Construction of LncRNA-Related ceRNA Networks in Longissimus Dorsi Muscle of Jinfen White Pigs at Different Developmental Stages

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and LDM Tissue Collection
2.2. RNA Extraction and cDNA Synthesis
2.3. Construction of cDNA Library and Illumina Sequencing
2.4. Analysis of RNA-Seq Data
2.5. Identification of LncRNAs
2.6. Construction of LncRNA–miRNA–mRNA Networks
2.7. Validation of Sequencing Results Using Quantitative Real-Time PCR (qRT-PCR)
2.8. Statistical Analysis
3. Results
3.1. Landscape of the RNA Transcriptomes
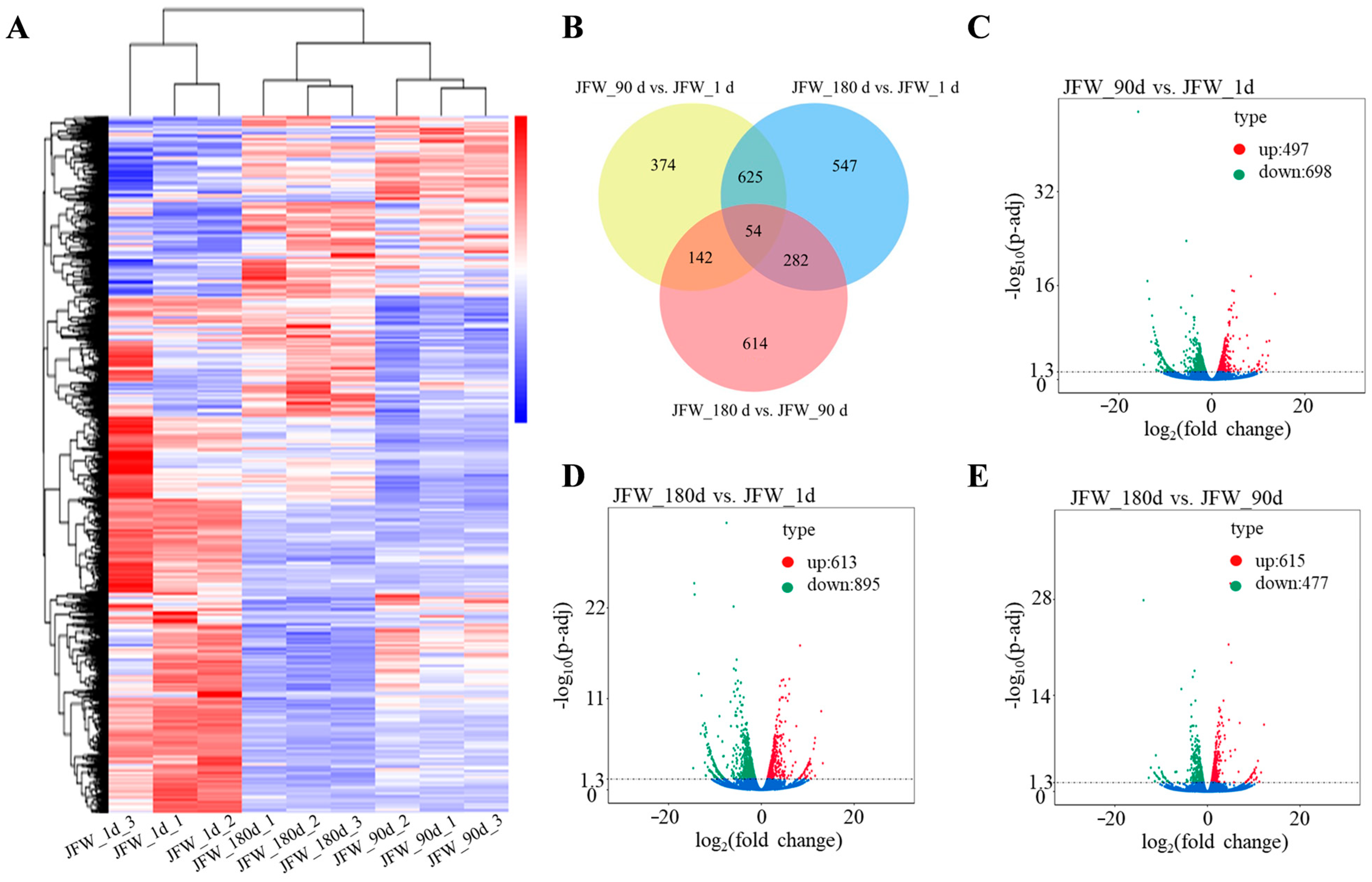
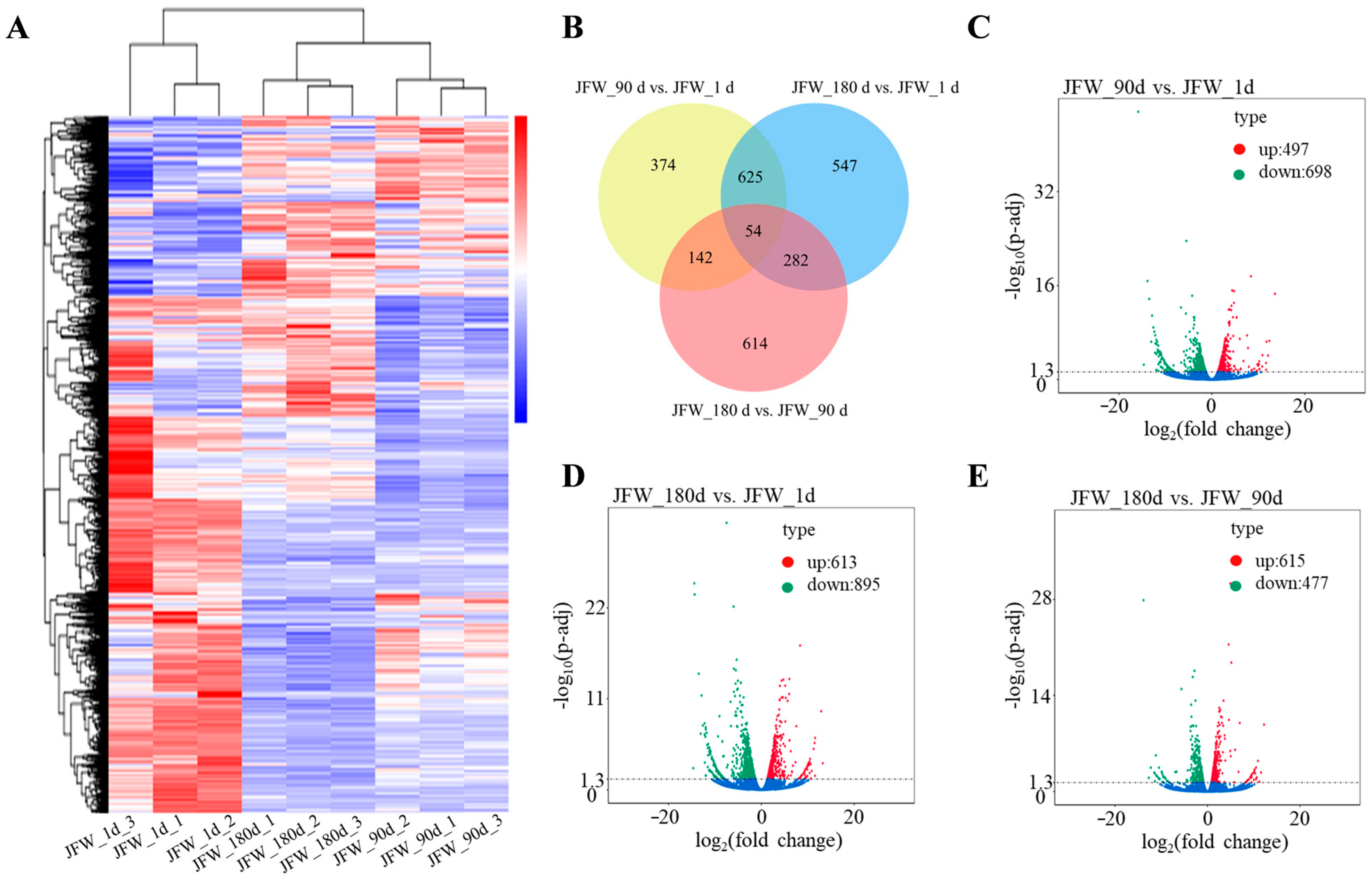
3.2. Differentially Expressed mRNAs
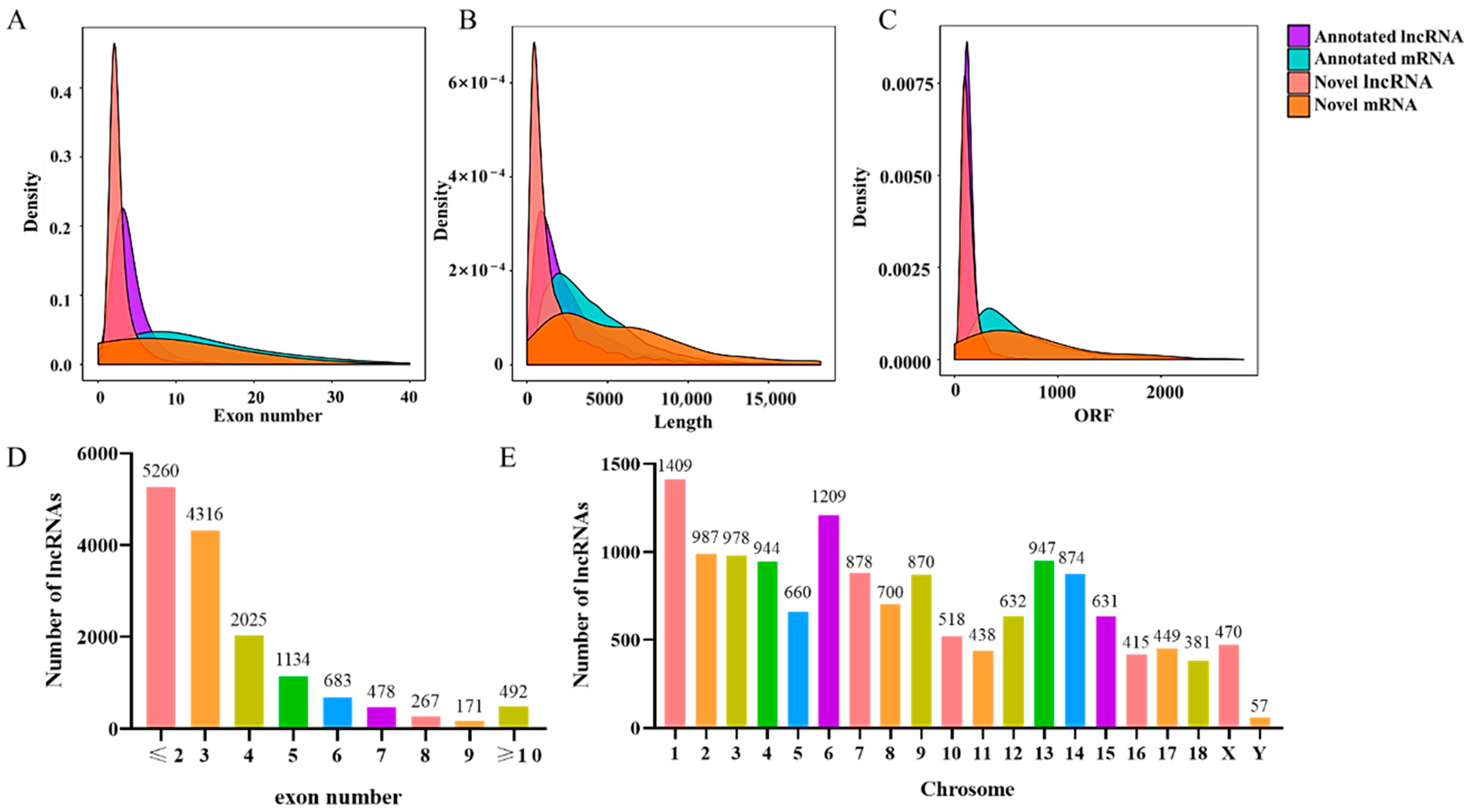
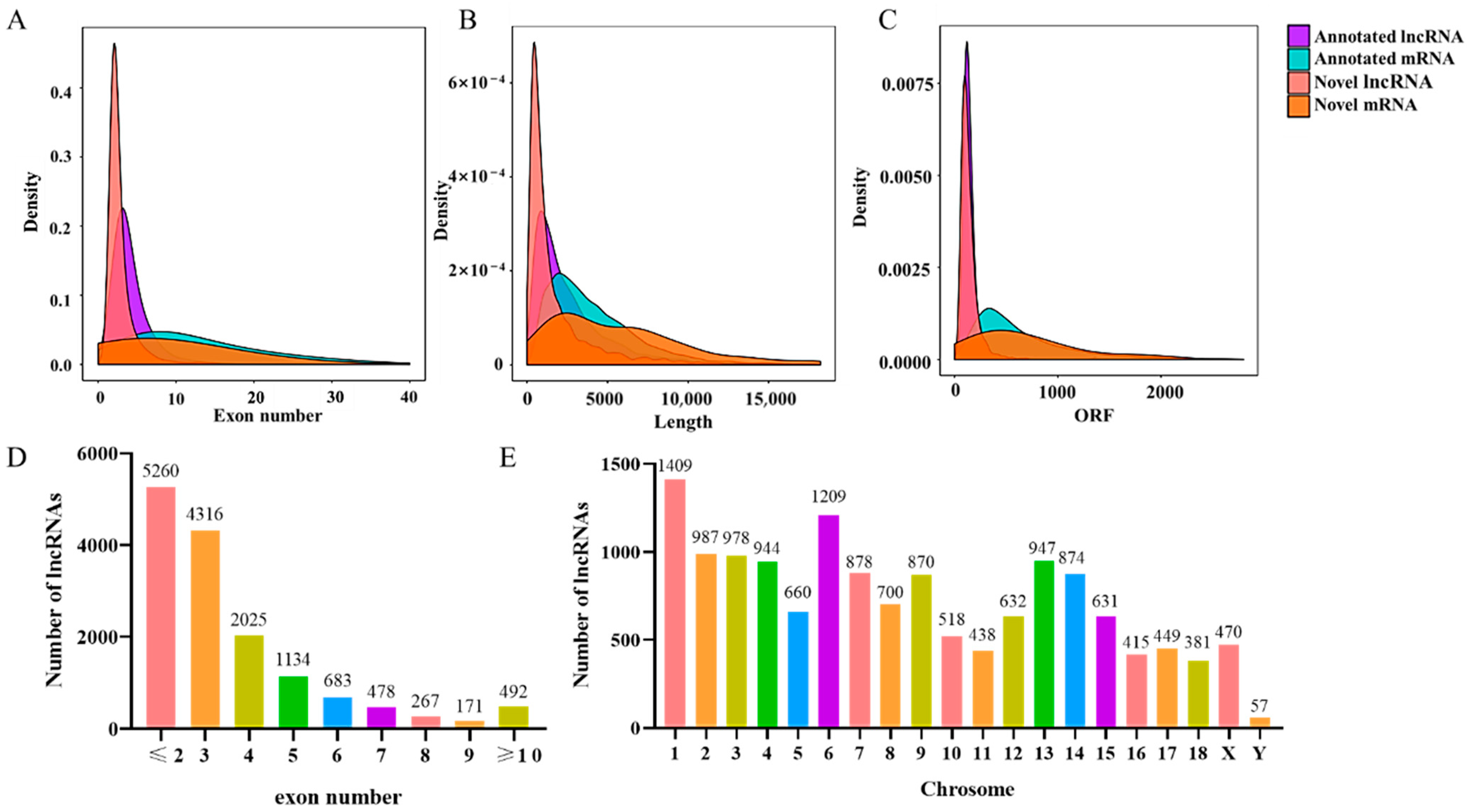
3.3. Genomic Features of LncRNAs
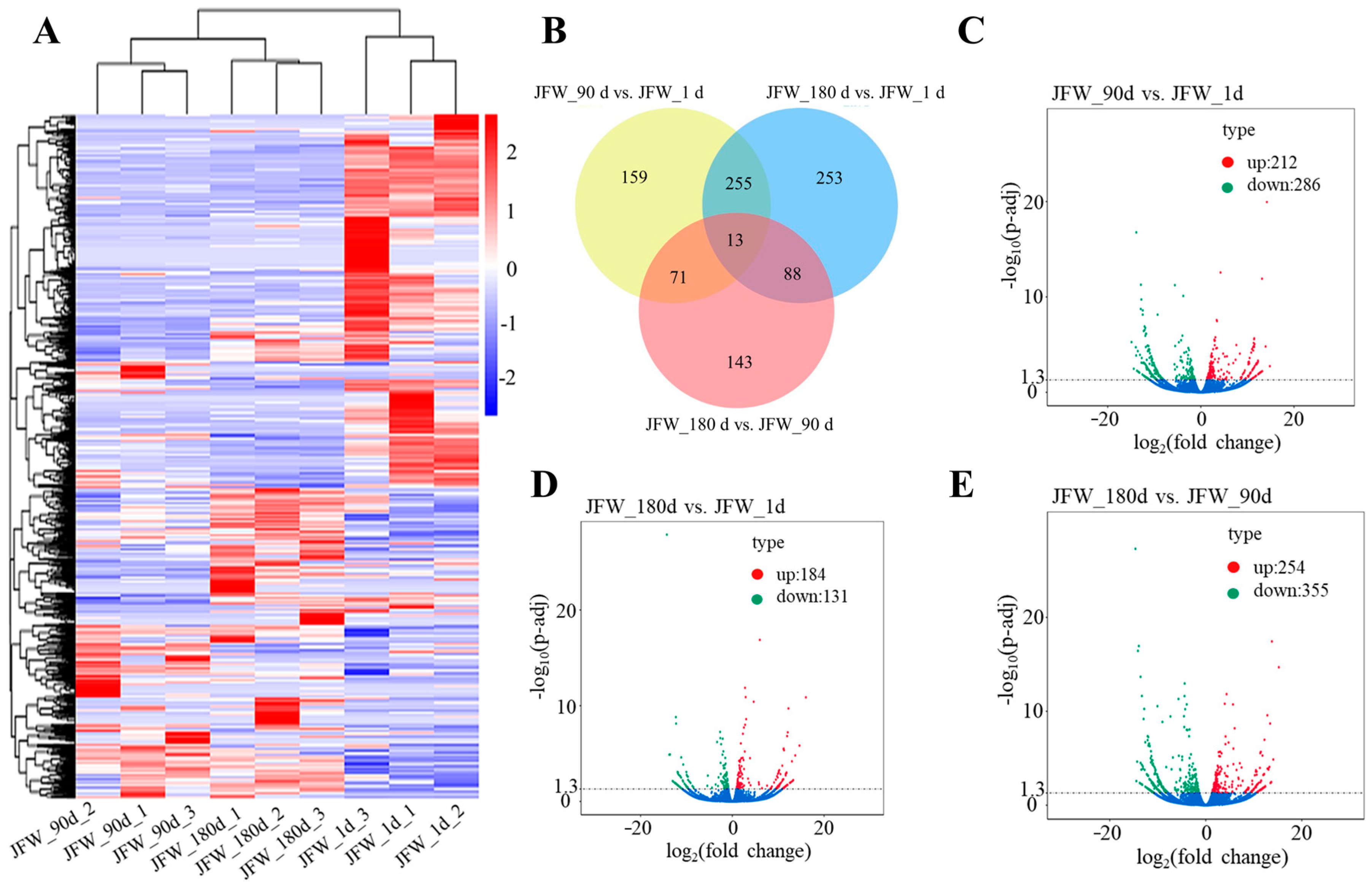
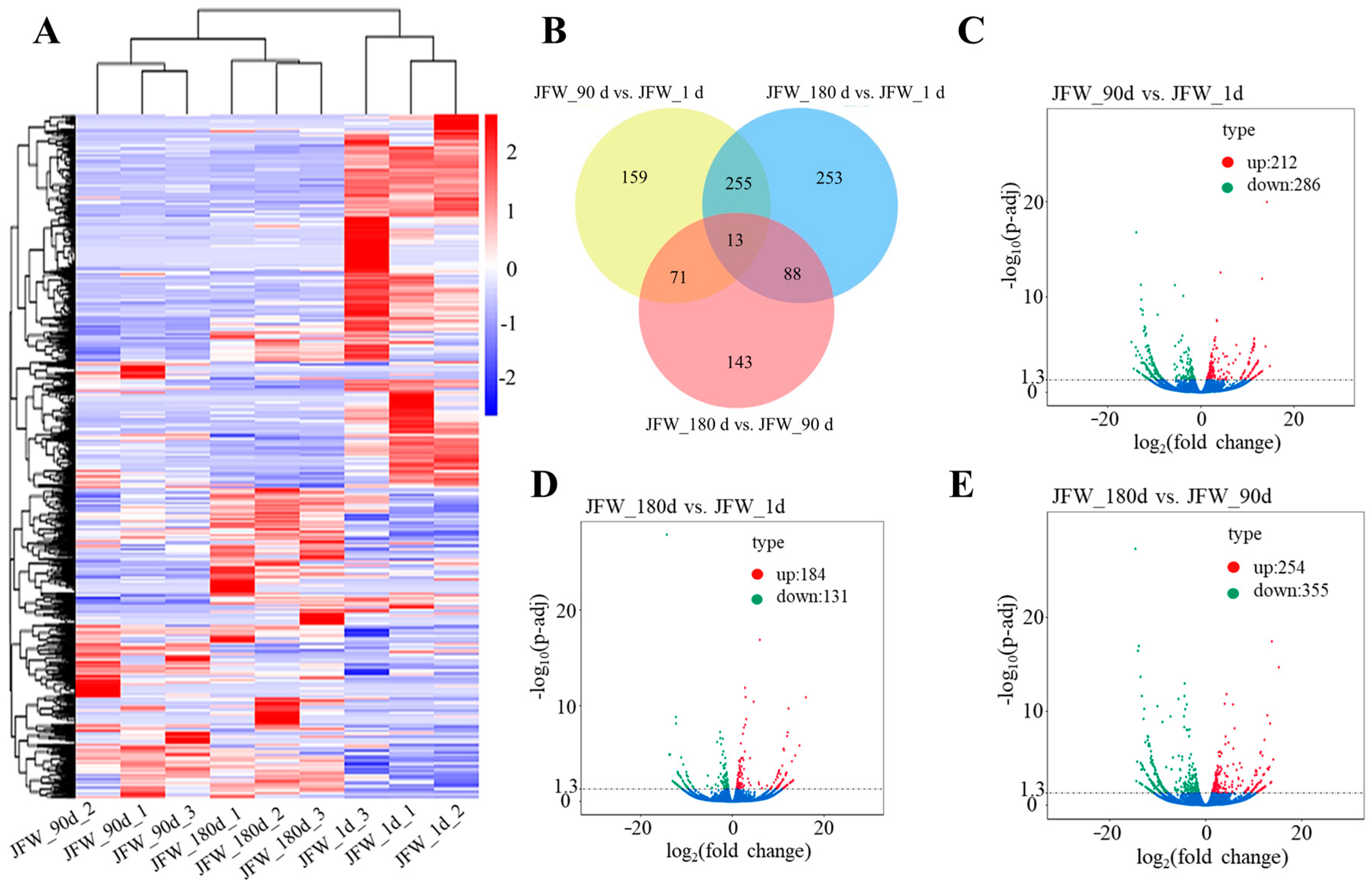
3.4. Differentially Expressed lncRNAs
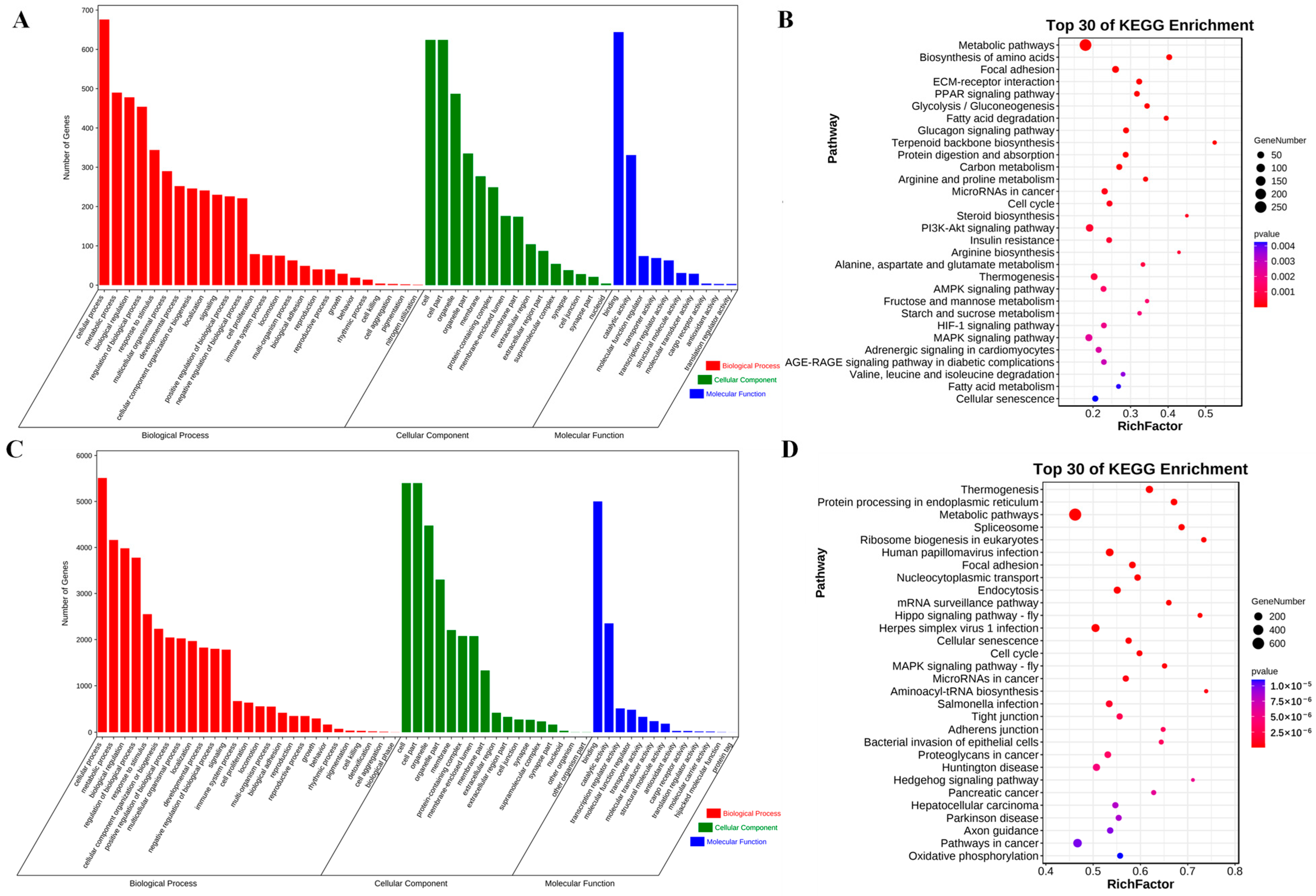
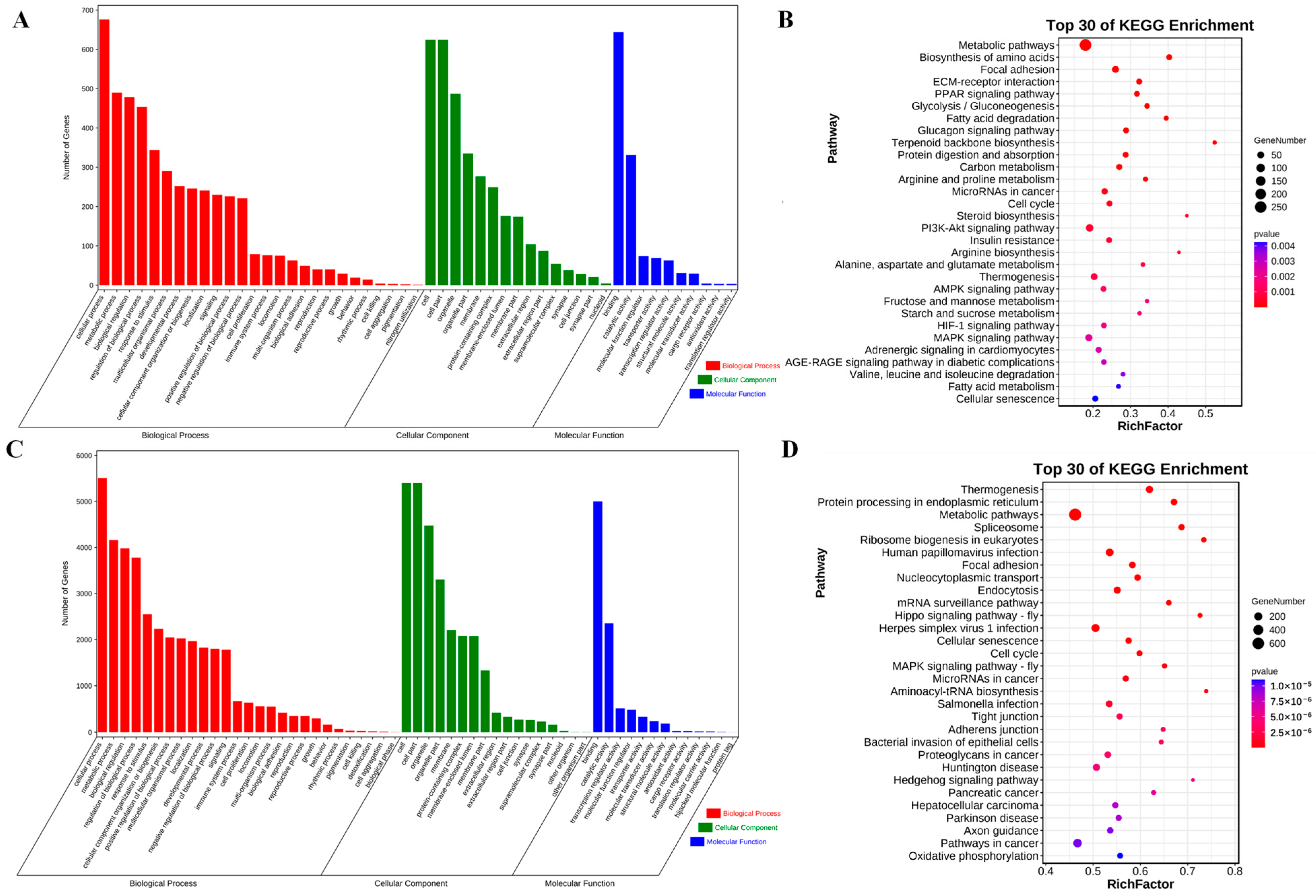
3.5. Functional Enrichment Analysis
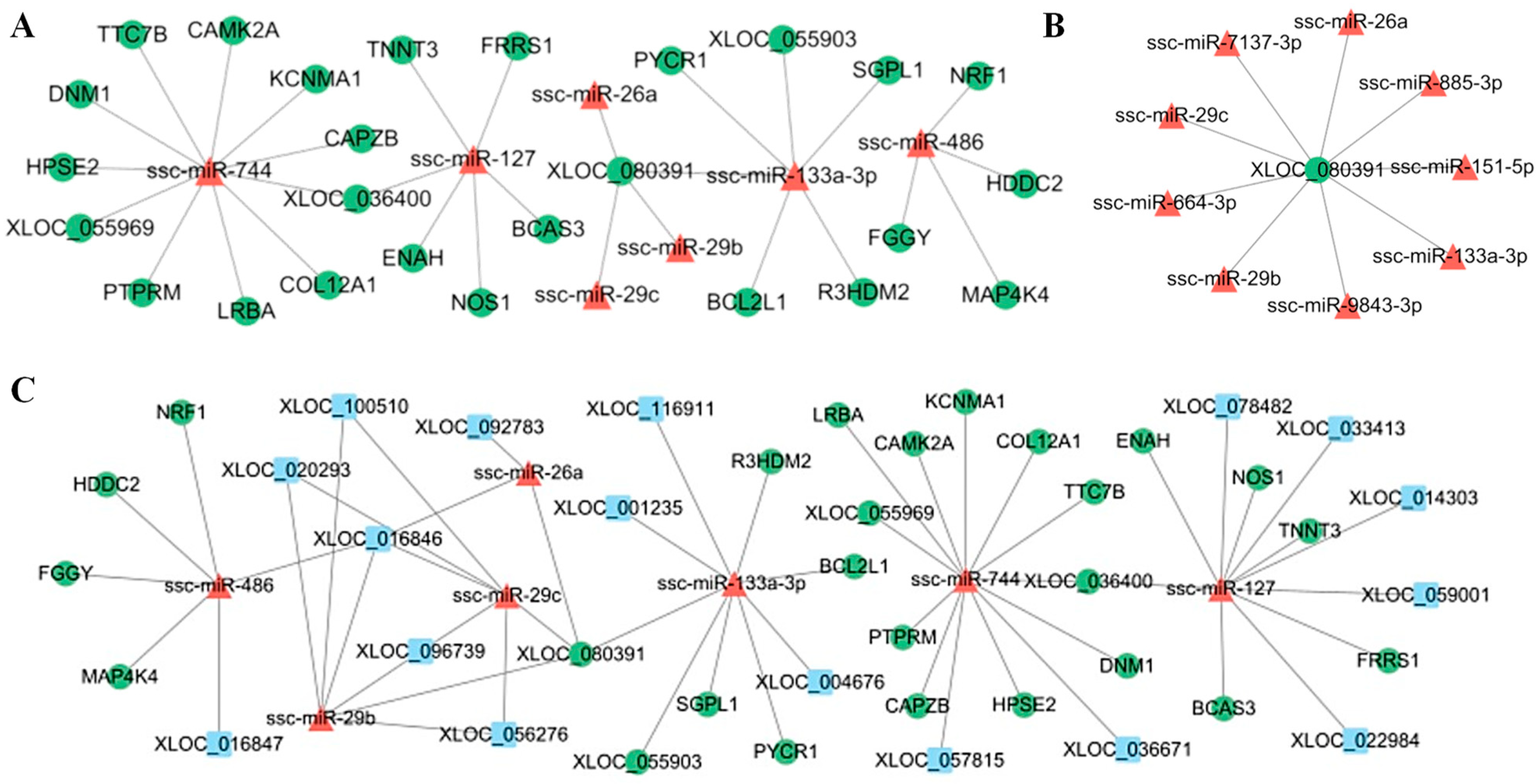
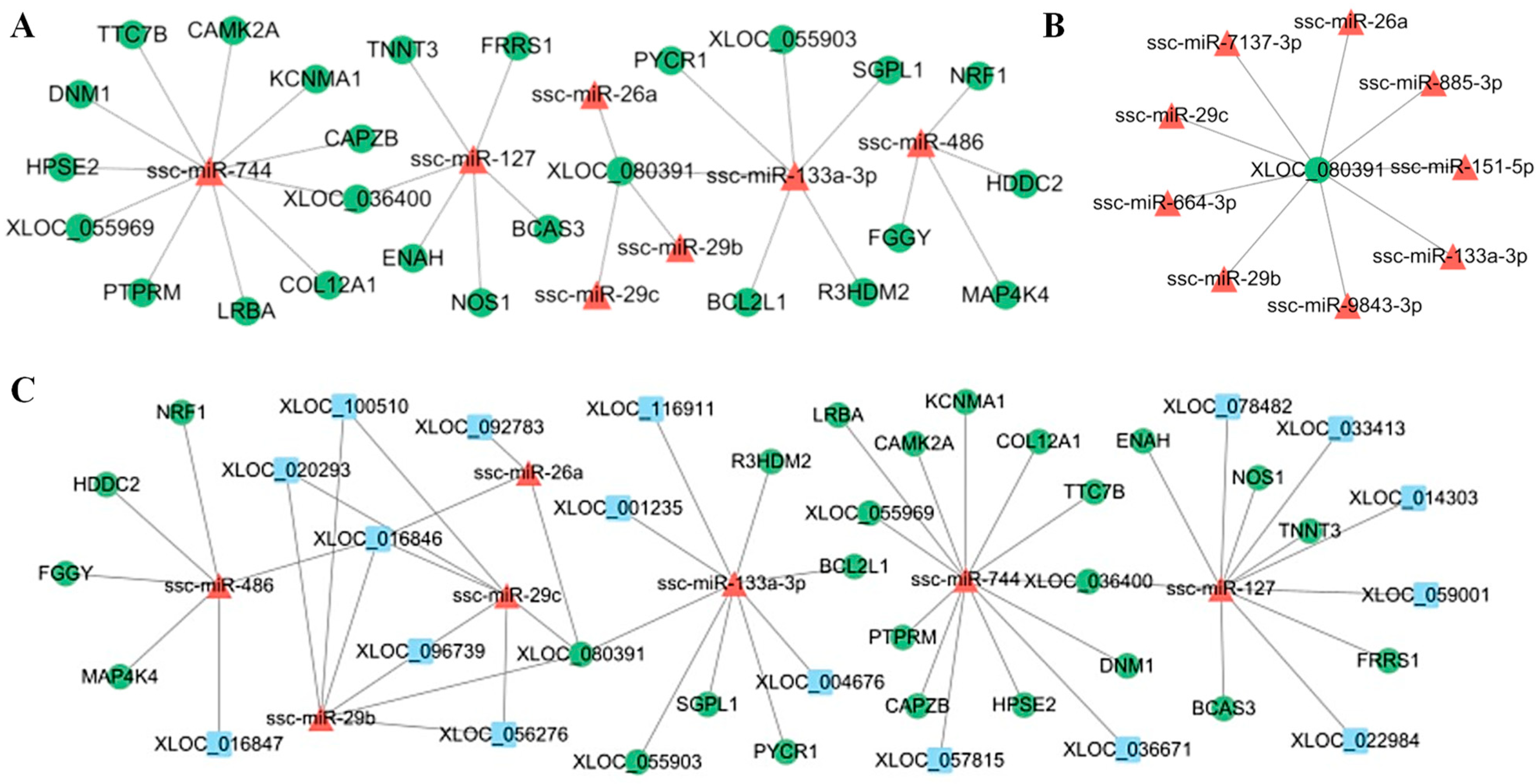
3.6. Construction of LncRNA–miRNA–mRNA Regulatory Networks
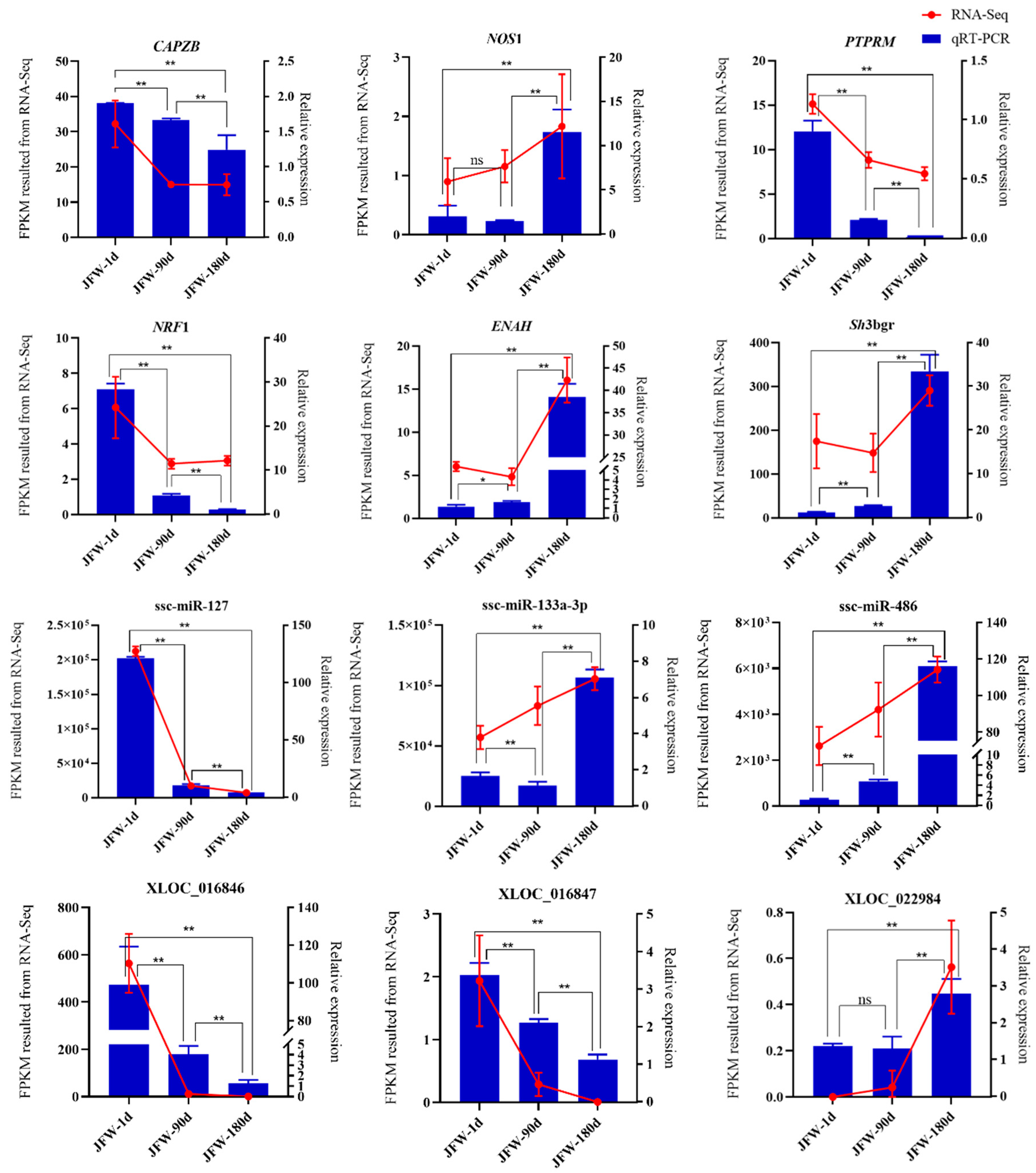
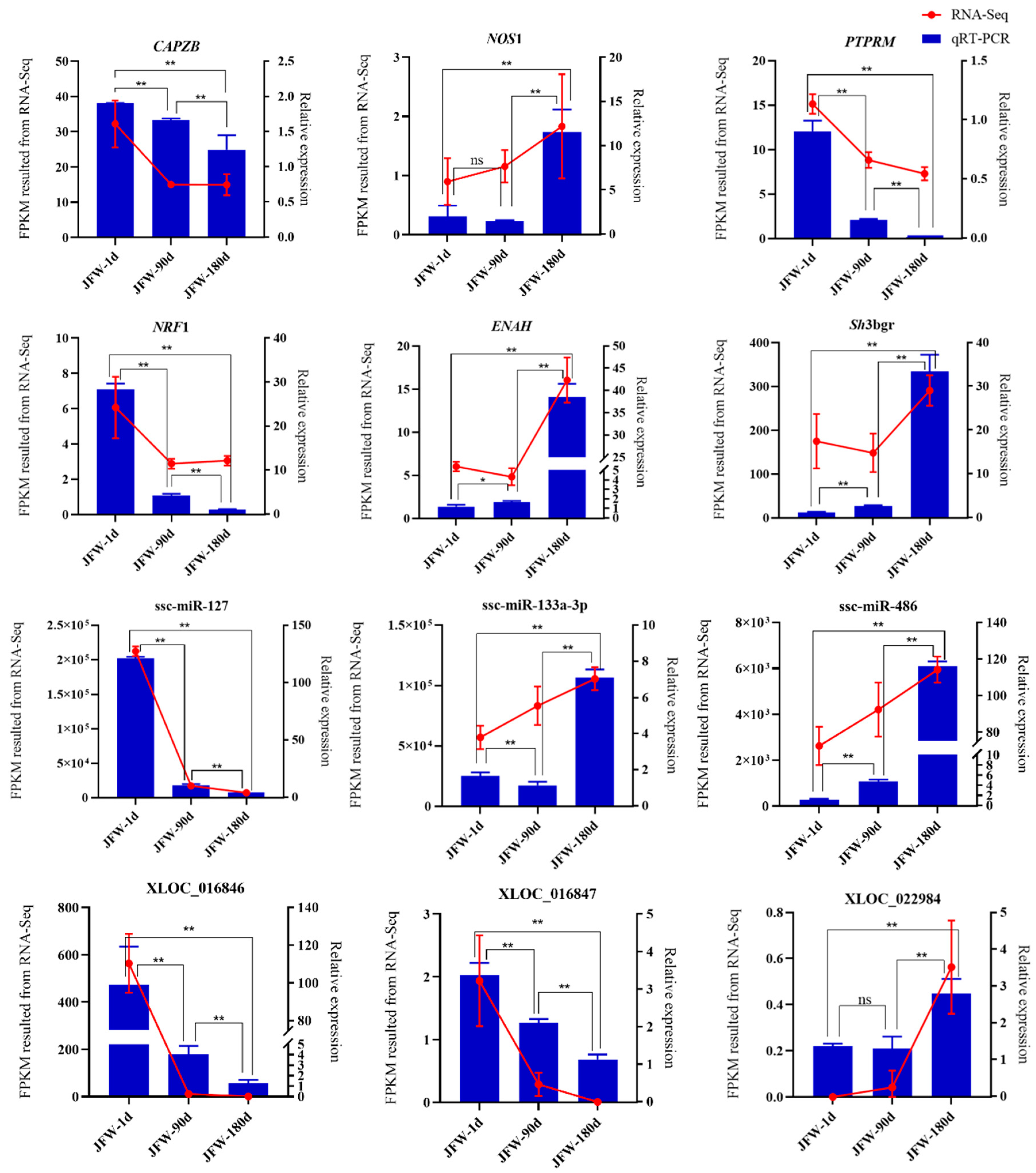
3.7. Validation of Sequencing Results via Quantitative Real-Time PCR (qRT-PCR)
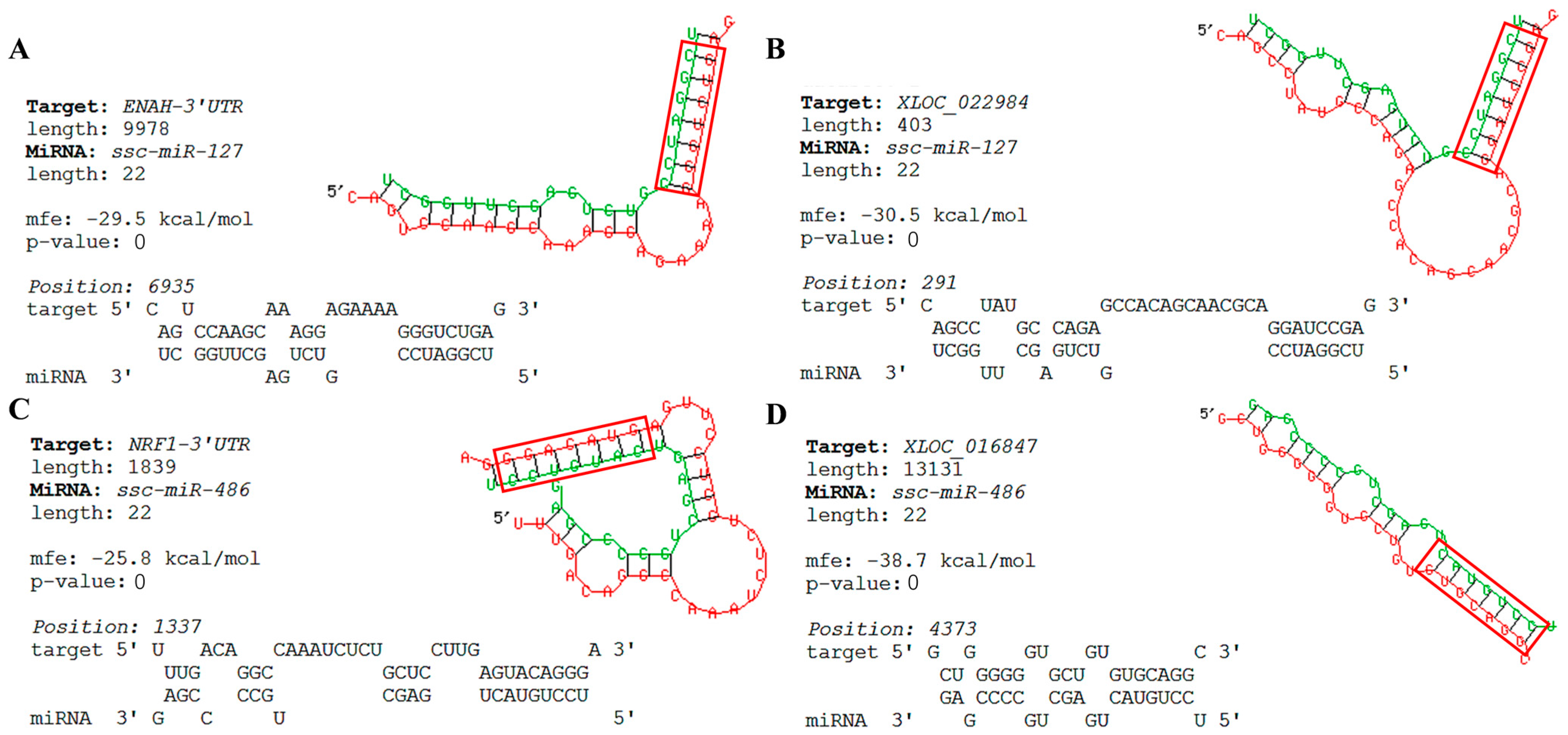
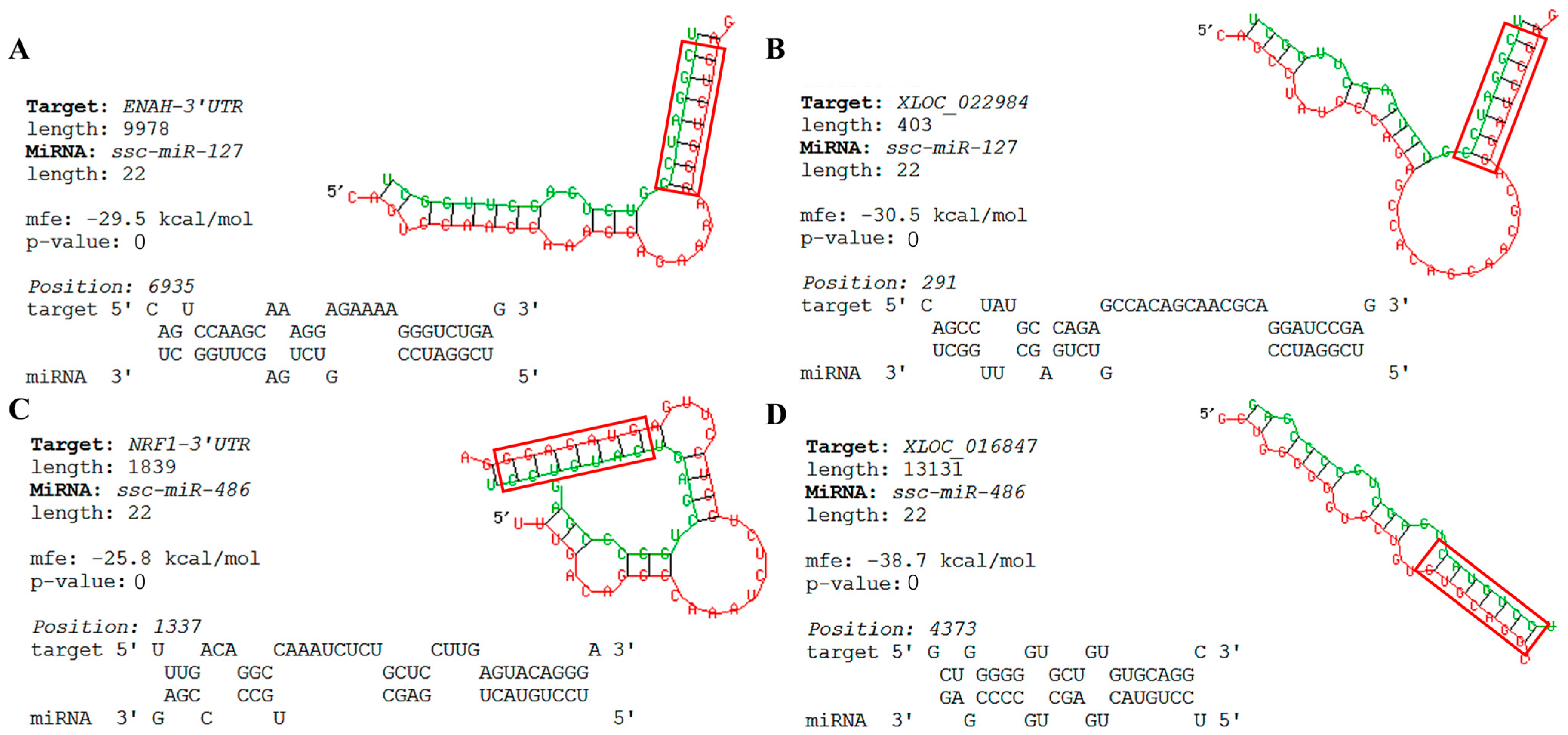
3.8. Bioinformatics Predicts Key Nodes of the Network
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baskin, K.K.; Winders, B.R.; Olson, E.N. Muscle as a “mediator” of systemic metabolism. Cell Metab. 2015, 21, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Mirzoev, T.M. Skeletal muscle recovery from disuse atrophy: Protein turnover signaling and strategies for accelerating muscle regrowth. Int. J. Mol. Sci. 2020, 21, 7049. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Sandri, M.; Murgia, M. Activity-dependent signaling pathways controlling muscle diversity and plasticity. Physiology 2007, 22, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, H.; Davis, R.; Tapscott, S.; Thayer, M.; Krause, M.; Benezra, R.; Blackwell, T.K.; Turner, D.; Rupp, R.; Hollenberg, S.; et al. The myoD gene family: Nodal point during specification of the muscle cell lineage. Science 1991, 251, 761–766. [Google Scholar] [CrossRef]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar] [CrossRef]
- Salhi, A.; Essack, M.; Alam, T.; Bajic, V.P.; Ma, L.; Radovanovic, A.; Marchand, B.; Schmeier, S.; Zhang, Z.; Bajic, V.B. DES-ncRNA: A knowledgebase for exploring information about human micro and long noncoding RNAs based on literature-mining. RNA Biol. 2017, 14, 963–971. [Google Scholar] [CrossRef]
- Zhao, Y.; Samal, E.; Srivastava, D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature 2005, 436, 214–220. [Google Scholar] [CrossRef]
- Kim, H.K.; Lee, Y.S.; Sivaprasad, U.; Malhotra, A.; Dutta, A. Muscle-specific microRNA miR-206 promotes muscle differentiation. J. Cell Biol. 2006, 174, 677–687. [Google Scholar] [CrossRef]
- Rosenberg, M.I.; Georges, S.A.; Asawachaicharn, A.; Analau, E.; Tapscott, S.J. MyoD inhibits Fstl1 and Utrn expression by inducing transcription of miR-206. J. Cell Biol. 2006, 175, 77–85. [Google Scholar] [CrossRef]
- Liu, N.; Williams, A.H.; Kim, Y.; McAnally, J.; Bezprozvannaya, S.; Sutherland, L.B.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. An intragenic MEF2-dependent enhancer directs muscle-specific expression of microRNAs 1 and 133. Proc. Natl. Acad. Sci. USA 2007, 104, 20844–20849. [Google Scholar] [CrossRef]
- Knoll, M.; Lodish, H.F.; Sun, L. Long non-coding RNAs as regulators of the endocrine system. Nat. Rev. Endocrinol. 2015, 11, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Endo, T. Molecular mechanisms of skeletal muscle development, regeneration, and osteogenic conversion. Bone 2015, 80, 2–13. [Google Scholar] [CrossRef]
- Tao, H.; Zhang, J.-G.; Qin, R.-H.; Dai, C.; Shi, P.; Yang, J.-J.; Deng, Z.-Y.; Shi, K.-H. LncRNA GAS5 controls cardiac fibroblast activation and fibrosis by targeting miR-21 via PTEN/MMP-2 signaling pathway. Toxicology 2017, 386, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, X.; Sun, H.; Wang, H. Long non-coding RNAs in the regulation of skeletal myogenesis and muscle diseases. Cancer Lett. 2018, 417, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.F.; Li, Y.; Ding, X.B.; Li, X.; Zhang, L.L.; Liu, X.F.; Guo, H. lnc133b, a novel, long non-coding RNA, regulates bovine skeletal muscle satellite cell proliferation and differentiation by mediating miR-133b. Gene 2017, 630, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Zhou, B.; Shi, L.; Wang, H.; Chu, Q.; Xu, F.; Li, Y.; Chen, R.; Shen, C.; Schinckel, A.P. lncRNA AK017368 promotes proliferation and suppresses differentiation of myoblasts in skeletal muscle development by attenuating the function of miR-30c. FASEB J. 2018, 32, 377–389. [Google Scholar] [CrossRef]
- Cong, W.; Liu, B.; Liu, S.; Sun, M.; Liu, H.; Yang, Y.; Wang, R.; Xiao, J. Implications of the Wnt5a/CaMKII pathway in retinoic acid-induced myogenic tongue abnormalities of developing mice. Sci. Rep. 2014, 4, 6082. [Google Scholar] [CrossRef]
- Xu, X.; Mishra, B.; Qin, N.; Sun, X.; Zhang, S.; Yang, J.; Xu, R. Differential Transcriptome Analysis of Early Postnatal Developing Longissimus Dorsi Muscle from Two Pig Breeds Characterized in Divergent Myofiber Traits and Fatness. Anim. Biotechnol. 2019, 30, 63–74. [Google Scholar] [CrossRef]
- Shang, P.; Wang, Z.; Chamba, Y.; Zhang, B.; Zhang, H.; Wu, C. A comparison of prenatal muscle transcriptome and proteome profiles between pigs with divergent growth phenotypes. J. Cell Biochem. 2019, 120, 5277–5286. [Google Scholar] [CrossRef]
- Miao, W.; Ma, Z.; Tang, Z.; Yu, L.; Liu, S.; Huang, T.; Wang, P.; Wu, T.; Song, Z.; Zhang, H.; et al. Integrative ATAC-seq and RNA-seq Analysis of the Longissimus Muscle of Luchuan and Duroc Pigs. Front. Nutr. 2021, 8, 742672. [Google Scholar] [CrossRef]
- Wang, D.; Pu, Y.; Li, Y.; Pan, D.; Wang, S.; Tian, W.; Ma, Y.; Jiang, L. Comprehensive analysis of lncRNAs involved in skeletal muscle development in ZBED6-knockout Bama pigs. BMC Genom. 2021, 22, 593. [Google Scholar] [CrossRef]
- Hou, X.; Wang, L.; Zhao, F. Genome-Wide Expression Profiling of mRNAs, lncRNAs and circRNAs in Skeletal Muscle of Two Different Pig Breeds. Animals 2021, 11, 3169. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.-Q.; Liu, X.-Q.; Zhao, S.-Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.-P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef]
- Rajewsky, N. microRNA target predictions in animals. Nat. Genet. 2006, 38, S8–S13. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 254, 402–408. [Google Scholar] [CrossRef]
- Kaya-Çopur, A.; Marchiano, F.; Hein, M.Y.; Alpern, D.; Russeil, J.; Luis, N.M.; Mann, M.; Deplancke, B.; Habermann, B.H.; Schnorrer, F.; et al. The Hippo pathway controls myofibril assembly and muscle fiber growth by regulating sarcomeric gene expression. Elife 2021, 10, e63726. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Oshima, T.; Morimoto, T.; Ikeda, S.; Yoshikawa, H.; Shiwa, Y.; Ishikawa, S.; Linak, M.C.; Hirai, A.; Takahashi, H.; et al. Sequence-specific error profile of Illumina sequencers. Nucleic Acids Res. 2011, 39, e90. [Google Scholar] [CrossRef] [PubMed]
- Meacham, F.; Boffelli, D.; Dhahbi, J.; Martin, D.I.; Singer, M.; Pachter, L. Identification and correction of systematic error in high-throughput sequence data. BMC Bioinform. 2011, 12, 451. [Google Scholar] [CrossRef] [PubMed]
- Watt, K.I.; Goodman, C.A.; Hornberger, T.A.; Gregorevic, P. The Hippo Signaling Pathway in the Regulation of Skeletal Muscle Mass and Function. Exerc. Sport Sci. Rev. 2018, 46, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Delafontaine, P. Mechanisms of IGF-1-Mediated Regulation of Skeletal Muscle Hypertrophy and Atrophy. Cells 2020, 9, 1970. [Google Scholar] [CrossRef]
- Alman, B.A. The role of hedgehog signalling in skeletal health and disease. Nat. Rev. Rheumatol. 2015, 11, 552–560. [Google Scholar] [CrossRef]
- Chen, M.; Ji, C.; Yang, Q.; Gao, S.; Peng, Y.; Li, Z.; Gao, X.; Li, Y.; Jiang, N.; Zhang, Y.; et al. AKT2 regulates development and metabolic homeostasis via AMPK-depedent pathway in skeletal muscle. Clin. Sci. 2020, 134, 2381–2398. [Google Scholar] [CrossRef]
- Yu, Z.; Xu, X.; Ai, N.; Wang, K.; Zhang, P.; Li, X.; LiuFu, S.; Liu, X.; Jiang, J.; Gu, J.; et al. Integrated analysis of circRNA, lncRNA, miRNA and mRNA to reveal the ceRNA regulatory network of postnatal skeletal muscle development in Ningxiang pig. Front. Cell Dev. Biol. 2023, 11, 1185823. [Google Scholar] [CrossRef] [PubMed]
- Suhr, F.; Gehlert, S.; Grau, M.; Bloch, W. Skeletal muscle function during exercise-fine-tuning of diverse subsystems by nitric oxide. Int. J. Mol. Sci. 2013, 14, 7109–7139. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xu, Y.; Wang, Y.; Zhong, T.; Tang, G.; Li, L.; Zhang, H.; Xiong, Y. Identification and characterization of a differentially expressed protein (CAPZB) in skeletal muscle between Meishan and Large White pigs. Gene 2014, 544, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wu, P.; Chen, D.; Zhou, J.; Yang, X.; Jiang, A.; Xiao, W.; Qiu, X.; Zeng, Y.; Xu, X.; et al. Detecting the selection signatures in Chinese Duroc, Landrace, Yorkshire, Liangshan, and Qingyu pigs. Funct. Integr. Genom. 2021, 21, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Atmanli, A.; Morales, M.G.; Tan, W.; Chen, K.; Xiao, X.; Xu, L.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Nrf1 promotes heart regeneration and repair by regulating proteostasis and redox balance. Nat. Commun. 2021, 12, 5270. [Google Scholar] [CrossRef] [PubMed]
- Sekine, H.; Motohashi, H. Roles of CNC Transcription Factors NRF1 and NRF2 in Cancer. Cancers 2021, 13, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Widenmaier, S.B.; Snyder, N.A.; Nguyen, T.B.; Arduini, A.; Lee, G.Y.; Arruda, A.P.; Saksi, J.; Bartelt, A.; Hotamisligil, G.S. NRF1 Is an ER Membrane Sensor that Is Central to Cholesterol Homeostasis. Cell 2017, 171, 1094–1109. [Google Scholar] [CrossRef] [PubMed]
- Rovito, D.; Rerra, A.-I.; Ueberschlag-Pitiot, V.; Joshi, S.; Karasu, N.; Dacleu-Siewe, V.; Ben Rayana, K.; Ghaibour, K.; Parisotto, M.; Ferry, A.; et al. Myod1 and GR coordinate myofiber-specific transcriptional enhancers. Nucleic Acids Res. 2021, 49, 4472–4492. [Google Scholar] [CrossRef]
- Bear, J.E.; Gertler, F.B. Ena/VASP: Towards resolving a pointed controversy at the barbed end. J. Cell Sci. 2009, 122, 1947–1953. [Google Scholar] [CrossRef]
- Kragtorp, K.A.; Miller, J.R. Regulation of somitogenesis by Ena/VASP proteins and FAK during Xenopus development. Development 2006, 133, 685–695. [Google Scholar] [CrossRef]
- Jang, D.G.; Sim, H.J.; Song, E.K.; Medina-Ruiz, S.; Seo, J.K.; Park, T.J. A thioredoxin fold protein Sh3bgr regulates Enah and is necessary for proper sarcomere formation. Dev. Biol. 2015, 405, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Zhang, L.; Jiang, X.; Sheng, Y.; Xu, N. Differential miRNA expression profiles in the longissimus dorsi muscle between intact and castrated male pigs. Res. Vet. Sci. 2015, 99, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Qi, R.; Wang, J.; Huang, W.; Wu, Y.; Huang, X.; Yang, F.; Huang, J. Differential expression profile of miRNAs in porcine muscle and adipose tissue during development. Gene 2017, 618, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ma, J.; Qiu, W.; Zhang, J.; Feng, S.; Zhou, X.; Wang, X.; Jin, L.; Long, K.; Liu, L.; et al. Guanidinoacetic Acid Regulates Myogenic Differentiation and Muscle Growth Through miR-133a-3p and miR-1a-3p Co-mediated Akt/mTOR/S6K Signaling Pathway. Int. J. Mol. Sci. 2018, 19, 2837. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.; Wu, R.; Han, W.; Zhang, Y.; Zhu, D. miR-127 enhances myogenic cell differentiation by targeting S1PR3. Cell Death Dis. 2017, 8, e2707. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, C.; Dou, M.; Sun, Y.; Yu, T.; Pang, W.; Yang, G. Deciphering the miRNA transcriptome of Rongchang pig longissimus dorsi at weaning and slaughter time points. J. Anim. Physiol. Anim. Nutr. 2020, 104, 954–964. [Google Scholar] [CrossRef]
- Peng, S.; Song, C.; Li, H.; Cao, X.; Ma, Y.; Wang, X.; Huang, Y.; Lan, X.; Lei, C.; Chaogetu, B.; et al. Circular RNA SNX29 Sponges miR-744 to Regulate Proliferation and Differentiation of Myoblasts by Activating the Wnt5a/Ca(2+) Signaling Pathway. Mol. Ther. Nucleic Acids 2019, 16, 481–493. [Google Scholar] [CrossRef]
- Hitachi, K.; Nakatani, M.; Tsuchida, K. Myostatin signaling regulates Akt activity via the regulation of miR-486 expression. Int. J. Biochem. Cell Biol. 2014, 47, 93–103. [Google Scholar] [CrossRef]
- Small, E.M.; O’rourke, J.R.; Moresi, V.; Sutherland, L.B.; McAnally, J.; Gerard, R.D.; Richardson, J.A.; Olson, E.N. Regulation of PI3-kinase/Akt signaling by muscle-enriched microRNA-486. Proc. Natl. Acad. Sci. USA 2010, 107, 4218–4223. [Google Scholar] [CrossRef]
- Zhu, J.G.; Shen, Y.H.; Liu, H.L.; Liu, M.; Shen, Y.Q.; Kong, X.Q.; Song, G.X.; Qian, L.M. Long noncoding RNAs expression profile of the developing mouse heart. J. Cell. Biochem. 2014, 115, 910–918. [Google Scholar] [CrossRef]
- Malouf, G.G.; Zhang, J.; Yuan, Y.; Compérat, E.; Rouprêt, M.; Cussenot, O.; Chen, Y.; Thompson, E.J.; Tannir, N.M.; Weinstein, J.N.; et al. Characterization of long non-coding RNA transcriptome in clear-cell renal cell carcinoma by next-generation deep sequencing. Mol. Oncol. 2015, 9, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.F.; Guo, X.H.; Du, M.; Cao, G.Q.; Yang, Q.C.; Pu, Z.D.; Wang, Z.Y.; Zhang, Q.; Li, M.; Jin, Y.S.; et al. LncRNA profiling of skeletal muscles in Large White pigs and Mashen pigs during development. J. Anim. Sci. 2017, 95, 4239–4250. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ren, Q.; Hua, L.; Chen, J.; Zhang, J.; Bai, H.; Li, H.; Xu, B.; Shi, Z.; Cao, H.; et al. Comprehensive Analysis of Differentially Expressed mRNA, lncRNA and circRNA and Their ceRNA Networks in the Longissimus Dorsi Muscle of Two Different Pig Breeds. Int. J. Mol. Sci. 2019, 20, 1107. [Google Scholar] [CrossRef] [PubMed]
- Thomson, D.W.; Dinger, M.E. Endogenous microRNA sponges: Evidence and controversy. Nat. Rev. Genet. 2016, 17, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-K.; Li, J.; Guan, D.; Liang, C.; Zhuo, Z.; Liu, J.; Lu, A.; Zhang, G.; Zhang, B.-T. Long Noncoding RNA lncMUMA Reverses Established Skeletal Muscle Atrophy following Mechanical Unloading. Mol. Ther. 2018, 26, 2669–2680. [Google Scholar] [CrossRef] [PubMed]
- Giovarelli, M.; Bucci, G.; Ramos, A.; Bordo, D.; Wilusz, C.J.; Chen, C.-Y.; Puppo, M.; Briata, P.; Gherzi, R. H19 long noncoding RNA controls the mRNA decay promoting function of KSRP. Proc. Natl. Acad. Sci. USA 2014, 111, E5023–E5028. [Google Scholar] [CrossRef]
- Liu, Y.; Li, G.; Zhang, J.-F. The role of long non-coding RNA H19 in musculoskeletal system: A new player in an old game. Exp. Cell Res. 2017, 360, 61–65. [Google Scholar] [CrossRef]
- Wu, P.; Wang, K.; Zhou, J.; Chen, D.; Yang, X.; Jiang, A.; Shen, L.; Zhang, S.; Xiao, W.; Jiang, Y.; et al. Whole-genome sequencing association analysis reveals the genetic architecture of meat quality traits in Chinese Qingyu pigs. Genome 2020, 63, 503–515. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Raw Reads | Clean Reads | Q20 (%) | Q30 (%) | GC Content (%) |
|---|---|---|---|---|---|
| JFW_1d_1 | 92,655,706 | 91,942,766 | 98.62 | 95.72 | 55.62 |
| JFW_1d_2 | 107,050,232 | 106,444,968 | 98.64 | 95.78 | 57.33 |
| JFW_1d_3 | 104,392,040 | 103,814,406 | 98.58 | 95.45 | 47.55 |
| JFW_90d_1 | 96,202,328 | 95,550,798 | 98.08 | 94.20 | 55.77 |
| JFW_90d_2 | 106,311,518 | 105,563,842 | 98.56 | 95.75 | 60.57 |
| JFW_90d_3 | 95,622,436 | 94,984,938 | 98.67 | 95.89 | 56.13 |
| JFW_180 d_1 | 94,853,354 | 94,375,978 | 97.81 | 93.78 | 57.86 |
| JFW_180 d_2 | 104,189,042 | 103,407,232 | 98.59 | 95.54 | 54.04 |
| JFW_180 d_3 | 107,380,946 | 106,567,824 | 98.40 | 95.04 | 55.09 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Shi, M.; Zhang, Y.; Niu, J.; Li, W.; Yuan, J.; Cai, C.; Yang, Y.; Gao, P.; Guo, X.; et al. Construction of LncRNA-Related ceRNA Networks in Longissimus Dorsi Muscle of Jinfen White Pigs at Different Developmental Stages. Curr. Issues Mol. Biol. 2024, 46, 340-354. https://doi.org/10.3390/cimb46010022
Wang S, Shi M, Zhang Y, Niu J, Li W, Yuan J, Cai C, Yang Y, Gao P, Guo X, et al. Construction of LncRNA-Related ceRNA Networks in Longissimus Dorsi Muscle of Jinfen White Pigs at Different Developmental Stages. Current Issues in Molecular Biology. 2024; 46(1):340-354. https://doi.org/10.3390/cimb46010022
Chicago/Turabian StyleWang, Shouyuan, Mingyue Shi, Yunting Zhang, Jin Niu, Wenxia Li, Jiale Yuan, Chunbo Cai, Yang Yang, Pengfei Gao, Xiaohong Guo, and et al. 2024. "Construction of LncRNA-Related ceRNA Networks in Longissimus Dorsi Muscle of Jinfen White Pigs at Different Developmental Stages" Current Issues in Molecular Biology 46, no. 1: 340-354. https://doi.org/10.3390/cimb46010022
APA StyleWang, S., Shi, M., Zhang, Y., Niu, J., Li, W., Yuan, J., Cai, C., Yang, Y., Gao, P., Guo, X., Li, B., Lu, C., & Cao, G. (2024). Construction of LncRNA-Related ceRNA Networks in Longissimus Dorsi Muscle of Jinfen White Pigs at Different Developmental Stages. Current Issues in Molecular Biology, 46(1), 340-354. https://doi.org/10.3390/cimb46010022