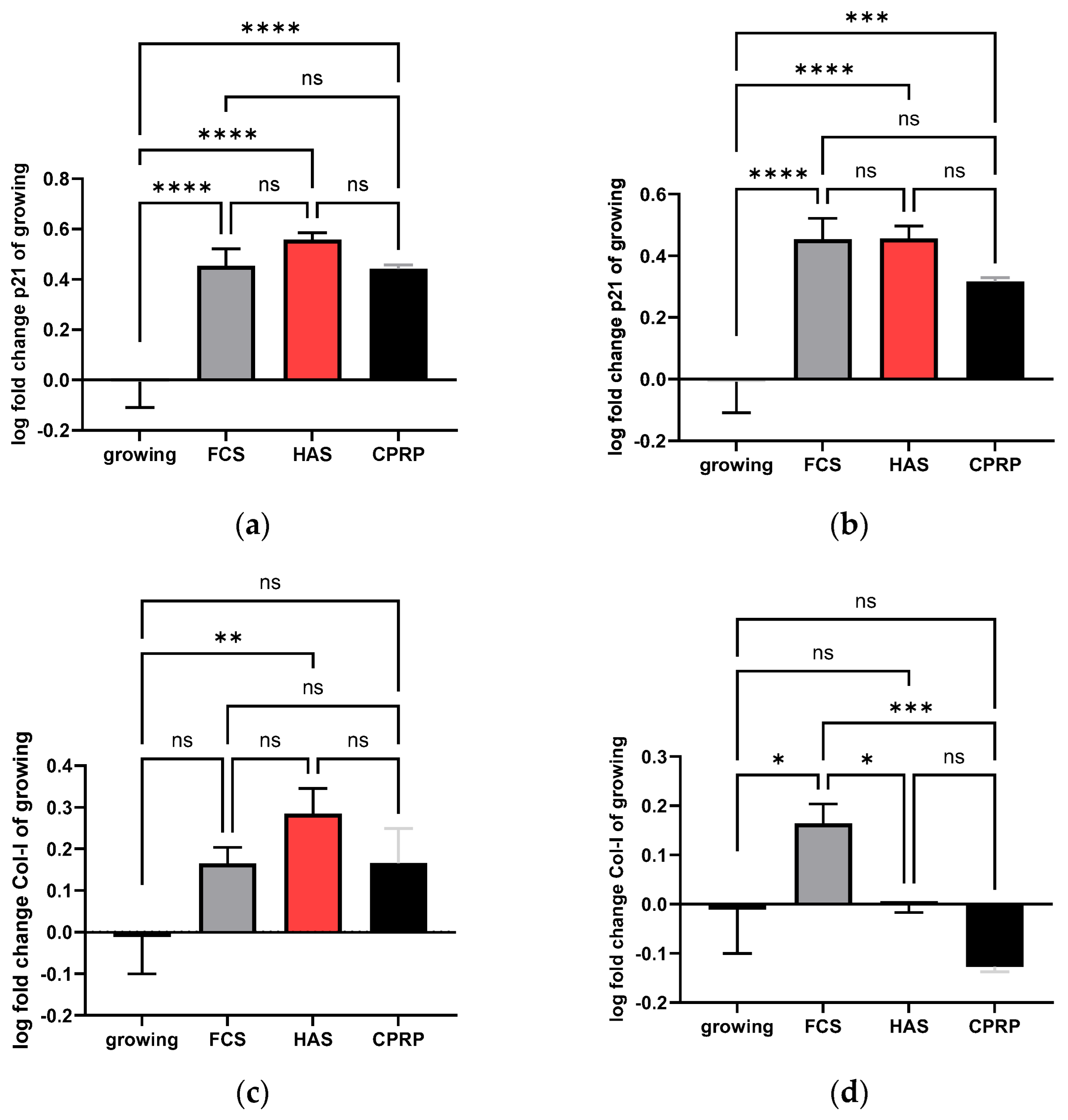
3.4.1. p21 Expression
3.4.2. Type I Collagen Expression
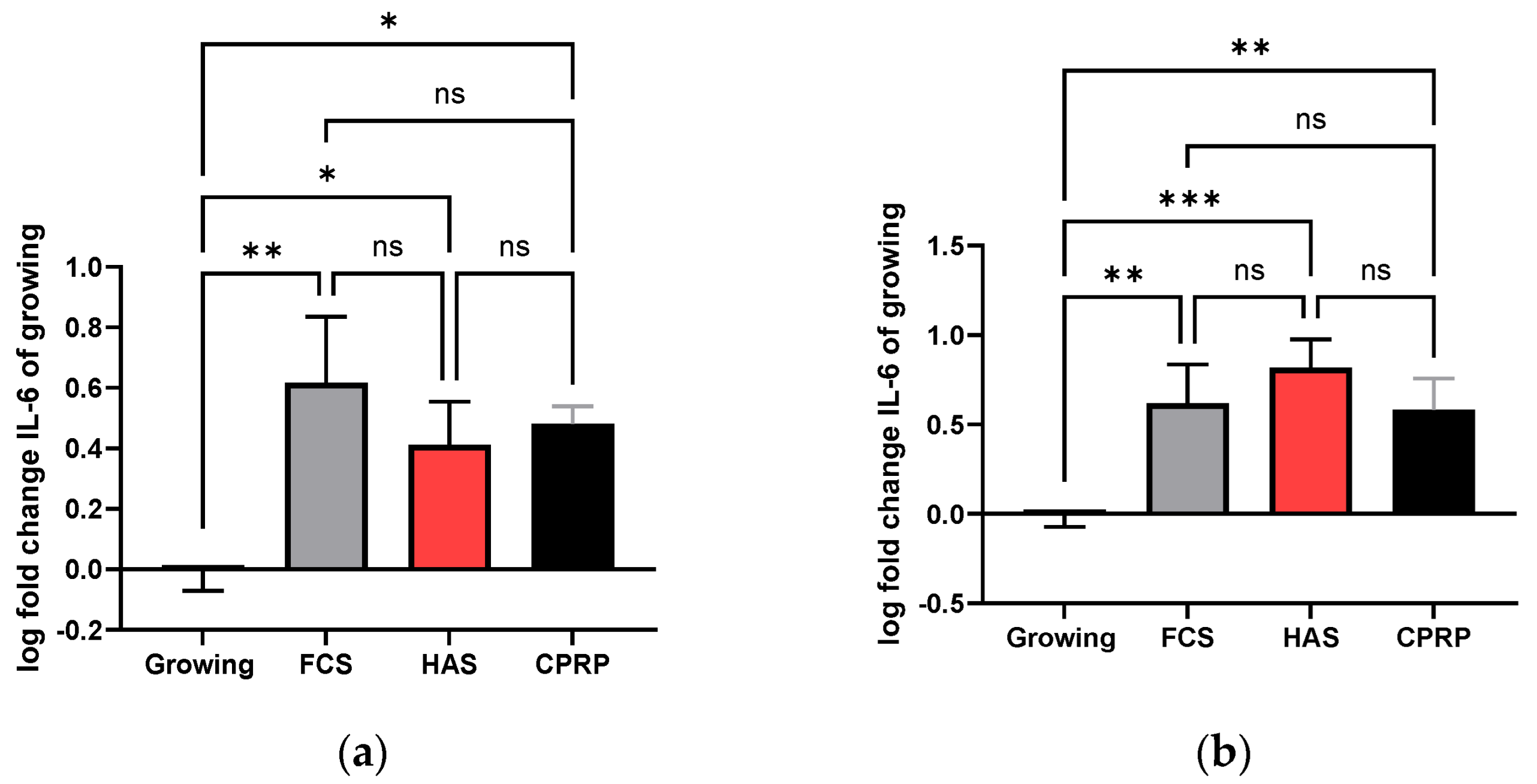
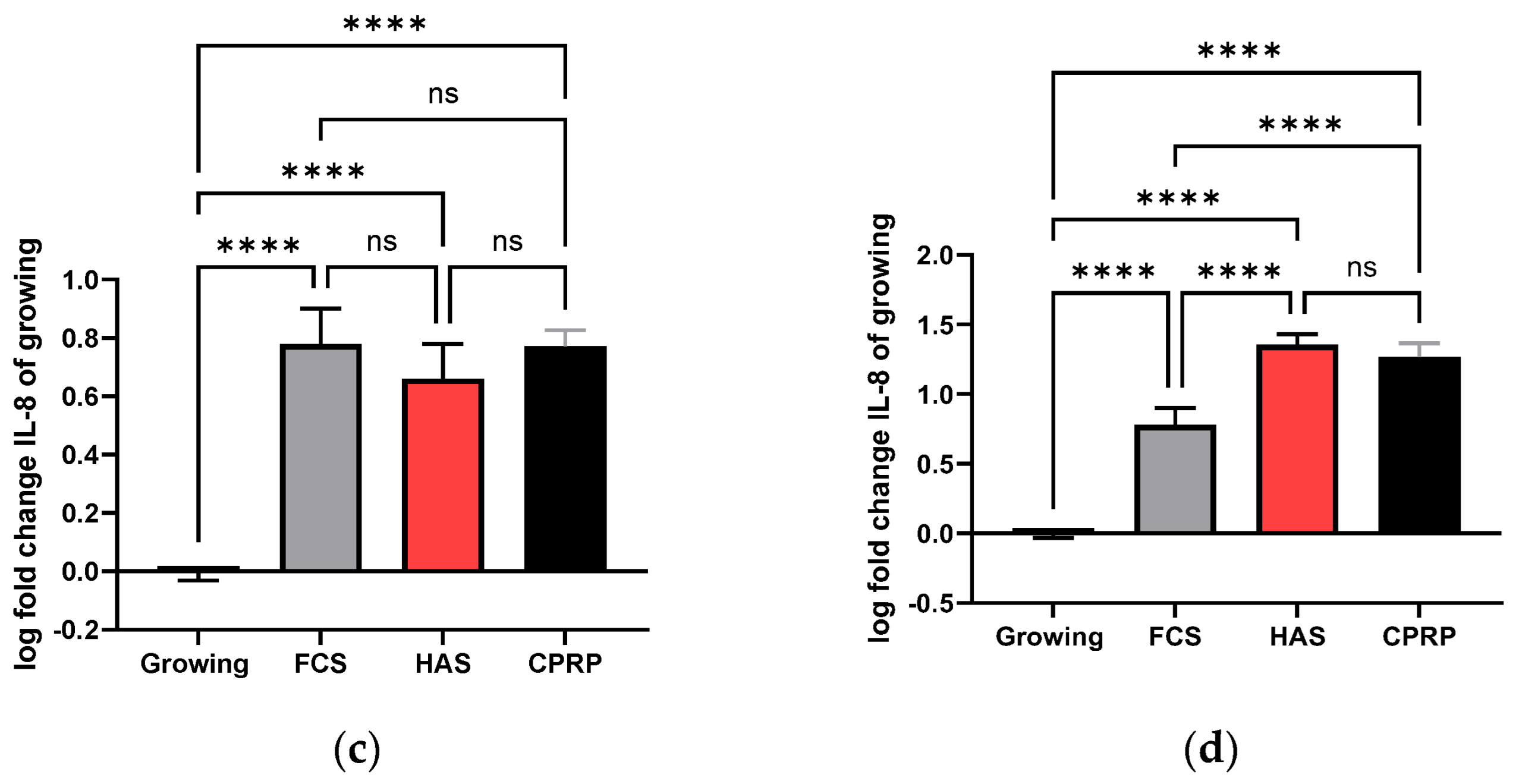
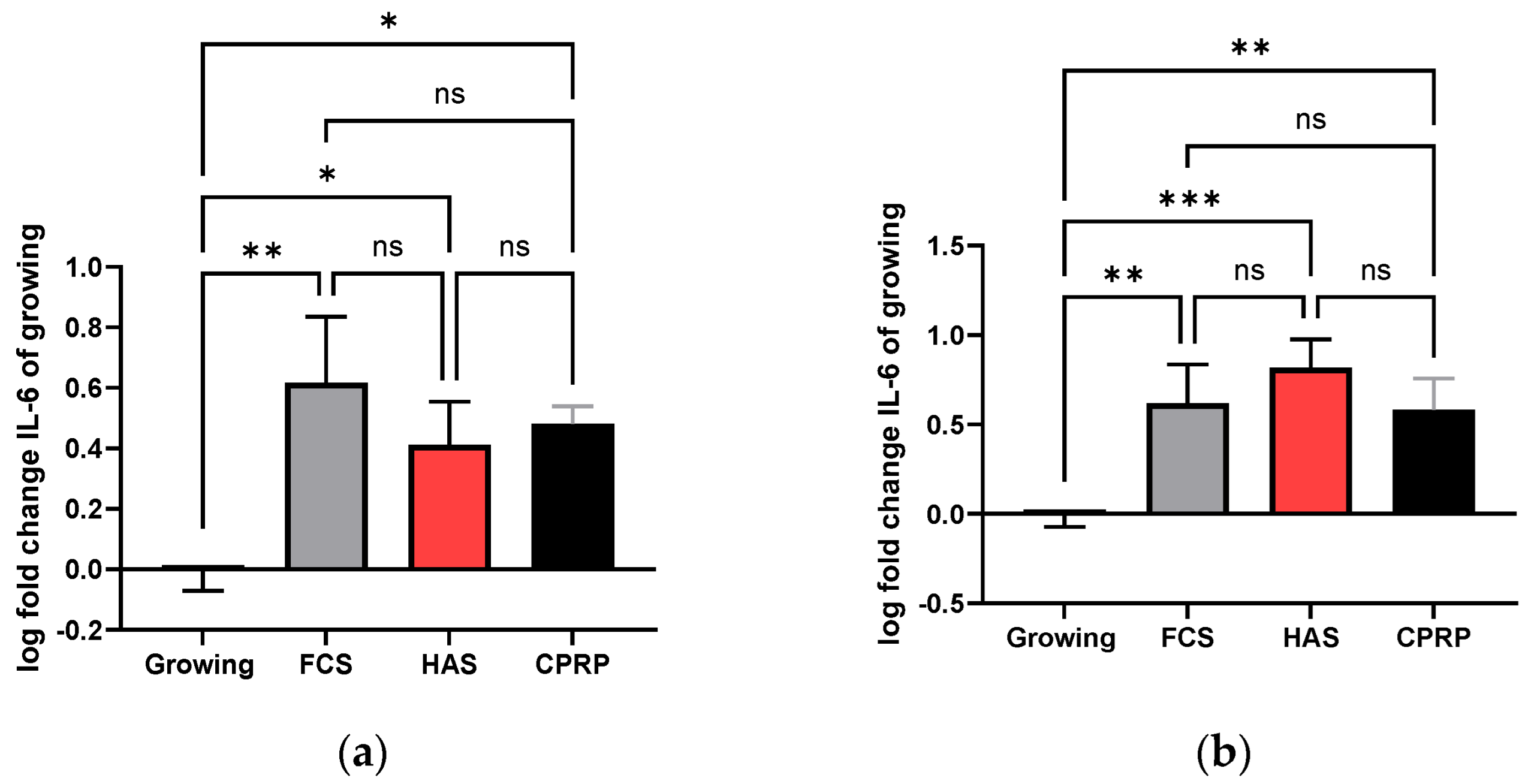
3.4.3. IL-6 Concentration in Supernatant
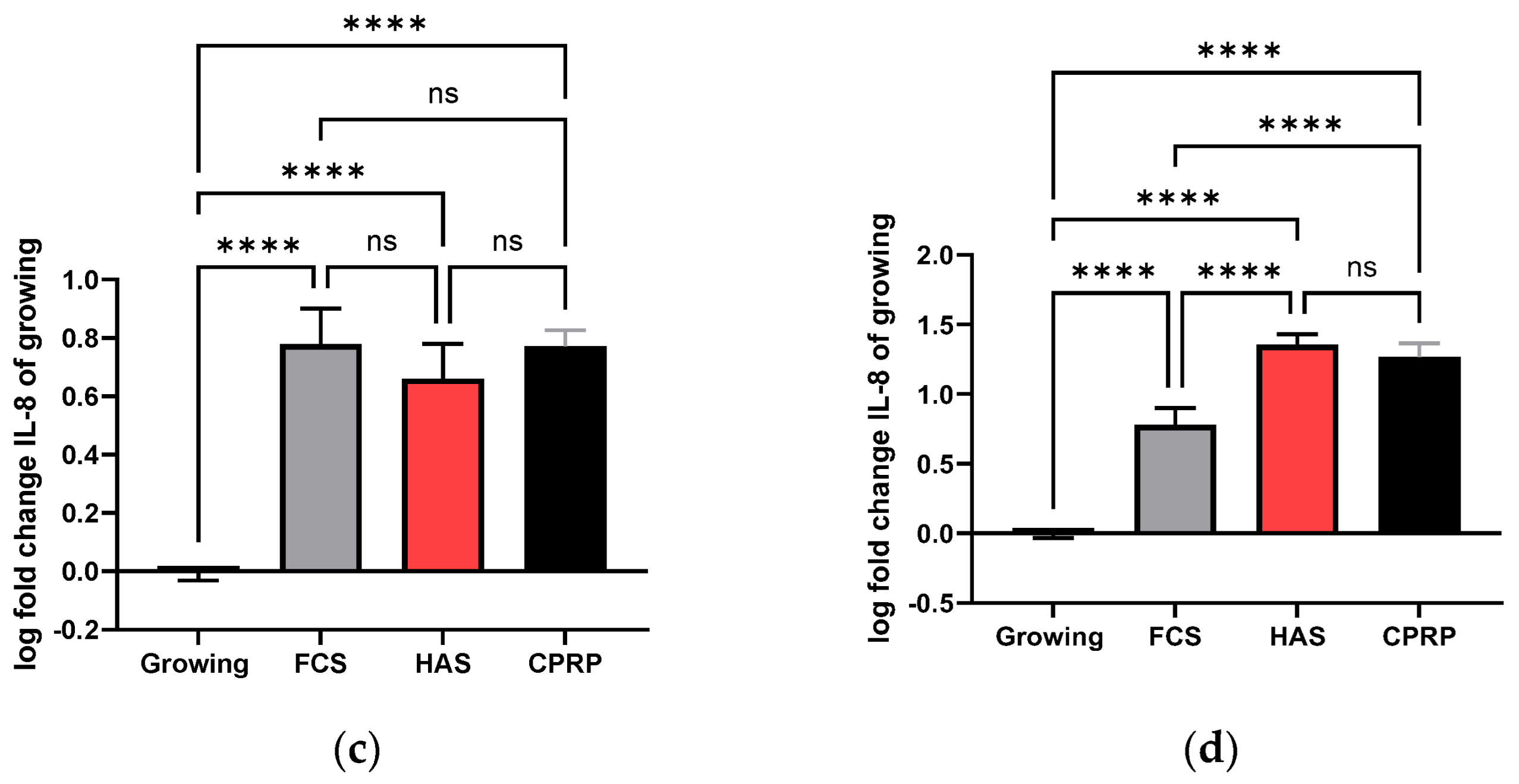
3.4.4. IL-8 Concentration in Supernatant
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4apositive senescent cells delays ageingassociated disorders. Nature 2011, 479, 232. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Toussaint, O.; Dumont, P.; Remacle, J.; Dierick, J.-F.; Pascal, T.; Frippiat, C.; Magalhaes, J.P.; Zdanov, S.; Chainiaux, F. Stress-induced premature senescence or stress-induced senescence-like phenotype: One in vivo reality, two possible definitions? Sci. World J. 2002, 2, 230–247. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef]
- Velarde, M.C.; Demaria, M.; Campisi, J. Senescent cells and their secretory phenotype as targets for cancer therapy. Cancer Aging Bench Clin. 2013, 38, 17–21. [Google Scholar]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front. Immunol. 2018, 9, 586. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Marin, I.; Serrano, M.; Pietrocola, F. Recent insights into the crosstalk between senescent cells and CD8 T lymphocytes. Npj Aging 2023, 9, 8. [Google Scholar] [CrossRef]
- Loughery, J.; Cox, M.; Smith, L.M.; Meek, D.W. Critical role for p53-serine 15 phosphorylation in stimulating transactivation at p53-responsive promoters. Nucleic Acids Res. 2014, 42, 7666–7680. [Google Scholar] [CrossRef]
- Rodier, F.; Kim, S.-H.; Nijjar, T.; Yaswen, P.; Campisi, J. Cancer and aging: The importance of telomeres in genome maintenance. Int. J. Biochem. Cell Biol. 2005, 37, 977–990. [Google Scholar] [CrossRef]
- Baldwin, E.L.; Osheroff, N. Etoposide, topoisomerase II and cancer. Curr. Med. Chem. Anticancer. Agents 2005, 5, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Petrova, N.V.; Velichko, A.K.; Razin, S.V.; Kantidze, O.L. Small molecule compounds that induce cellular senescence. Aging Cell 2016, 15, 999–1017. [Google Scholar] [CrossRef] [PubMed]
- Probin, V.; Wang, Y.; Bai, A.; Zhou, D. Busulfan selectively induces cellular senescence but not apoptosis in WI38 fibroblasts via a p53-independent but extracellular signal-regulated kinase-p38 mitogen-activated protein kinase-dependent mechanism. J. Pharmacol. Exp. Ther. 2006, 319, 551–560. [Google Scholar] [CrossRef]
- Odeh, A.; Dronina, M.; Domankevich, V.; Shams, I.; Manov, I. Downregulation of the inflammatory network in senescent fibroblasts and aging tissues of the long-lived and cancer-resistant subterranean wild rodent, Spalax. Aging Cell 2020, 19, e13045. [Google Scholar] [CrossRef]
- Griffiths, T.W.; Watson, R.E.B.; Langton, A.K. Skin ageing and topical rejuvenation strategies. Br. J. Dermatol. 2023, 189, i17–i23. [Google Scholar] [CrossRef]
- Farage, M.A.; Miller, K.W.; Elsner, P.; Maibach, H.I. Characteristics of the Aging Skin. Adv. Wound Care 2013, 2, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Boukamp, P. Skin aging: A role for telomerase and telomere dynamics? Curr. Mol. Med. 2005, 5, 171–177. [Google Scholar] [CrossRef]
- Waldera Lupa, D.M.; Kalfalah, F.; Safferling, K.; Boukamp, P.; Poschmann, G.; Volpi, E.; Götz-Rösch, C.; Bernerd, F.; Haag, L.; Huebenthal, U.; et al. Characterization of Skin Aging-Associated Secreted Proteins (SAASP) Produced by Dermal Fibroblasts Isolated from Intrinsically Aged Human Skin. J. Investig. Dermatol. 2015, 135, 1954–1968. [Google Scholar] [CrossRef]
- Ghosh, K.; Capell, B.C. The Senescence-Associated Secretory Phenotype: Critical Effector in Skin Cancer and Aging. J. Investig. Dermatol. 2016, 136, 2133–2139. [Google Scholar] [CrossRef]
- Charles-de-Sá, L.; Gontijo-de-Amorim, N.F.; Takiya, C.M.; Borojevic, R.; Benati, D.; Bernardi, P.; Sbarbati, A.; Rigotti, G. Effect of Use of Platelet-Rich Plasma (PRP) in Skin with Intrinsic Aging Process. Aesthet. Surg. J. 2018, 38, 321–328. [Google Scholar] [CrossRef]
- Maisel-Campbell, A.L.; Ismail, A.; Reynolds, K.A.; Poon, E.; Serrano, L.; Grushchak, S.; Farid, C.; West, D.P.; Alam, M. A systematic review of the safety and effectiveness of platelet-rich plasma (PRP) for skin aging. Arch. Dermatol. Res. 2019, 312, 301–315. [Google Scholar] [CrossRef]
- Kardos, D.; Simon, M.; Vácz, G.; Hinsenkamp, A.; Holczer, T.; Cseh, D.; Sárközi, A.; Szenthe, K.; Bánáti, F.; Szathmary, S.; et al. The Composition of Hyperacute Serum and Platelet-Rich Plasma Is Markedly Different despite the Similar Production Method. Int. J. Mol. Sci. 2019, 20, 721. [Google Scholar] [CrossRef]
- Jia, C.; Lu, Y.; Bi, B.; Chen, L.; Yang, Q.; Yang, P.; Guo, Y.; Zhu, J.; Zhu, N.; Liu, T. Platelet-rich plasma ameliorates senescence-like phenotypes in a cellular photoaging model. RSC Adv. 2017, 7, 3152–3160. [Google Scholar] [CrossRef]
- Kuten, O.; Simon, M.; Hornyák, I.; de Luna-Preitschopf, A.; Nehrer, S.; Lacza, Z. The Effects of Hyperacute Serum on Adipogenesis and Cell Proliferation of Mesenchymal Stromal Cells. Tissue Eng. Part A 2018, 24, 1011–1021. [Google Scholar] [CrossRef] [PubMed]
- Jeyakumar, V.; Niculescu-Morzsa, E.; Bauer, C.; Lacza, Z.; Nehrer, S. Platelet-Rich Plasma Supports Proliferation and Redifferentiation of Chondrocytes during In Vitro Expansion. Front. Bioeng. Biotechnol. 2017, 5, 75. [Google Scholar] [CrossRef]
- Fabi, S.; Sundaram, H. The potential of topical and injectable growth factors and cytokines for skin rejuvenation. Facial Plast. Surg. 2014, 30, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Passaretti, F.; Tia, M.; D’Esposito, V.; de Pascale, M.; Del Corso, M.; Sepulveres, R.; Liguoro, D.; Valentino, R.; Beguinot, F.; Formisano, P.; et al. Growth-promoting action and growth factor release by different platelet derivatives. Platelets 2014, 25, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Lammermann, I.; Terlecki-Zaniewicz, L.; Weinmullner, R.; Schosserer, M.; Dellago, H.; de Matos Branco, A.D.; Autheried, D.; Sevcnikar, B.; Kleissl, L.; Berlin, I.; et al. Blocking negative effects of senescence in human skin fibroblasts with a plant extract. NPJ Aging Mech. Dis. 2018, 4, 4. [Google Scholar] [CrossRef]
- Neubauer, M.; Kuten, O.; Stotter, C.; Kramer, K.; de Luna, A.; Muellner, T.; Lacza, Z.; Nehrer, S. The Effect of Blood-Derived Products on the Chondrogenic and Osteogenic Differentiation Potential of Adipose-Derived Mesenchymal Stem Cells Originated from Three Different Locations. Stem Cells Int. 2019, 2019, 1358267. [Google Scholar] [CrossRef]
- Otahal, A.; Kuten-Pella, O.; Kramer, K.; Neubauer, M.; Lacza, Z.; Nehrer, S.; de Luna, A. Functional repertoire of EV-associated miRNA profiles after lipoprotein depletion via ultracentrifugation and size exclusion chromatography from autologous blood products. Sci. Rep. 2021, 11, 5823. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Chahla, J.; Cinque, M.E.; Piuzzi, N.S.; Mannava, S.; Geeslin, A.G.; Murray, I.R.; Dornan, G.J.; Muschler, G.F.; LaPrade, R.F. A Call for Standardization in Platelet-Rich Plasma Preparation Protocols and Composition Reporting: A Systematic Review of the Clinical Orthopaedic Literature. J. Bone Jt. Surg. Am. 2017, 99, 1769–1779. [Google Scholar] [CrossRef]
- Martínez, C.E.; Smith, P.C.; Palma Alvarado, V.A. The influence of platelet-derived products on angiogenesis and tissue repair: A concise update. Front. Physiol. 2015, 6, 290. [Google Scholar] [CrossRef]
- Alessio, N.; Aprile, D.; Cappabianca, S.; Peluso, G.; Di Bernardo, G.; Galderisi, U. Different Stages of Quiescence, Senescence, and Cell Stress Identified by Molecular Algorithm Based on the Expression of Ki67, RPS6, and Beta-Galactosidase Activity. Int. J. Mol. Sci. 2021, 22, 3102. [Google Scholar] [CrossRef] [PubMed]
- Soubeyrand, S.; Pope, L.; Haché, R.J.G. Topoisomerase IIalpha-dependent induction of a persistent DNA damage response in response to transient etoposide exposure. Mol. Oncol. 2010, 4, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Biran, A.; Zada, L.; Abou Karam, P.; Vadai, E.; Roitman, L.; Ovadya, Y.; Porat, Z.; Krizhanovsky, V. Quantitative identification of senescent cells in aging and disease. Aging Cell 2017, 16, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Wyld, L.; Bellantuono, I.; Tchkonia, T.; Morgan, J.; Turner, O.; Foss, F.; George, J.; Danson, S.; Kirkland, J.L. Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies. Cancers 2020, 12, 2134. [Google Scholar] [CrossRef] [PubMed]
- Liguori, T.T.A.; Liguori, G.R.; Moreira, L.F.P.; Harmsen, M.C. Fibroblast growth factor-2, but not the adipose tissue-derived stromal cells secretome, inhibits TGF-β1-induced differentiation of human cardiac fibroblasts into myofibroblasts. Sci. Rep. 2018, 8, 16633. [Google Scholar] [CrossRef] [PubMed]
- Iocono, J.A.; Colleran, K.R.; Remick, D.G.; Gillespie, B.W.; Ehrlich, H.P.; Garner, W.L. Interleukin-8 levels and activity in delayed-healing human thermal wounds. Wound Repair. Regen. 2000, 8, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Quan, W.-Y.; Ko, J.-A.; Yanai, R.; Nakamura, Y.; Nishida, T. Integrin-mediated inhibition of interleukin-8 secretion from human neutrophils by collagen type I. J. Leukoc. Biol. 2010, 87, 487–491. [Google Scholar] [CrossRef] [PubMed]
- PubMed Central. Can Platelet-Rich Plasma Be Used for Skin Rejuvenation? Evaluation of Effects of Platelet-rich Plasma on Human Dermal Fibroblast. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3229934/ (accessed on 23 February 2024).
- Hernandez-Segura, A.; Rubingh, R.; Demaria, M. Identification of stable senescence-associated reference genes. Aging Cell 2019, 18, e12911. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kühnel, H.; Pasztorek, M.; Kuten-Pella, O.; Kramer, K.; Bauer, C.; Lacza, Z.; Nehrer, S. Effects of Blood-Derived Products on Cellular Senescence and Inflammatory Response: A Study on Skin Rejuvenation. Curr. Issues Mol. Biol. 2024, 46, 1865-1885. https://doi.org/10.3390/cimb46030122
Kühnel H, Pasztorek M, Kuten-Pella O, Kramer K, Bauer C, Lacza Z, Nehrer S. Effects of Blood-Derived Products on Cellular Senescence and Inflammatory Response: A Study on Skin Rejuvenation. Current Issues in Molecular Biology. 2024; 46(3):1865-1885. https://doi.org/10.3390/cimb46030122
Chicago/Turabian StyleKühnel, Harald, Markus Pasztorek, Olga Kuten-Pella, Karina Kramer, Christoph Bauer, Zsombor Lacza, and Stefan Nehrer. 2024. "Effects of Blood-Derived Products on Cellular Senescence and Inflammatory Response: A Study on Skin Rejuvenation" Current Issues in Molecular Biology 46, no. 3: 1865-1885. https://doi.org/10.3390/cimb46030122
APA StyleKühnel, H., Pasztorek, M., Kuten-Pella, O., Kramer, K., Bauer, C., Lacza, Z., & Nehrer, S. (2024). Effects of Blood-Derived Products on Cellular Senescence and Inflammatory Response: A Study on Skin Rejuvenation. Current Issues in Molecular Biology, 46(3), 1865-1885. https://doi.org/10.3390/cimb46030122