
Decoding the Genetic Basis of Mast Cell Hypersensitivity and Infection Risk in Hypermobile Ehlers-Danlos Syndrome



Abstract
1. Introduction
2. Materials and Methods
2.1. Study Participants and Ethical Considerations
2.2. Clinical Assessment
2.3. DNA Extraction and Whole-Genome Sequencing
2.4. Data Processing and Quality Control
2.5. Data Analysis
2.6. Gene Selection and Variant Filtering
3. Results
3.1. Sequencing Quality and Coverage
3.2. Genetic Variants and Filtering Approaches
3.3. Pathways Potentially Involved in Hypermobile Ehlers-Danlos Syndrome (hEDS)
3.4. Identifying Relevant Genetic Variations in Hypermobile Ehlers-Danlos Syndrome (hEDS)
3.5. Identifying Relevant Genetic Variations in Association with MCAS in hEDS
3.6. Key Genes and Variations Associated with hEDS
4. Discussion
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beighton, P.; De Paepe, A.; Steinmann, B.; Tsipouras, P.; Wenstrup, R.J. Ehlers-Danlos syndromes: Revised nosology, Villefranche, 1997. Am. J. Med. Genet. 1998, 77, 31–37. [Google Scholar] [CrossRef]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef]
- Tinkle, B.; Castori, M.; Berglund, B.; Cohen, H.; Grahame, R.; Kazkaz, H.; Levy, H. Hypermobile Ehlers-Danlos syndrome (a.k.a. Ehlers-Danlos syndrome Type III and Ehlers-Danlos syndrome hypermobility type): Clinical description and natural history. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 48–69. [Google Scholar] [CrossRef]
- Malfait, F.; Hakim, A.J.; De Paepe, A.; Grahame, R. The genetic basis of the joint hypermobility syndromes. Rheumatology 2006, 45, 502–507. [Google Scholar] [CrossRef]
- Molderings, G.J.; Brettner, S.; Homann, J.; Afrin, L.B. Mast cell activation disease: A concise practical guide for diagnostic workup and therapeutic options. J. Hematol. Oncol. 2011, 4, 10. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Alysandratos, K.D.; Angelidou, A.; Delivanis, D.A.; Sismanopoulos, N.; Zhang, B.; Asadi, S.; Vasiadi, M.; Weng, Z.; Miniati, A.; et al. Mast cells and inflammation. Biochim. Biophys. Acta 2012, 1822, 21–33. [Google Scholar] [CrossRef]
- Seneviratne, S.L.; Maitland, A.; Afrin, L. Mast cell disorders in Ehlers-Danlos syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 226–236. [Google Scholar] [CrossRef]
- Kucharik, A.H.; Chang, C. The Relationship Between Hypermobile Ehlers-Danlos Syndrome (hEDS), Postural Orthostatic Tachycardia Syndrome (POTS), and Mast Cell Activation Syndrome (MCAS). Clin. Rev. Allergy Immunol. 2020, 58, 273–297. [Google Scholar] [CrossRef]
- Monaco, A.; Choi, D.; Uzun, S.; Maitland, A.; Riley, B. Association of mast-cell-related conditions with hypermobile syndromes: A review of the literature. Immunol. Res. 2022, 70, 419–431. [Google Scholar] [CrossRef]
- Brock, I.; Prendergast, W.; Maitland, A. Mast Cell Activation Disease and Immunoglobulin Deficiency in Patients with Hypermobile Ehlers-Danlos Syndrome/Hypermobility Spectrum Disorder. Am. J. Med. Genet. C Semin. Med. Genet. 2021, 187, 473–481. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C.; Arock, M.; Brockow, K.; Butterfield, J.H.; Carter, M.C.; Castells, M.; Escribano, L.; Hartmann, K.; Lieberman, P.; et al. Definitions, criteria and global classification of mast cell disorders with special reference to mast cell activation syndromes: A consensus proposal. Int. Arch. Allergy Immunol. 2012, 157, 215–225. [Google Scholar] [CrossRef]
- Castells, M.; Metcalfe, D.D.; Escribano, L. Diagnosis and treatment of cutaneous mastocytosis in children: Practical recommendations. Am. J. Clin. Dermatol. 2011, 12, 259–270. [Google Scholar] [CrossRef]
- Boileau, A.; Brierre, T.; Castel-Lacanal, É.; Soulié, M.; Gamé, X. Lower urinary tract involvement in Ehlers-Danlos and Joint Hypermobility syndromes: Review of the literature. Fr. J. Urol. 2024, 34, 102698. [Google Scholar] [CrossRef]
- Chiarelli, N.; Carini, G.; Zoppi, N.; Dordoni, C.; Ritelli, M.; Venturini, M.; Castori, M.; Colombi, M. Transcriptome-Wide Expression Profiling in Skin Fibroblasts of Patients with Joint Hypermobility Syndrome/Ehlers-Danlos Syndrome Hypermobility Type. PLoS ONE 2016, 11, e0161347. [Google Scholar] [CrossRef]
- Urb, M.; Sheppard, D.C. The role of mast cells in the defence against pathogens. PLoS Pathog. 2012, 8, e1002619. [Google Scholar] [CrossRef]
- Galli, S.J.; Tsai, M.; Piliponsky, A.M. The development of allergic inflammation. Nature 2008, 454, 445–454. [Google Scholar] [CrossRef]
- Akin, C. Mast cell activation syndromes. J. Allergy Clin. Immunol. 2017, 140, 349–355. [Google Scholar] [CrossRef]
- Jiménez, M.; Cervantes-García, D.; Córdova-Dávalos, L.E.; Pérez-Rodríguez, M.J.; Gonzalez-Espinosa, C.; Salinas, E. Responses of Mast Cells to Pathogens: Beneficial and Detrimental Roles. Front. Immunol. 2021, 12, 685865. [Google Scholar] [CrossRef]
- Leru, P.M.; Anton, V.F.; Ureche, C.; Zurac, S.; Bratu, O.; Neagoe, C.D. Mast cell activation syndromes—Evaluation of current diagnostic criteria and laboratory tools in clinical practice (Review). Exp. Ther. Med. 2020, 20, 2348–2351. [Google Scholar] [CrossRef]
- Frieri, M. Mast Cell Activation Syndrome. Clin. Rev. Allergy Immunol. 2018, 54, 353–365. [Google Scholar] [CrossRef]
- Mathias, K.; Mantha, A.; Mathias, L.; Arkfeld, D. The Relationship of Mast Cell Activation Syndrome and Hypermobile Ehlers-Danlos Syndrome in Hospitalized Patients in the United States. Ann. Rheum. Dis. 2021, 80 (Suppl. S1), 965. [Google Scholar] [CrossRef]
- Ritelli, M.; Chiarelli, N.; Cinquina, V.; Vezzoli, M.; Venturini, M.; Colombi, M. Looking back and beyond the 2017 diagnostic criteria for hypermobile Ehlers-Danlos syndrome: A retrospective cross-sectional study from an Italian reference center. Am. J. Med. Genet. A 2024, 194, 174–194. [Google Scholar] [CrossRef]
- Tofts, L.J.; Simmonds, J.; Schwartz, S.B.; Richheimer, R.M.; O’Connor, C.; Elias, E.; Engelbert, R.; Cleary, K.; Tinkle, B.T.; Kline, A.D.; et al. Pediatric joint hypermobility: A diagnostic framework and narrative review. Orphanet J. Rare Dis. 2023, 18, 104. [Google Scholar] [CrossRef]
- Nicholson, L.L.; Chan, C.; Tofts, L.; Pacey, V. Hypermobility syndromes in children and adolescents: Assessment, diagnosis and multidisciplinary management. Aust. J. Gen. Pract. 2022, 51, 409–414. [Google Scholar] [CrossRef]
- RCPCH Establishing a Correct Diagnosis of Ehlers Danlos Syndrome Hypermobility Type (hEDS) in Children. Available online: https://www.rcpch.ac.uk/resources/establishing-correct-diagnosis-ehlers-danlos-syndrome-hypermobility-type-heds-children#footnote5_wgt4z0j (accessed on 10 September 2024).
- Leone, V.; Tornese, G.; Zerial, M.; Locatelli, C.; Ciambra, R.; Bensa, M.; Pocecco, M. Joint hypermobility and its relationship to musculoskeletal pain in schoolchildren: A cross-sectional study. Arch. Dis. Child. 2009, 94, 627–632. [Google Scholar] [CrossRef]
- Yew, K.S.; Kamps-Schmitt, K.A.; Borge, R. Hypermobile Ehlers-Danlos Syndrome and Hypermobility Spectrum Disorders. Am. Fam. Physician 2021, 103, 481–492. [Google Scholar]
- Holick, M.F.; Shirvani, A.; Charoenngam, N. Fetal Fractures in an Infant with Maternal Ehlers-Danlos Syndrome, CCDC134 Pathogenic Mutation and a Negative Genetic Test for Osteogenesis Imperfecta. Children 2021, 17, 512. [Google Scholar] [CrossRef]
- Haenisch, B.; Nöthen, M.M.; Molderings, G.J. Systemic mast cell activation disease: The role of molecular genetic alterations in pathogenesis, heritability and diagnostics. Immunology 2012, 137, 197–205. [Google Scholar] [CrossRef]
- Christy, A.L.; Brown, M.A. The multitasking mast cell: Positive and negative roles in the progression of autoimmunity. J. Immunol. 2007, 179, 2673–2679. [Google Scholar] [CrossRef]
- Mayorga, C.; Ariza, A.; Muñoz-Cano, R.; Sabato, V.; Doña, I.; Torres, M.J. Biomarkers of immediate drug hypersensitivity. Allergy 2024, 79, 601–612. [Google Scholar] [CrossRef]
- Nedoszytko, B.; Arock, M.; Lyons, J.J.; Bachelot, G.; Schwartz, L.B.; Reiter, A.; Jawhar, M.; Schwaab, J.; Lange, M.; Greiner, G.; et al. Clinical Impact of Inherited and Acquired Genetic Variants in Mastocytosis. Int. J. Mol. Sci. 2021, 22, 411. [Google Scholar] [CrossRef]
- Voss, M.; Kotrba, J.; Gaffal, E.; Katsoulis-Dimitriou, K.; Dudeck, A. Mast Cells in the Skin: Defenders of Integrity or Offenders in Inflammation? Int. J. Mol. Sci. 2021, 22, 4589. [Google Scholar] [CrossRef]
- McCurdy, J.D.; Olynych, T.J.; Maher, L.H.; Marshall, J.S. Cutting edge: Distinct Toll-like receptor 2 activators selectively induce different classes of mediator production from human mast cells. J. Immunol. 2003, 170, 1625–1629. [Google Scholar] [CrossRef]
- Jayapal, M.; Tay, H.K.; Reghunathan, R.; Zhi, L.; Chow, K.K.; Rauff, M.; Melendez, A.J. Genome-wide gene expression profiling of human mast cells stimulated by IgE or FcepsilonRI-aggregation reveals a complex network of genes involved in inflammatory responses. BMC Genom. 2006, 7, 210. [Google Scholar] [CrossRef]
- Dileepan, K.N.; Raveendran, V.V.; Sharma, R.; Abraham, H.; Barua, R.; Singh, V.; Sharma, R.; Sharma, M. Mast cell-mediated immune regulation in health and disease. Front. Med. 2023, 10, 1213320. [Google Scholar] [CrossRef]
- Li, Y.; Gao, J.; Kamran, M.; Harmacek, L.; Danhorn, T.; Leach, S.M.; O’Connor, B.P.; Hagman, J.R.; Huang, H. GATA2 regulates mast cell identity and responsiveness to antigenic stimulation by promoting chromatin remodeling at super-enhancers. Nat. Commun. 2021, 12, 494. [Google Scholar] [CrossRef]
- Molderings, G.J. The genetic basis of mast cell activation disease—Looking through a glass darkly. Crit. Rev. Oncol. Hematol. 2015, 93, 75–89. [Google Scholar] [CrossRef]
- Zorzan, E.; Hanssens, K.; Giantin, M.; Dacasto, M.; Dubreuil, P. Mutational Hotspot of TET2, IDH1, IDH2, SRSF2, SF3B1, KRAS, and NRAS from Human Systemic Mastocytosis Are Not Conserved in Canine Mast Cell Tumors. PLoS ONE 2015, 10, e0142450. [Google Scholar] [CrossRef]
- Leoni, C.; Montagner, S.; Rinaldi, A.; Bertoni, F.; Polletti, S.; Balestrieri, C.; Monticelli, S. Dnmt3a restrains mast cell inflammatory responses. Proc. Natl Acad. Sci. USA 2017, 114, E1490–E1499. [Google Scholar] [CrossRef]
- Yang, Y.; Li, L.; Wong, G.W.; Krilis, S.A.; Madhusudhan, M.S.; Sali, A.; Stevens, R.L. RasGRP4, a new mast cell-restricted Ras guanine nucleotide-releasing protein with calcium- and diacylglycerol-binding motifs. Identification of defective variants of this signaling protein in asthma, mastocytosis, and mast cell leukemia patients and demonstration of the importance of RasGRP4 in mast cell development and function. J. Biol. Chem. 2002, 277, 25756–25774. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Adli, M.; LaFave, L.M.; Gao, J.; Hricik, T.; Shih, A.H.; Pandey, S.; Patel, J.P.; Chung, Y.R.; Koche, R.; et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 2012, 22, 180–193. [Google Scholar] [CrossRef]
- Kluin-Nelemans, H.C.; Jawhar, M.; Reiter, A.; van Anrooij, B.; Gotlib, J.; Hartmann, K.; Illerhaus, A.; Oude Elberink, H.N.G.; Gorska, A.; Niedoszytko, M.; et al. Cytogenetic and molecular aberrations and worse outcome for male patients in systemic mastocytosis. Theranostics 2021, 11, 292–303. [Google Scholar] [CrossRef]
- Kurashima, Y.; Amiya, T.; Fujisawa, K.; Shibata, N.; Suzuki, Y.; Kogure, Y.; Hashimoto, E.; Otsuka, A.; Kabashima, K.; Sato, S.; et al. The enzyme Cyp26b1 mediates inhibition of mast cell activation by fibroblasts to maintain skin-barrier homeostasis. Immunity 2014, 40, 530–541. [Google Scholar] [CrossRef]
- Lasho, T.L.; Finke, C.M.; Zblewski, D.; Patnaik, M.; Ketterling, R.P.; Chen, D.; Hanson, C.A.; Tefferi, A.; Pardanani, A. Novel recurrent mutations in ethanolamine kinase 1 (ETNK1) gene in systemic mastocytosis with eosinophilia and chronic myelomonocytic leukemia. Blood Cancer J. 2015, 5, e275. [Google Scholar] [CrossRef]
- Tkaczyk, C.; Okayama, Y.; Metcalfe, D.D.; Gilfillan, A.M. Fcgamma receptors on mast cells: Activatory and inhibitory regulation of mediator release. Int. Arch. Allergy Immunol. 2004, 133, 305–315. [Google Scholar] [CrossRef]
- Dendrou, C.; Petersen, J.; Rossjohn, J.; Fugger, L. HLA variation and disease. Nat. Rev. Immunol. 2018, 18, 325–339. [Google Scholar] [CrossRef]
- Poscente, M.; Tolomeo, D.; Arshadi, A.; Agostini, A.; L’Abbate, A.; Solimando, A.G.; Palumbo, O.; Carella, M.; Palumbo, P.; González, T.; et al. Aggressive systemic mastocytosis with the co-occurrence of PRKG2::PDGFRB, KAT6A::NCOA2, and RXRA::NOTCH1 fusion transcripts and a heterozygous RUNX1 frameshift mutation. Cancer Genet. 2024, 284–285, 5–11. [Google Scholar] [CrossRef]
- Lyons, J.J.; Yu, X.; Hughes, J.D.; Le, Q.T.; Jamil, A.; Bai, Y.; Ho, N.; Zhao, M.; Liu, Y.; O’Connell, M.P.; et al. Elevated basal serum tryptase identifies a multisystem disorder associated with increased TPSAB1 copy number. Nat. Genet. 2016, 48, 1564–1569. [Google Scholar] [CrossRef]
- Li, Y.; Wu, J. CD177 is a novel IgG Fc receptor and CD177 genetic variants affect IgG-mediated function. Front. Immunol. 2024, 15, 1418539. [Google Scholar] [CrossRef]
- Lévy, Y.; Wiedemann, A.; Hejblum, B.P.; Durand, M.; Lefebvre, C.; Surénaud, M.; Lacabaratz, C.; Perreau, M.; Foucat, E.; Déchenaud, M.; et al. CD177, a specific marker of neutrophil activation, is associated with coronavirus disease 2019 severity and death. iScience 2021, 24, 102711. [Google Scholar] [CrossRef]
- Demaret, J.; Venet, F.; Plassais, J.; Cazalis, M.A.; Vallin, H.; Friggeri, A.; Lepape, A.; Rimmelé, T.; Textoris, J.; Monneret, G. Identification of CD177 as the most dysregulated parameter in a microarray study of purified neutrophils from septic shock patients. Immunol. Lett. 2016, 178, 122–130. [Google Scholar] [CrossRef]
- Crux, N.B.; Elahi, S. Human Leukocyte Antigen (HLA) and Immune Regulation: How Do Classical and Non-Classical HLA Alleles Modulate Immune Response to Human Immunodeficiency Virus and Hepatitis C Virus Infections? Front. Immunol. 2017, 8, 832. [Google Scholar] [CrossRef]
- Medhasi, S.; Chantratita, N. Human Leukocyte Antigen (HLA) System: Genetics and Association with Bacterial and Viral Infections. J. Immunol. Res. 2022, 2022, 9710376. [Google Scholar] [CrossRef]
- Corfield, A.P. Mucins: A biologically relevant glycan barrier in mucosal protection. Biochim. Biophys. Acta 2015, 1850, 236–252. [Google Scholar] [CrossRef]
- Chelombitko, M.A.; Chernyak, B.V.; Fedorov, A.V.; Zinovkin, R.A.; Razin, E.; Paruchuru, L.B. The Role Played by Mitochondria in FcεRI-Dependent Mast Cell Activation. Front. Immunol. 2020, 11, 584210. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, M.J.; Ibarra-Sánchez, A.; Román-Figueroa, A.; Pérez-Severiano, F.; González-Espinosa, C. Mutant Huntingtin affects toll-like receptor 4 intracellular trafficking and cytokine production in mast cells. J. Neuroinflamm. 2020, 17, 95. [Google Scholar] [CrossRef]
- Chiarella, E.; Aloisio, A.; Scicchitano, S.; Lucchino, V.; Montalcini, Y.; Galasso, O.; Greco, M.; Gasparini, G.; Mesuraca, M.; Bond, H.M.; et al. ZNF521 Represses Osteoblastic Differentiation in Human Adipose-Derived Stem Cells. Int. J. Mol. Sci. 2018, 19, 4095. [Google Scholar] [CrossRef]
- Komori, T. Whole Aspect of Runx2 Functions in Skeletal Development. Int. J. Mol. Sci. 2022, 23, 5776. [Google Scholar] [CrossRef]
- Hardy, E.; Fernandez-Patron, C. Destroy to Rebuild: The Connection Between Bone Tissue Remodeling and Matrix Metalloproteinases. Front. Physiol. 2020, 11, 47. [Google Scholar] [CrossRef]
- Guo, Y.; Yang, T.L.; Liu, Y.Z.; Shen, H.; Lei, S.F.; Yu, N.; Chen, J.; Xu, T.; Cheng, Y.; Tian, Q.; et al. Mitochondria-wide association study of common variants in osteoporosis. Ann. Hum. Genet. 2011, 75, 569–574. [Google Scholar] [CrossRef]
- Douroudis, K.; Tarassi, K.; Athanassiades, T.; Giannakopoulos, F.; Kominakis, A.; Thalassinos, N.; Papasteriades, C.H. HLA alleles as predisposal factors for postmenopausal osteoporosis in a Greek population. Tissue Antigens 2007, 69, 592–596. [Google Scholar] [CrossRef]
- Ragipoglu, D.; Dudeck, A.; Haffner-Luntzer, M.; Voss, M.; Kroner, J.; Ignatius, A.; Fischer, V. The Role of Mast Cells in Bone Metabolism and Bone Disorders. Front. Immunol. 2020, 11, 163. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Gene | Associated Conditions/Functions |
|---|---|
| KIT (CD117) | Encodes the receptor for stem cell factor (SCF), crucial for mast cell development and function and mastocytosis, pathological mast cell activation [29] |
| JAK2 | Mast cell activation, V617F mutation associated with pathological activation [29,30] |
| MRGPRX2 | Mas-related G-protein coupled receptor, involved in non-IgE-mediated mast cell activation [31] |
| ADGRE2 | Mast cell activation, influence on mast cell reactivity [32] |
| PLCG2 | Signal transduction, involved in cold-induced urticaria [32] |
| TET2 | Systemic mastocytosis, candidate tumor suppressor gene [29] |
| NRAS | Aggressive systemic mastocytosis [29] |
| IL13 AND IL4 | Allergic diseases, polymorphism associated with increased risk of systemic mastocytosis [33] |
| TLR1 | Innate immunity, implicated in mast cell activation [34] |
| FCER1 | The high-affinity IgE receptor, plays a central role in allergic reactions and mast cell activation and high-affinity IgE receptor [35,36] |
| H1R, H2R, H3R, H4R | Histamine receptors, mediate various physiological responses including allergic reactions [36] |
| TNF RECEPTORS (TNFR1 AND TNFR2) | Receptors for tumor necrosis factor, involved in inflammatory signaling [30,33,36] |
| GATA2 | Hematopoiesis and immune regulation, transcription factor [37] |
| HRH4 | Histamine signaling, inhibition of full-length receptor function in mast cells [29] |
| NLRP3 | Inflammatory responses, associated with IL-1β production [38] |
| IDH1I AND DH2 | Cancer metabolism, involved in immune responses [39] |
| DNMT3A | Epigenetic regulation, associated with hematological malignancies [40] |
| TLR2 AND TLR4 | Toll-like receptors involved in recognizing bacterial components and mediating immune responses and their polymorphisms associated with systemic mastocytosis [33] |
| IL-4R AND IL6R | Receptor for interleukin-4, involved in promoting Th2 immune responses. Allergic diseases, gain-of-function polymorphism associated with mastocytosis [33]. Receptor for interleukin-6, involved in inflammatory responses and polymorphisms studied in mastocytosis [33] |
| RASGRP4 | Systemic mastocytosis, involved in mast cell activation [29,41] |
| ASXL1 | Chromatin modification, associated with hematological malignancies [42,43] |
| CYP26B1 | Mediates inhibition of mast cell activation by fibroblasts to maintain skin-barrier homeostasis [44] |
| ETNK1 | Phospholipid metabolism, involved in immune responses [45] |
| ZNF521 | Zinc finger transcription factor, involved in immune responses |
| SRSF2 | RNA splicing, associated with hematological malignancies [43] |
| PDGFRA | Pathological mast cell activation, associated with systemic mastocytosis [29] |
| CBL | Cytokine-independent mast cell activation, associated with systemic mastocytosis [29] |
| MITF | Transcription factor involved in mast cell development [36] |
| CXCR4 | Chemokine receptor involved in cell migration and immune responses [36] |
| PAR2 | Protease-activated receptor, involved in inflammation and pain signaling [36] |
| FCGR1A | Fc gamma receptors are widely expressed on a variety of immune cells and play a myriad of regulatory roles in the immune system [46] |
| HLA-B | This gene is part of the major histocompatibility complex (MHC) and plays a critical role in immune response. Variations in HLA-B have been associated with various immune-mediated conditions [47], which could influence mast cell activity through immune modulation. |
| RUNX1 | Variations in RUNX1 have been associated aggressive systemic mastocytosis [43,48] |
| TPSAB1 | Hereditary alpha-tryptasemia, mast cell activation syndrome [49] |
| CD177 | This is a specific neutrophil activation marker [50,51,52] |
| Clusters | Genes | hEDS Subjects with Variations % (n) |
|---|---|---|
| Antigen processing and presentation of endogenous peptide antigens and MHC protein complex | CYP21A2, CCR5, ERAP2, BTN1A1, TLR1, HLA-A, HLA-B, HLA-C, HLA-DRB1, and HLA-DRB5. | 88% (16) |
| Defective GALNT3 causes HFTC | MUC3A, MUC16, MUC19, and ZNF717. | 72% (13) |
| Retinoid and cholesterol metabolism | LPL, LRP2, and HP. | 27% (5) |
| Mitochondrial complex I assembly model OXPHOS system | MT-CYB, MT-ND1, EMC1, and ACAD9. | 55% (10) |
| Triplet repeat expansion | SPTA1, HTT, and ATN1. | 55% (10) |
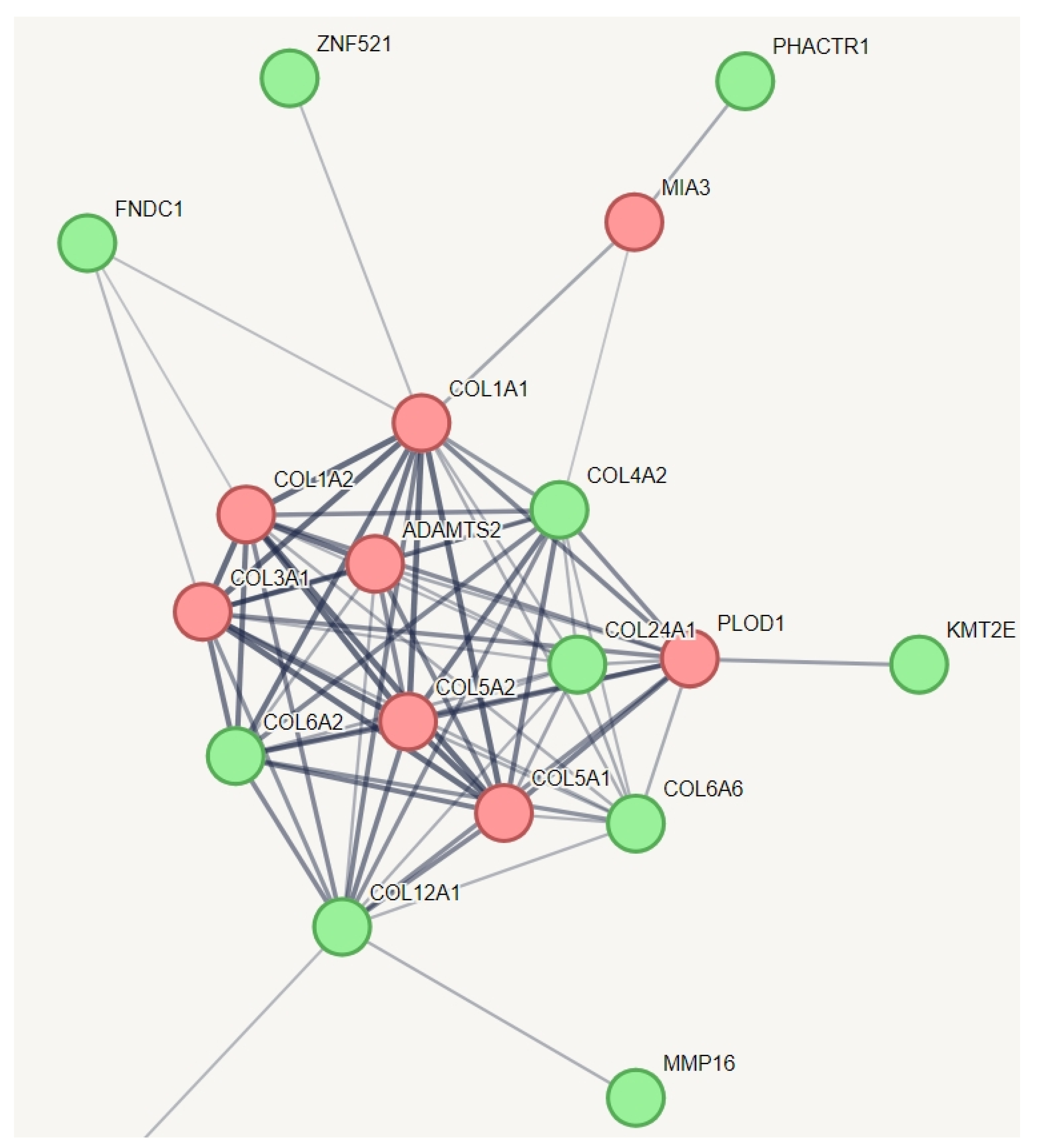
| Collagen chain trimerization | COL6A2, COL6A6, COL4A2, COL12A1, COL24A1, KMT2E, COL24A1, FNDC1, ZNF521, PHACTR1, and MMP16. | 77% (14) |
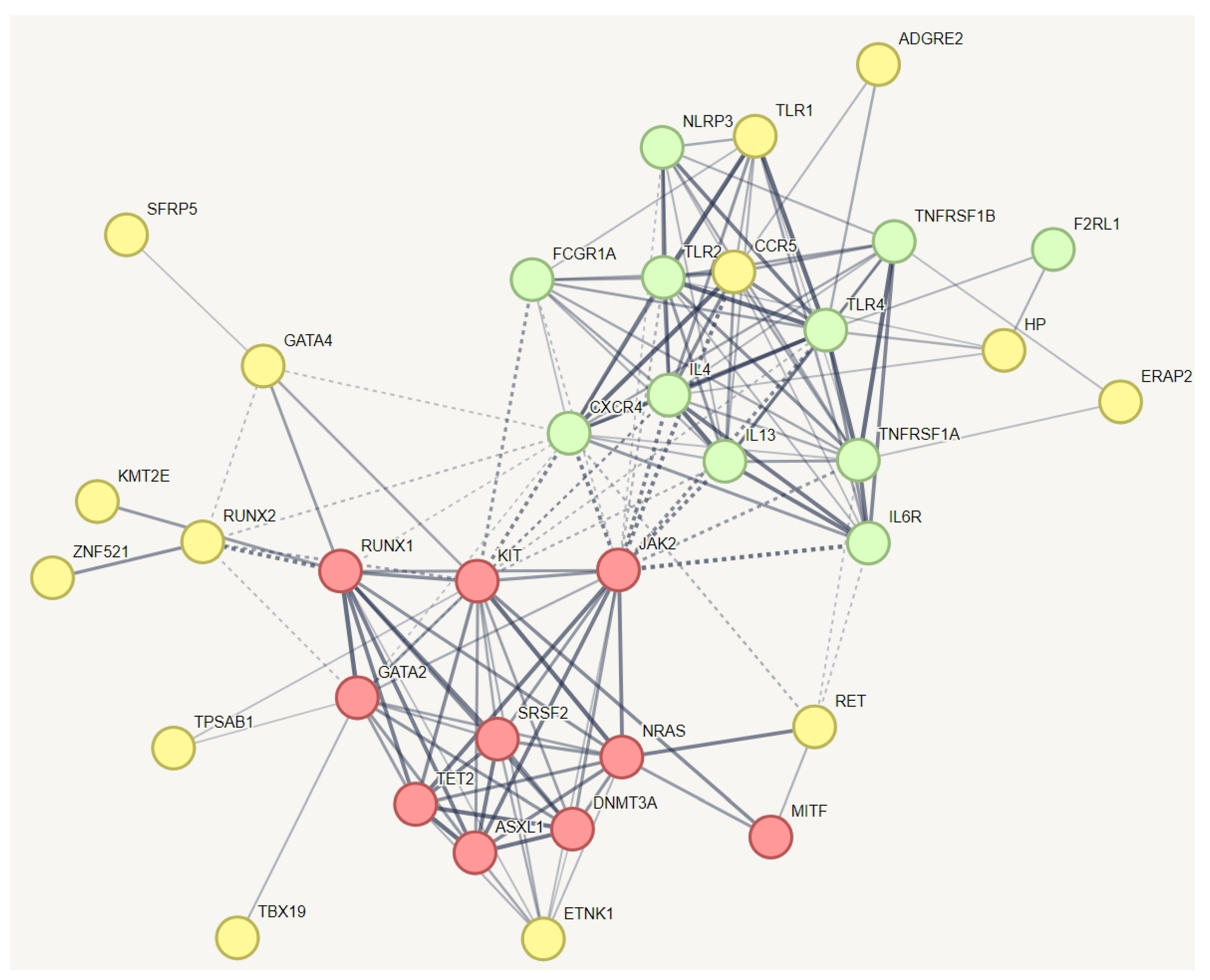
| Mast cell activation syndrome | ADGRE2, TLR1, CCR5, HP, ERAP2, SFRP5, GATA4, RUNX2, KMT2E, RET, TPSAB1, TBX19, and ZNF521. | 88% (16) |
| Selected genes with more variation among the hEDS | MT-CYB, HTT, MUC3A, HLA-B, and HLA-DRB1 | 94% (17) |
| Variations | VAF † in HEDS | Genes | Allele Frequency | HGVS/SNP | Consequence | PolyPhen Prediction †† |
|---|---|---|---|---|---|---|
| MT:15607:G | 0.11 | MT-CYB | G = 0.08 | c.861A>G(p.(Lys287=)) rs193302996 | Synonymous | likely pathogenic/benign * |
| MT:15452:A | 0.13 | MT-CYB | A = 0.17 | p.(Leu236Ile) rs193302994 | Missense | tolerated— low confidence likely pathogenic/benign |
| MT:15043:A | 0.22 | MT-CYB | A = 0.06 | c.297G>A(p.(Gly99=)) rs193302985 | Synonymous | likely pathogenic/benign * |
| MT:14905:A | 0.11 | MT-CYB | A = 0.07 | c.159G>A(p.(Met53=)) rs193302983 | Synonymous | likely pathogenic/benign * |
| MT:14783:C | 0.16 | MT-CYB | C = 0.02 | c.37T>C(p.(Leu13=)) rs193302982 | Synonymous | likely pathogenic * |
| 7:100957985: 100957984: CCAGCCAGACCC | 0.22 | MUC3A | Unknown | p.(Ser2070_His2071 insThrLysThrProSer) | Inframe insertion | Not Reported |
| 7:100954094:G | 0.11 | MUC3A | G = 0.008 | p.(His772Arg) rs933695519 | Missense | deleterious— low confidence |
| 7:100953370:A | 0.08 | MUC3A | A = 0.07 | p.(Ala531Thr) rs1490623108 | Missense | deleterious— low confidence |
| 6:32589726:G | 0.02 | HLA-DRB1 | G = 0.04 | p.(Leu6Pro) rs201726340 | Missense | deleterious |
| 6:32584379:T | 0.02 | HLA-DRB1 | T = 0.01 | rs775308685 | Splice acceptor | Not Reported |
| 6:32584218: 32584219: CT | 0.02 | HLA-DRB1 | Unknown | p.(Ala87Glu) | Missense | Deleterious |
| 6:32584185: 32584184:TC | 0.02 | HLA-DRB1 | Unknown | p.(Gln99AspfsTer31) | Frameshift | Not Reported |
| 6:32584175: 32584176 | 0.02 | HLA-DRB1 | Unknown | p.(Ala102ArgfsTer25) | Frameshift | Not Reported |
| 6:32584158:T | 0.13 | HLA-DRB1 | T = 0.00 | p.(Tyr107Ter) rs11554463 | Stop gained | Not Reported |
| 6:32584108:T | 0.02 | HLA-DRB1 | T = 0.1 | rs200689965 | Splice donor | |
| 6:32581834: 32581836:CTC | 0.02 | HLA-DRB1 | Unknown | p.(Gln125Glu) | Missense/ Splice region | Tolerated |
| 6:32581575:C | 0.02 | HLA-DRB1 | C = 0.02 | p.(Pro212Ala) rs1136846 | Missense | deleterious |
| 6:32580276:G | 0.02 | HLA-DRB1 | G = 0.03 | rs28732251 | Splice region/ Intron variant | Not Reported |
| 6:31356429: 31356431:CCA | 0.25 | HLA-B | Unknown | p.(Leu119Trp) | Missense | deleterious— low confidence |
| 6:31356280:A | 0.08 | HLA-B | A = 0.01 | p.(Arg169Leu) rs12697943 | Missense | deleterious— low confidence |
| 4:3160307:T | 0.05 | HTT | T = 0.002 | p.(Thr1260Met) rs34315806 | Missense | Deleterious/pathogenic/benign |
| 4:3074939: 3074939 | 0.05 | HTT | delG = 0.00 | p.(Gln38HisfsTer63) rs1560535226 | Frameshift | Not Reported |
| 4:3074932: 3074936 | 0.05 | HTT | 0.004 | p.(Gln36ProfsTer45) rs1338001820 | Frameshift | Not Reported |
| 4:3074930: 3074929:ACA | 0.05 | HTT | 0.00 | p.(Gln38dup) rs1553909026 | Inframe insertion | Not Reported |
| 4:3074883: 3074882: CAGCAGCAGCAG | 0.11 | HTT | Unknown | p.(Gln35_Gln38dup) | Inframe insertion | Not Reported |
| 4:3074880: 3074879: CAGCAGCAG | 0.02 | HTT | Unknown | p.(Gln36_Gln38dup) | Inframe insertion | Not Reported |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shirvani, P.; Shirvani, A.; Holick, M.F. Decoding the Genetic Basis of Mast Cell Hypersensitivity and Infection Risk in Hypermobile Ehlers-Danlos Syndrome. Curr. Issues Mol. Biol. 2024, 46, 11613-11629. https://doi.org/10.3390/cimb46100689
Shirvani P, Shirvani A, Holick MF. Decoding the Genetic Basis of Mast Cell Hypersensitivity and Infection Risk in Hypermobile Ehlers-Danlos Syndrome. Current Issues in Molecular Biology. 2024; 46(10):11613-11629. https://doi.org/10.3390/cimb46100689
Chicago/Turabian StyleShirvani, Purusha, Arash Shirvani, and Michael F. Holick. 2024. "Decoding the Genetic Basis of Mast Cell Hypersensitivity and Infection Risk in Hypermobile Ehlers-Danlos Syndrome" Current Issues in Molecular Biology 46, no. 10: 11613-11629. https://doi.org/10.3390/cimb46100689
APA StyleShirvani, P., Shirvani, A., & Holick, M. F. (2024). Decoding the Genetic Basis of Mast Cell Hypersensitivity and Infection Risk in Hypermobile Ehlers-Danlos Syndrome. Current Issues in Molecular Biology, 46(10), 11613-11629. https://doi.org/10.3390/cimb46100689