Targeting CDK9 in Cancer: An Integrated Approach of Combining In Silico Screening with Experimental Validation for Novel Degraders



Abstract
1. Introduction
2. Materials and Methods
2.1. Computational Methods
2.1.1. Compound Selection and Analysis
2.1.2. Target Preparation
2.1.3. Molecular Docking
2.1.4. ADMET Properties
2.1.5. Molecular Dynamics Calculations
2.2. Experimental Methods
2.2.1. Chemical Compounds
2.2.2. Biochemical Assays
Adapta Assay
Z-Lyte Assay
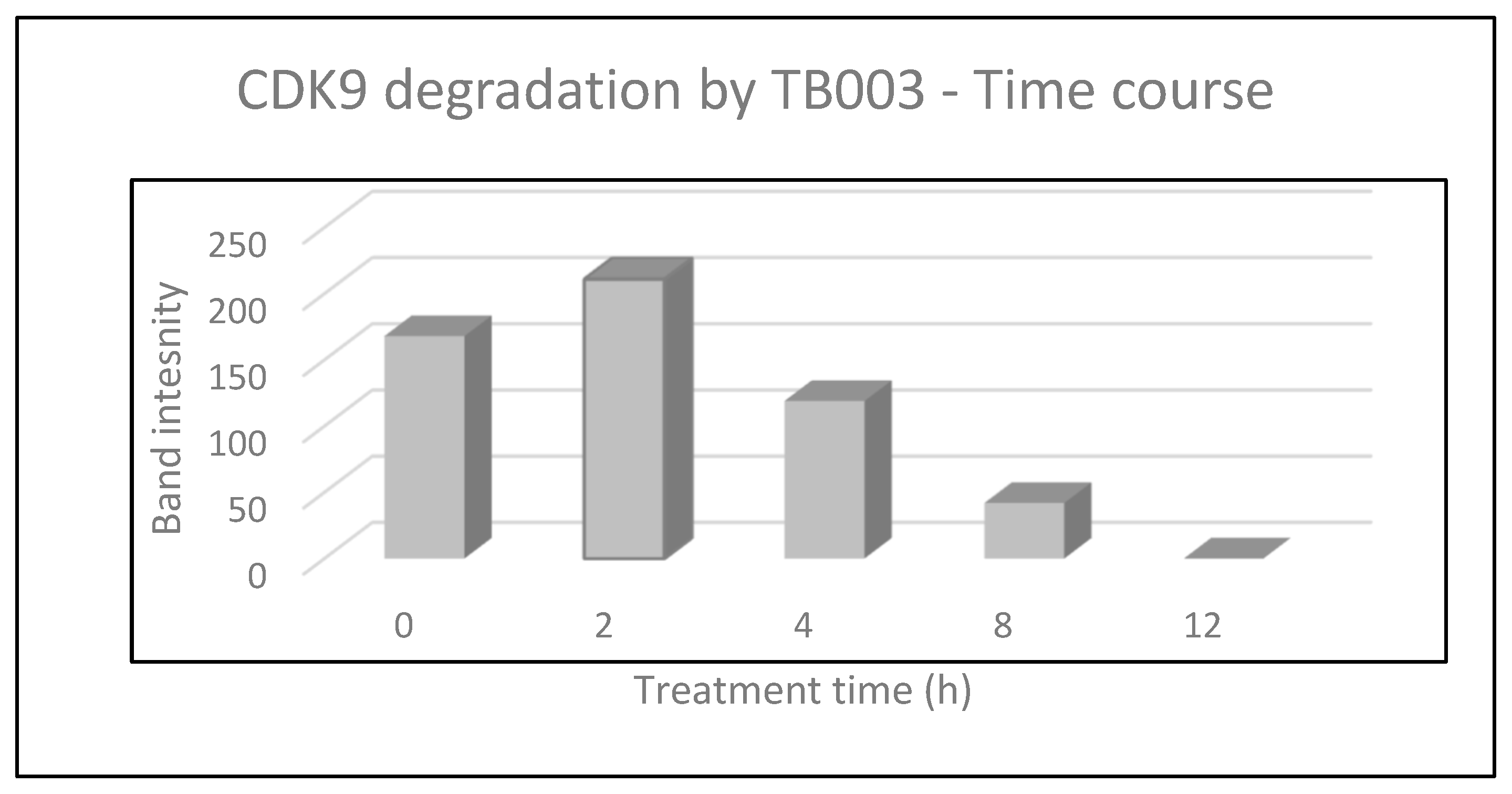
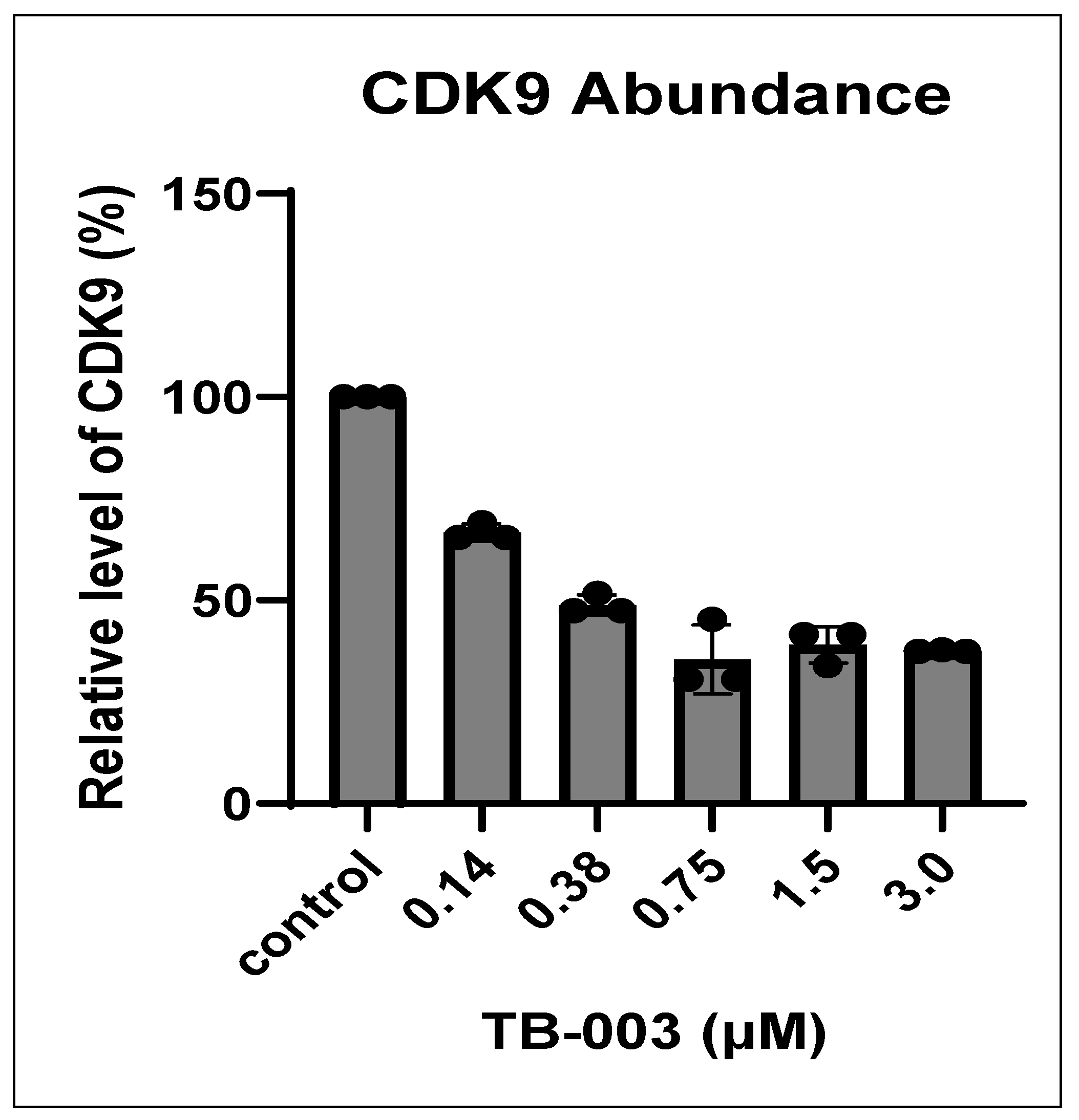
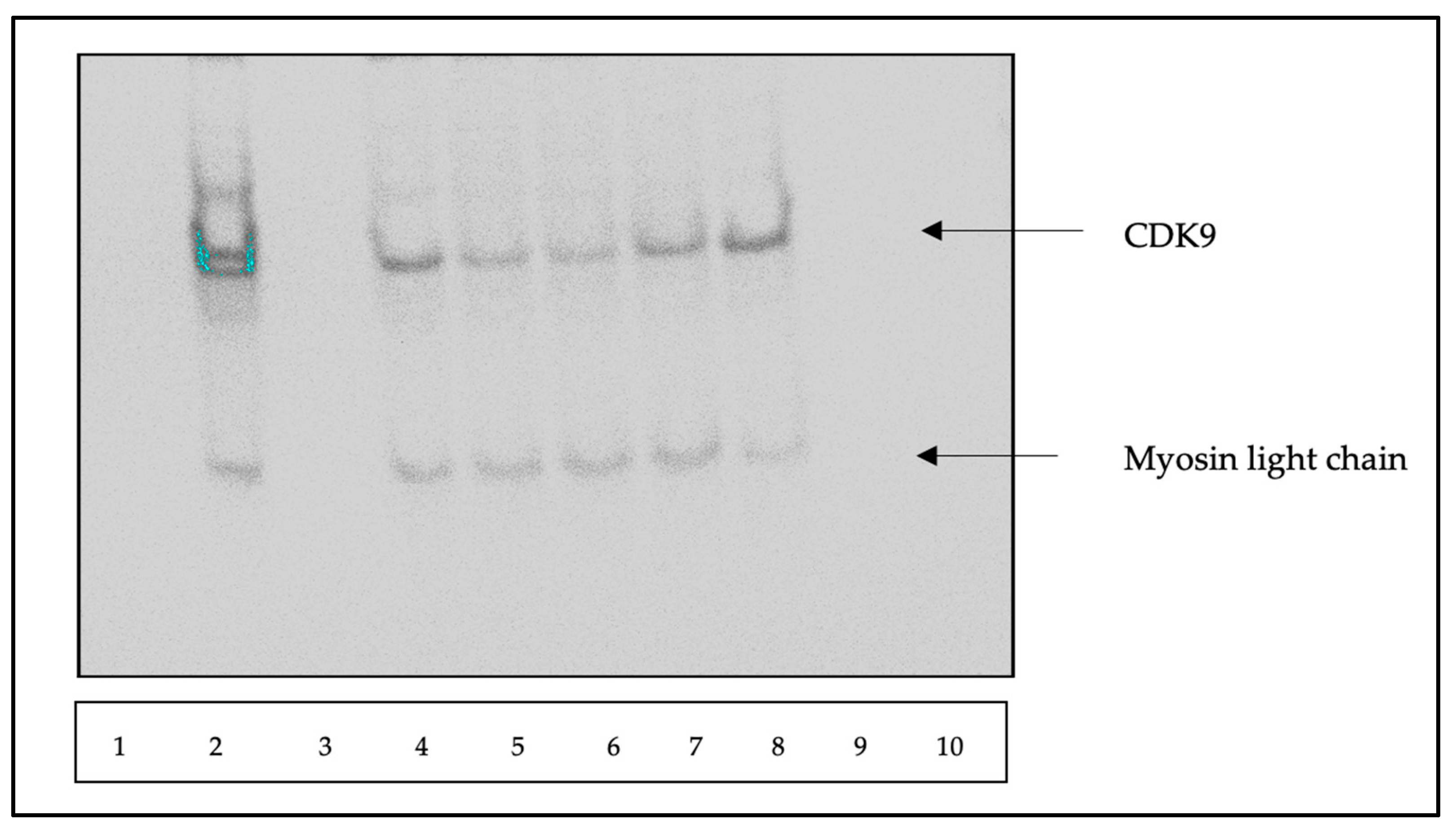
2.2.3. CDK9 Degradation with TB003 and TB008
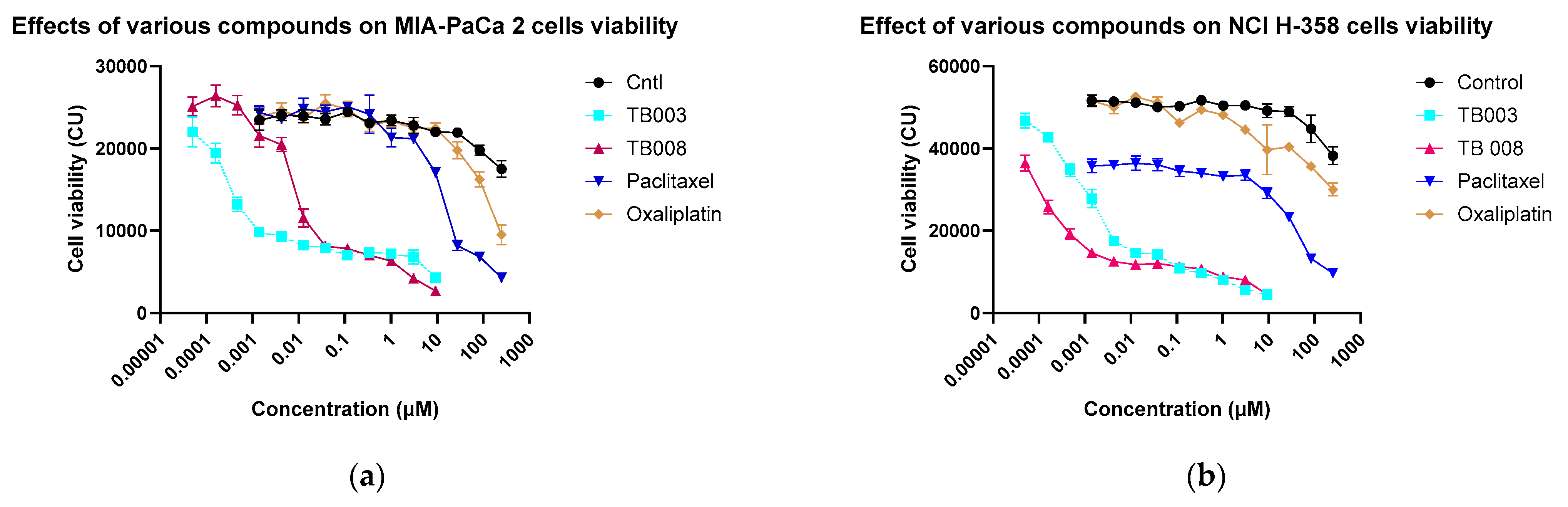
2.2.4. Effect of Degrader on Cell Viability
2.2.5. Graphical Presentations and Statistical Analysis
3. Results
3.1. Computational Results
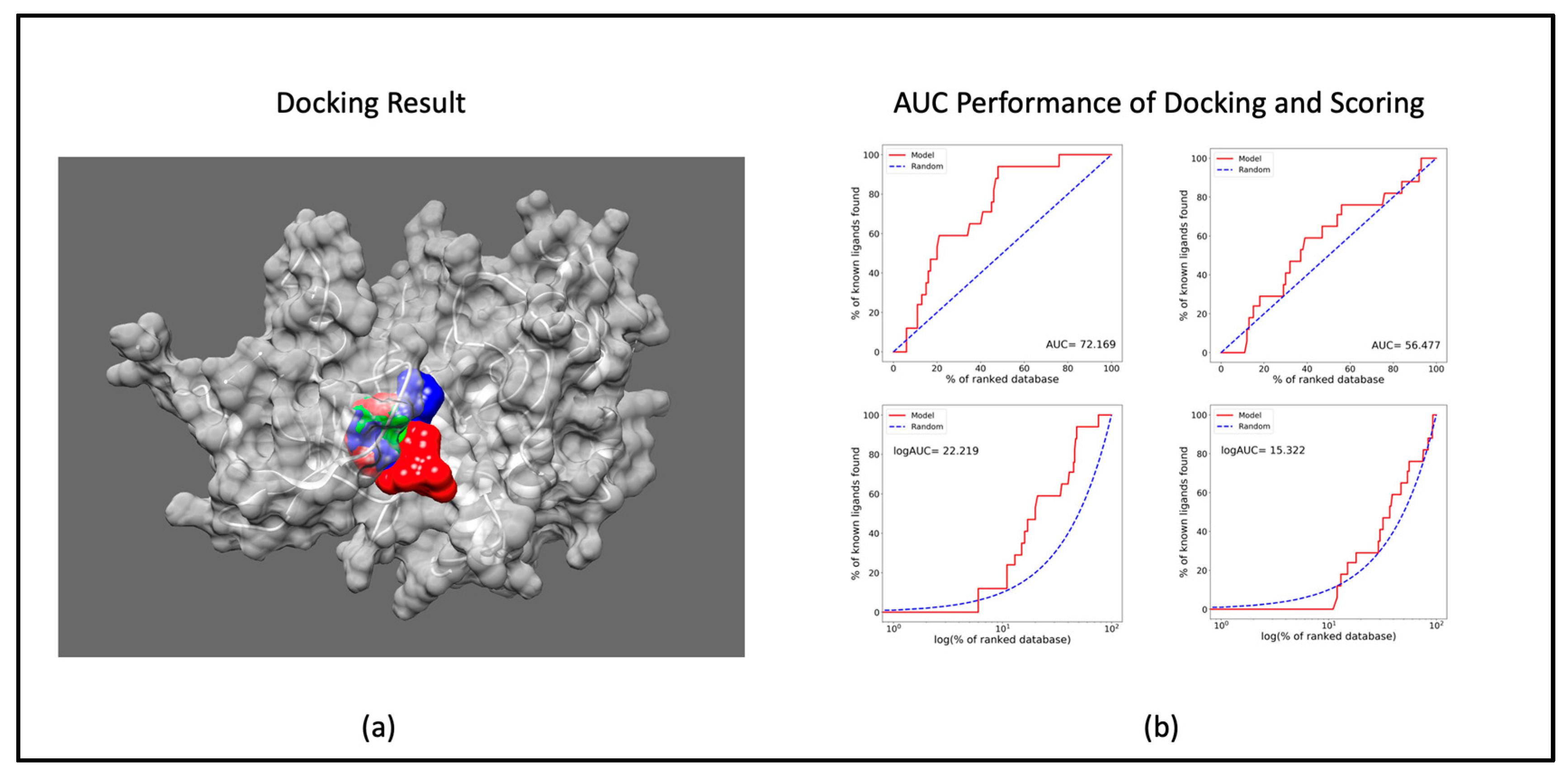
3.1.1. Docking Study
3.1.2. ADMET Analysis
3.1.3. MD Simulations
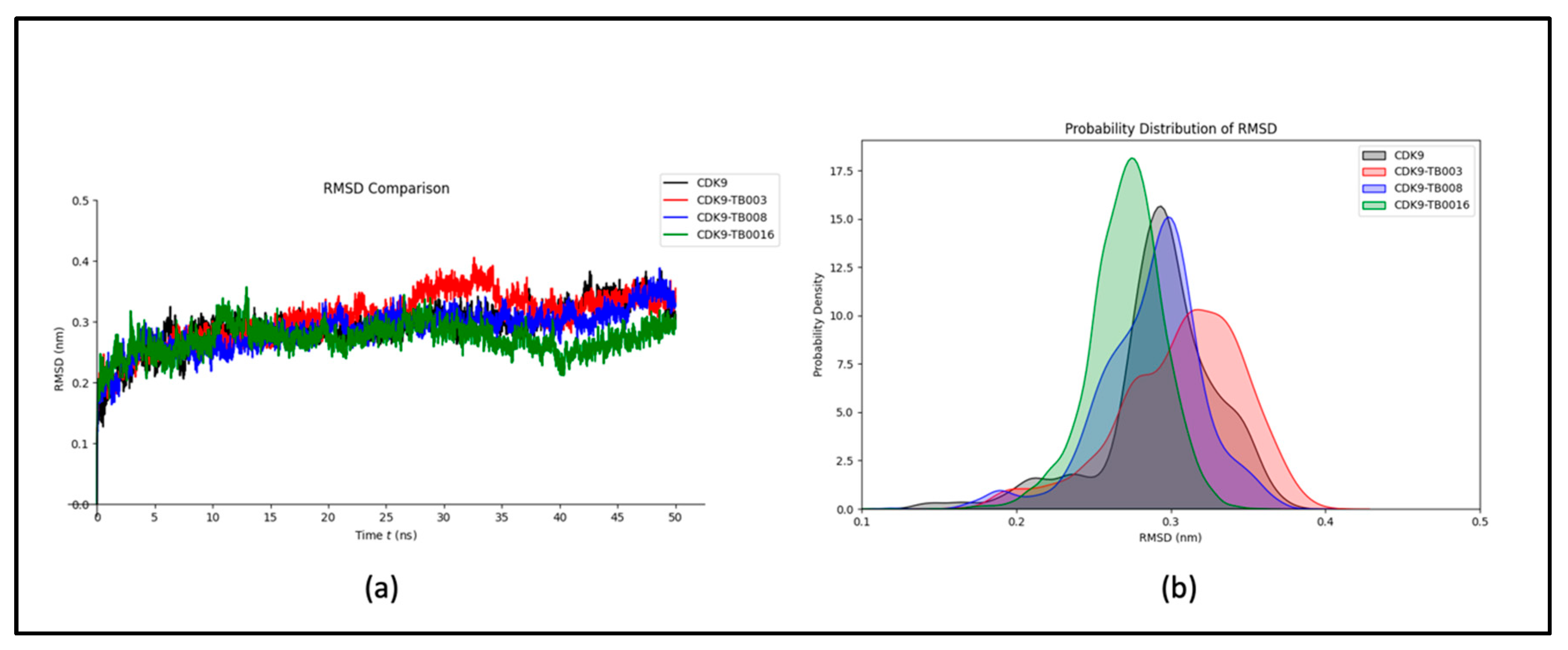
RMSD Analysis
RMSF Analysis
Radius of Gyration Analysis
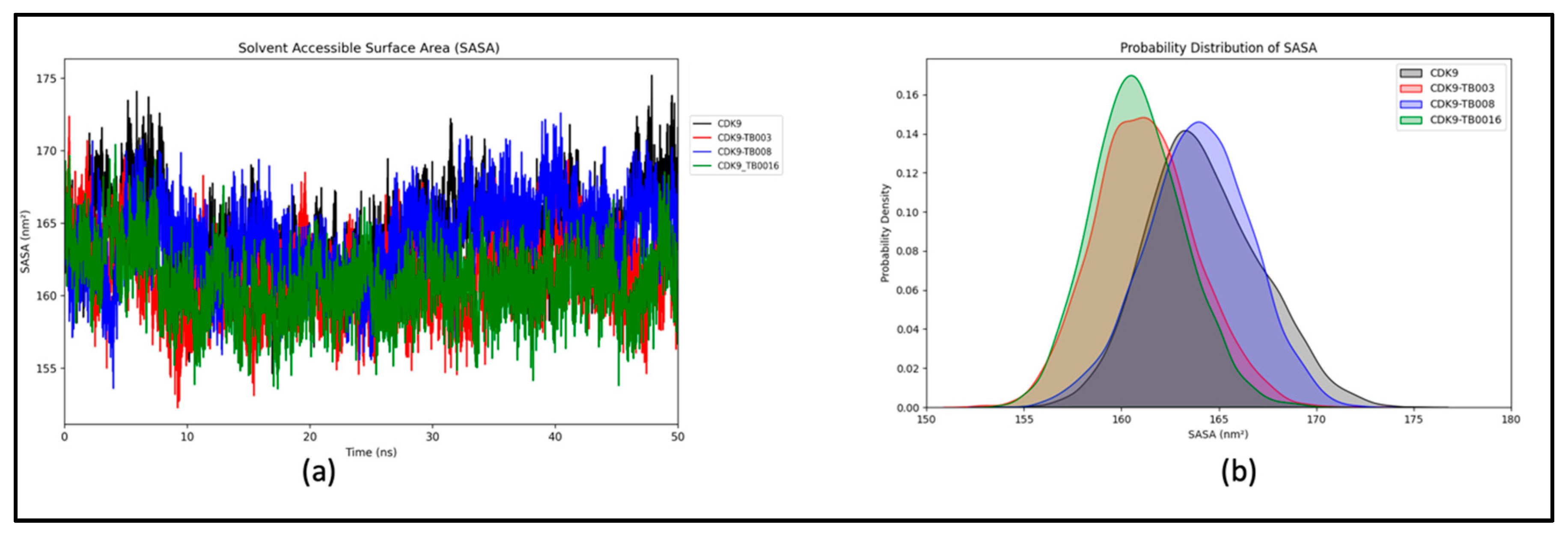
Analysis of Solvent-Accessible Surface Area (SASA)
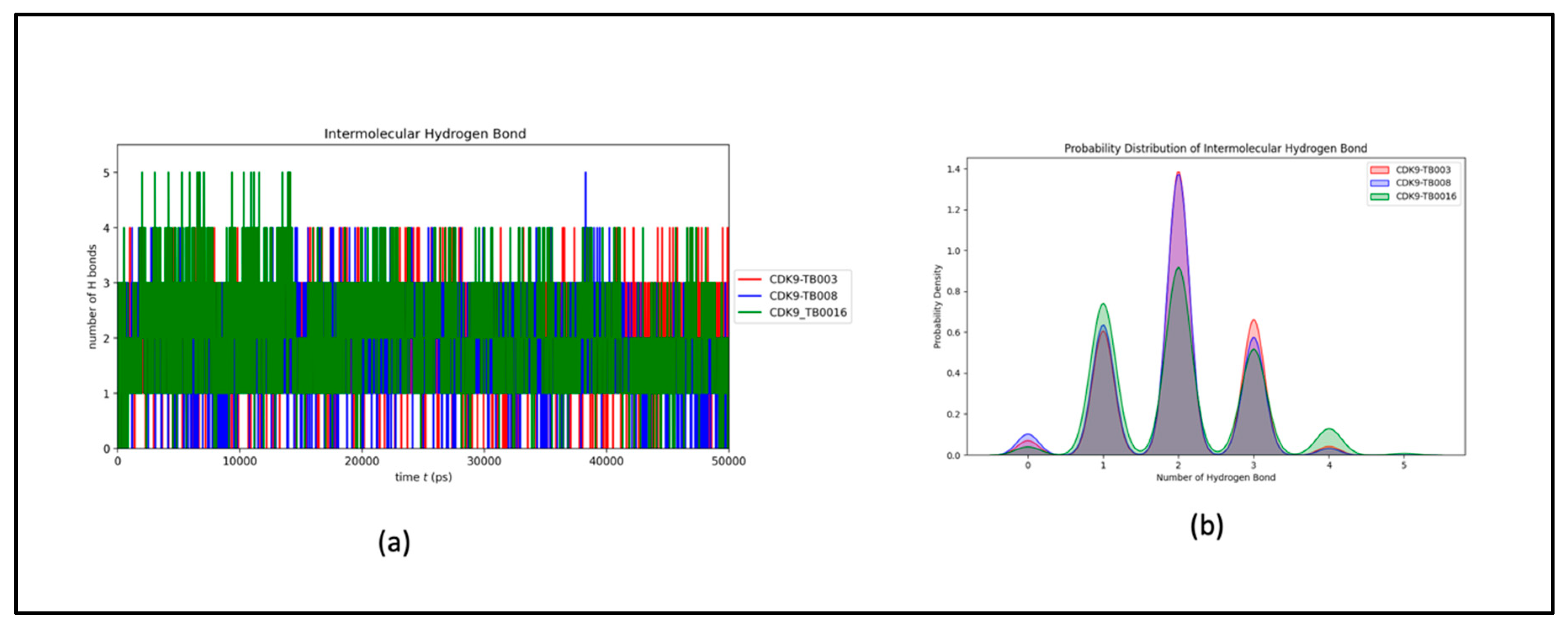
Hydrogen Bonds Dynamics
3.2. Experimental Results
3.2.1. Assay
3.2.2. CDK9 Degradation by Western Blot
3.2.3. Cell Viability
4. Discussions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tai, W.; Mahato, R.; Cheng, K. The role of HER2 in cancer therapy and targeted drug delivery. J. Control. Release 2010, 146, 264–275. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Mares, A.; Smith, I.E.D.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Garriga, J.; Graña, X. CDK9 inhibition strategy defines distinct sets of target genes. BMC Res. Notes 2014, 7, 301. [Google Scholar] [CrossRef] [PubMed]
- Sajjadi-Dokht, M.; Mohamad, T.A.M.; Rahman, H.S.; Maashi, M.S.; Danshina, S.; Shomali, N.; Solali, S.; Marofi, F.; Zeinalzadeh, E.; Akbari, M.; et al. MicroRNAs and JAK/STAT3 signaling: A new promising therapeutic axis in blood cancers. Genes Dis. 2022, 9, 849–867. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Li, T.; Price, D.H. RNA polymerase II elongation control. Annu. Rev. Biochem. 2012, 81, 119–143. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Geyer, M.; Zhou, Q. The control of HIV transcription: Keeping RNA polymerase II on track. Cell Host Microbe 2011, 10, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Blachly, J.S.; Byrd, J.C.; Grever, M. Cyclin-dependent kinase inhibitors for the treatment of chronic lymphocytic leukemia. In Seminars in Oncology; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Blake, D.R.; Vaseva, A.V.; Hodge, R.G.; Kline, M.P.; Gilbert, T.S.K.; Tyagi, V.; Huang, D.; Whiten, G.C.; Larson, J.E.; Wang, X.; et al. Application of a MYC degradation screen identifies sensitivity to CDK9 inhibitors in KRAS-mutant pancreatic cancer. Sci. Signal. 2019, 12, eaav7259. [Google Scholar] [CrossRef]
- Lyu, J.; Yang, E.J.; Zhang, B.; Wu, C.; Pardeshi, L.; Shi, C.; Mou, P.K.; Liu, Y.; Tan, K.; Shim, J.S. Synthetic lethality of RB1 and aurora A is driven by stathmin-mediated disruption of microtubule dynamics. Nat. Commun. 2020, 11, 5105. [Google Scholar] [CrossRef] [PubMed]
- Knight, Z.A.; Shokat, K.M. Chemical genetics: Where genetics and pharmacology meet. Cell 2007, 128, 425–430. [Google Scholar] [CrossRef]
- Lu, H.; Xue, Y.; Yu, G.K.; Arias, C.; Lin, J.; Fong, S.; Faure, M.; Weisburd, B.; Ji, X.; Mercier, A.; et al. Compensatory induction of MYC expression by sustained CDK9 inhibition via a BRD4-dependent mechanism. Elife 2015, 4, e06535. [Google Scholar] [CrossRef]
- Morales, F.; Giordano, A. Overview of CDK9 as a target in cancer research. Cell Cycle 2016, 15, 519–527. [Google Scholar] [CrossRef]
- Rahaman, M.H.; Kumarasiri, M.; Mekonnen, L.B.; Yu, M.; Diab, S.; Albrecht, H.; Milne, R.W.; Wang, S. Targeting CDK9: A promising therapeutic opportunity in prostate cancer. Endocr.-Relat. Cancer 2016, 23, T211–T226. [Google Scholar] [CrossRef] [PubMed]
- Burslem, G.M.; Crews, C.M. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell 2020, 181, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Qin, Z.; Tian, Y.; Wang, J.; Xu, C.; Li, Z.; Bian, J. Recent developments in the biology and medicinal chemistry of CDK9 inhibitors: An update. J. Med. Chem. 2020, 63, 13228–13257. [Google Scholar] [CrossRef]
- Olson, C.M.; Jiang, B.; Erb, M.A.; Liang, Y.; Doctor, Z.M.; Zhang, Z.; Zhang, T.; Kwiatkowski, N.; Boukhali, M.; Green, J.L.; et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat. Chem. Biol. 2018, 14, 163–170. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Mukherjee, S.; Yang, J.W.; Hoffmann, S.; List, B. Asymmetric enamine catalysis. Chem. Rev. 2007, 107, 5471–5569. [Google Scholar] [CrossRef]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Consortium, U. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Baumli, S.; Lolli, G.; Lowe, E.D.; Troiani, S.; Rusconi, L.; Bullock, A.N.; Debreczeni, J.É.; Knapp, S.; Johnson, L.N. The structure of P-TEFb (CDK9/cyclin T1), its complex with flavopiridol and regulation by phosphorylation. EMBO J. 2008, 27, 1907–1918. [Google Scholar] [CrossRef]
- Marchand, J.-R.; Pirard, B.; Ertl, P.; Sirockin, F. CAVIAR: A method for automatic cavity detection, description and decomposition into subcavities. J. Comput. Aided Mol. Des. 2021, 35, 737–750. [Google Scholar] [CrossRef]
- Huey, R.; Morris, G.M.; Forli, S. Using AutoDock 4 and AutoDock vina with AutoDockTools: A tutorial. Scripps Res. Inst. Mol. Graph. Lab. 2012, 10550, 1000. [Google Scholar]
- Masters, L.; Eagon, S.; Heying, M. Evaluation of consensus scoring methods for AutoDock Vina, smina and idock. J. Mol. Graph. Model. 2020, 96, 107532. [Google Scholar] [CrossRef]
- Quiroga, R.; Villarreal, M.A. Vinardo: A scoring function based on autodock vina improves scoring, docking, and virtual screening. PLoS ONE 2016, 11, e0155183. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Berendsen, H.J.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Shukla, R.; Tripathi, T. Molecular dynamics simulation of protein and protein–ligand complexes. In Computer-Aided Drug Design; Springer: Berlin/Heidelberg, Germany, 2020; pp. 133–161. [Google Scholar]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) I: Bond perception and atom typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [Google Scholar] [CrossRef] [PubMed]
- Bjørkøy, G.; Lamark, T.; Pankiv, S.; Øvervatn, A.; Brech, A.; Johansen, T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar] [PubMed]
- Jang, J.; To, C.; De Clercq, D.J.; Park, E.; Ponthier, C.M.; Shin, B.H.; Mushajiang, M.; Nowak, R.P.; Fischer, E.S.; Eck, M.J.; et al. Mutant-selective allosteric EGFR degraders are effective against a broad range of drug-resistant mutations. Angew. Chem. 2020, 132, 14589–14597. [Google Scholar] [CrossRef]
- He, S.; Fang, X.; Xia, X.; Hou, T.; Zhang, T. Targeting CDK9: A novel biomarker in the treatment of endometrial cancer. Oncol. Rep. 2020, 44, 1929–1938. [Google Scholar] [CrossRef]
- Ranjan, A.; Pang, Y.; Butler, M.; Merchant, M.; Kim, O.; Yu, G.; Su, Y.-T.; Gilbert, M.R.; Levens, D.; Wu, J. Targeting CDK9 for the treatment of glioblastoma. Cancers 2021, 13, 3039. [Google Scholar] [CrossRef]
- Rowland, T.; Paiva, C.; Rowley, J.; Chen, A.; Drew, L.; Hurlin, P.; Chang, B.H.; Danilov, A.V. Selective targeting cyclin-dependent kinase-9 (CDK9) downmodulates c-MYC and induces apoptosis in diffuse large B-cell lymphoma (DLBCL) cells. Blood 2016, 128, 289. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand Name | Binding Affinity (kcal/mol) | Docking Method |
|---|---|---|
| TB003 | −7.8 | Vina |
| TB003 | −8.4 | Smina |
| TB008 | −9.6 | Vina |
| TB008 | −9.8 | Smina |
| TB0016 | −9.7 | Vina |
| TB0016 | −8.8 | Smina |
| Compound | Lipophilicity (LogP) | Absorption | Solubility (LogS) |
|---|---|---|---|
| TB003 | 5.09 (High) | Low | −6.29 (Poorly Soluble) |
| TB008 | 2.75 (Moderate) | High | −3.83 (Soluble) |
| TB0016 | 2.55 (Moderate) | High | −4.28 (Moderately Soluble) |
| TB003 PROTAC | 4.28 (High) | Low | −6.65 (Poorly Soluble) |
| TB008 PROTAC | 3.19 (High) | Low | −5.59 (Moderately Soluble) |
| Compound | TB003 | TB0016 | TB008 | |
|---|---|---|---|---|
| Modality | CDK9 degrader | CDK9 ligand | CDK9 degrader | |
| Potency (IC50) | 5 nM | >1 µM | 3.5 nM | |
| Fold selectivity CDK9 vs. other CDK family members | CDK7 | >200 | >1 µM | >500 |
| CDK5 | NT | >1 µM | >500 | |
| CDK4 | NT | >1 µM | >500 | |
| CDK2 | >200 | >1 µM | >500 | |
| CDK1 | >200 | >1 µM | >500 | |
| Route of administration | IP/Oral | IP/Oral | IP/Oral | |
| Chemical Entity | Cell Line | |
|---|---|---|
| NIH H-358 | MIA-PaCa 2 | |
| TB003 | 20 nM | 0.8 nM |
| TB008 | 0.3 nM | 9 nM |
| Paclitaxel | 75 µM | 10 µM |
| Oxaliplatin | ND | 100 µM |
| Cell Type | Test Compound | ||||
|---|---|---|---|---|---|
| TB003 | TB008 | Paclitaxel | Oxaliplatin | ||
| H358 | Control | <0.001 | <0.001 | <0.001 | <0.1; No SS at concentrations < 0.11 µM |
| MIA-PaCa | Control | <0.001 | <0.001 | <0.067; No SS at concentrations < 1 µM | <0.026; No SS at concentrations < 28 µM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koirala, M.; DiPaola, M. Targeting CDK9 in Cancer: An Integrated Approach of Combining In Silico Screening with Experimental Validation for Novel Degraders. Curr. Issues Mol. Biol. 2024, 46, 1713-1730. https://doi.org/10.3390/cimb46030111
Koirala M, DiPaola M. Targeting CDK9 in Cancer: An Integrated Approach of Combining In Silico Screening with Experimental Validation for Novel Degraders. Current Issues in Molecular Biology. 2024; 46(3):1713-1730. https://doi.org/10.3390/cimb46030111
Chicago/Turabian StyleKoirala, Mahesh, and Mario DiPaola. 2024. "Targeting CDK9 in Cancer: An Integrated Approach of Combining In Silico Screening with Experimental Validation for Novel Degraders" Current Issues in Molecular Biology 46, no. 3: 1713-1730. https://doi.org/10.3390/cimb46030111
APA StyleKoirala, M., & DiPaola, M. (2024). Targeting CDK9 in Cancer: An Integrated Approach of Combining In Silico Screening with Experimental Validation for Novel Degraders. Current Issues in Molecular Biology, 46(3), 1713-1730. https://doi.org/10.3390/cimb46030111