
Hypoxia and HIF-1α Regulate the Activity and Expression of Na,K-ATPase Subunits in H9c2 Cardiomyoblasts

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. HIF-1α Silencing Experiments and In Vitro Hypoxia
2.3. RNA Isolation and Quantitative RT-PCR
2.4. Preparation of Nuclear Extracts
2.5. Cell Surface Biotinylation Experiments
2.6. Preparation of Total Cell Lysates
2.7. Western Blotting
2.8. NKA Activity Measurements
2.9. Intracellular ATP Measurements
2.10. Statistical Analysis
3. Results
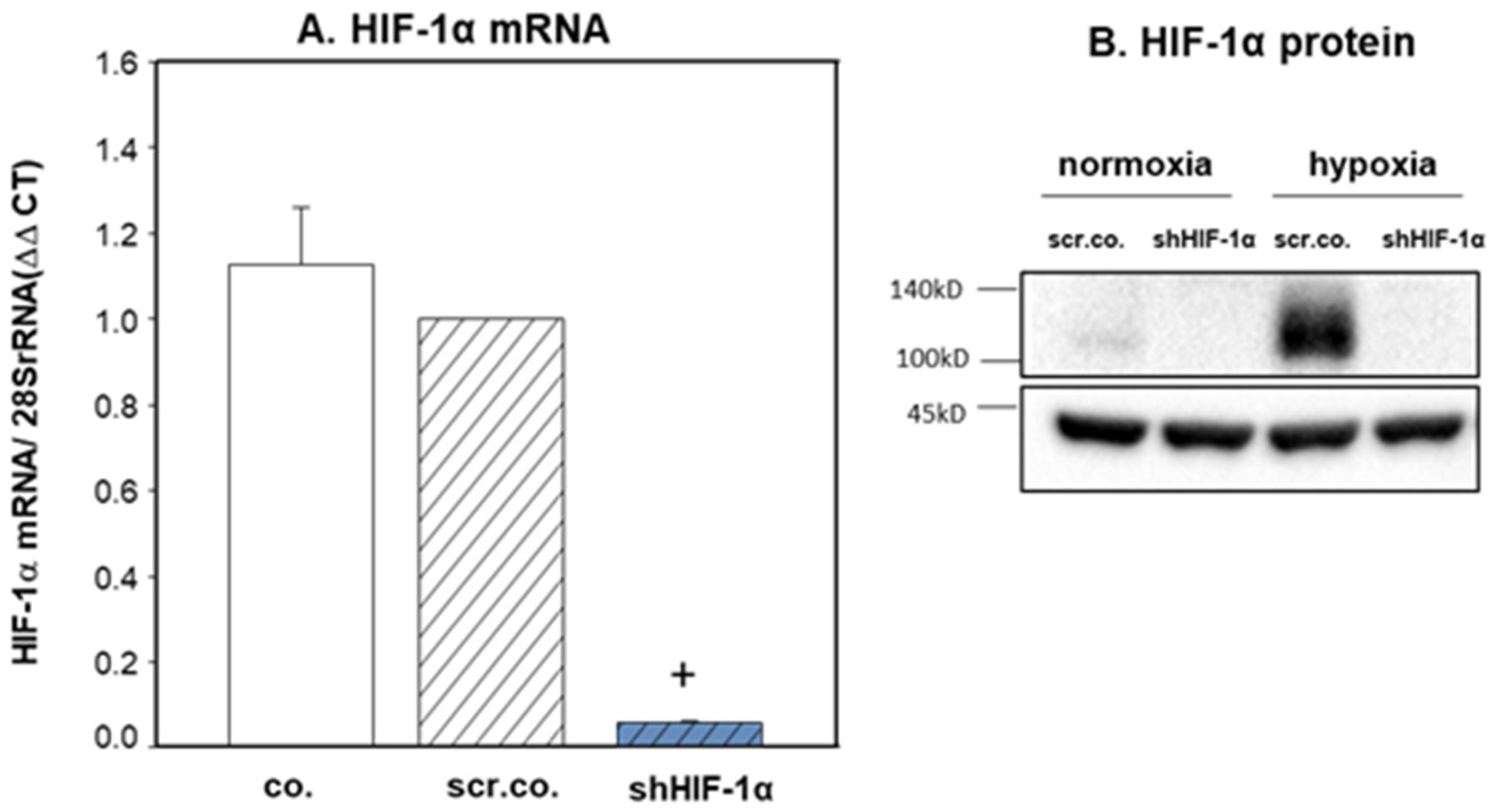
3.1. Silencing Efficiency of HIF-1α on the mRNA and Protein Expression of HIF-1α
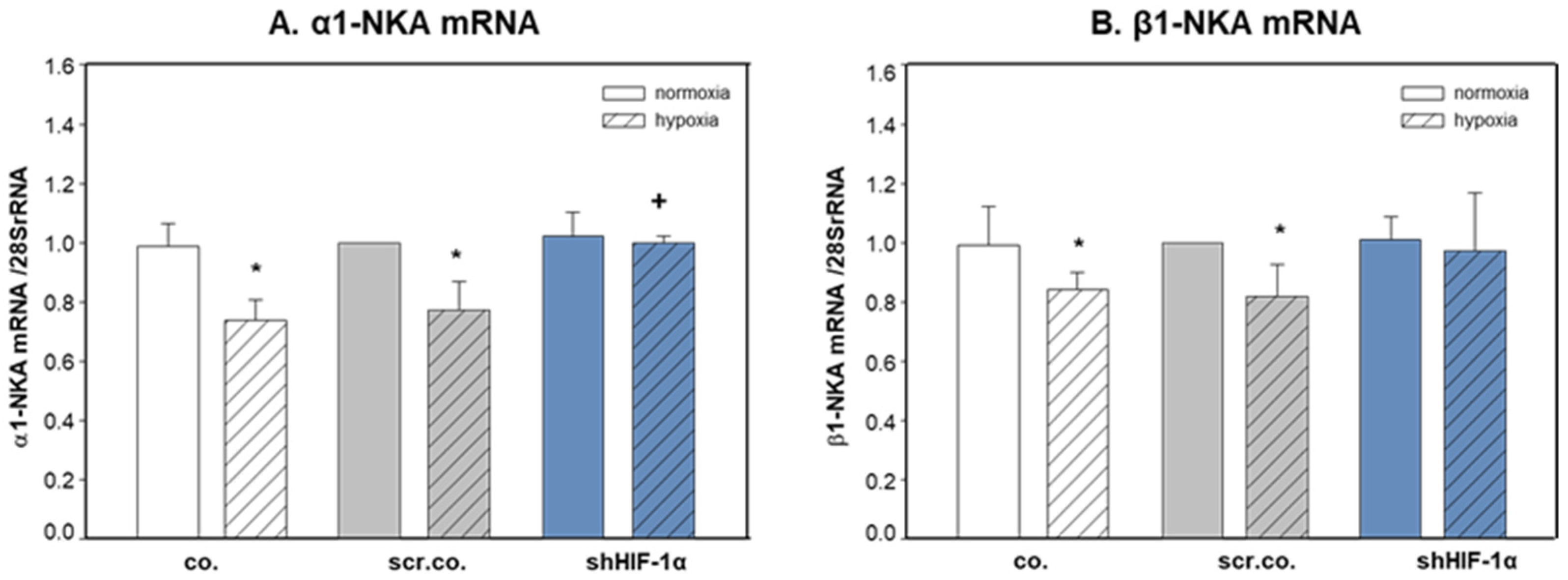
3.2. Effect of Hypoxia and HIF-1α Silencing on the mRNA Expression of α1- and β1-NKA
3.3. Effect of Hypoxia and HIF-1α Silencing on α1-NKA and β1-NKA Membrane, Intracellular, and Total Expression
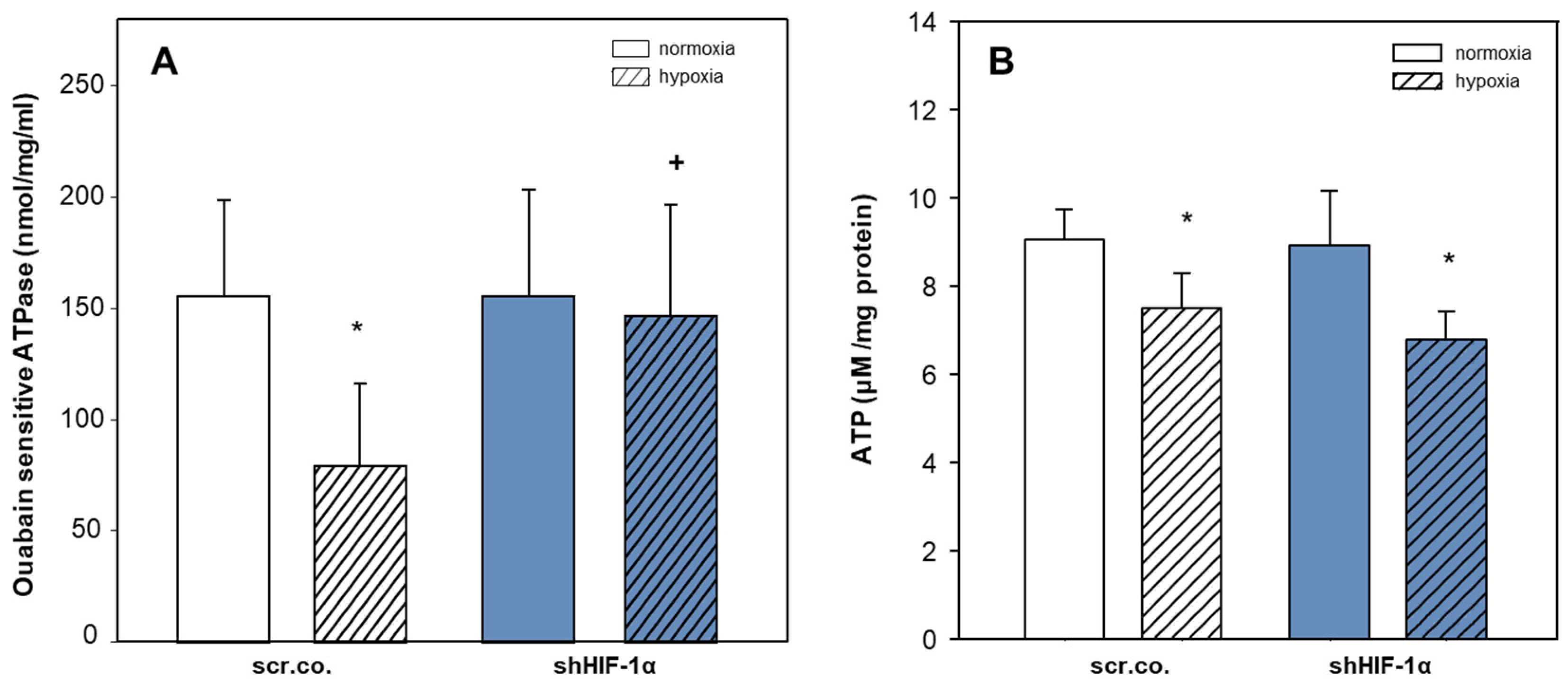
3.4. Effect of Hypoxia and HIF-1α Silencing on NKA Activity and Total ATP Levels
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Skou, J.C. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim. Biophys. Acta 1957, 23, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Geering, K. Fxyd proteins: New regulators of Na-K-atpase. Am. J. Physiol. Renal Physiol. 2006, 290, F241–F250. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Shapiro, J.I. The physiological and clinical importance of sodium potassium atpase in cardiovascular diseases. Curr. Opin. Pharmacol. 2016, 27, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.J.; Xie, J. The Na/K-atpase-mediated signal transduction as a target for new drug development. Front. Biosci-Landmrk 2005, 10, 3100–3109. [Google Scholar] [CrossRef] [PubMed]
- Bouzinova, E.V.; Hangaard, L.; Staehr, C.; Mazur, A.; Ferreira, A.; Chibalin, A.V.; Sandow, S.L.; Xie, Z.; Aalkjaer, C.; Matchkov, V.V. The alpha2 isoform Na,K-atpase modulates contraction of rat mesenteric small artery via csrc-dependent Ca(2+) sensitization. Acta Physiol. 2018, 224, e13059. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Huang, L.; Xie, Z.; Huang, W.H.; Askari, A. Partial inhibition of Na+/K+-atpase by ouabain induces the Ca2+-dependent expressions of early-response genes in cardiac myocytes. J. Biol. Chem. 1996, 271, 10372–10378. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Askari, A. Na(+)/K(+)-atpase as a signal transducer. Eur. J. Biochem. 2002, 269, 2434–2439. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Askari, A.; Xie, Z. Involvement of src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-atpase. J. Biol. Chem. 2000, 275, 27832–27837. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tian, J.; Haas, M.; Shapiro, J.I.; Askari, A.; Xie, Z. Ouabain interaction with cardiac Na+/K+-atpase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J. Biol. Chem. 2000, 275, 27838–27844. [Google Scholar] [CrossRef] [PubMed]
- Kometiani, P.; Li, J.; Gnudi, L.; Kahn, B.B.; Askari, A.; Xie, Z. Multiple signal transduction pathways link Na+/K+-atpase to growth-related genes in cardiac myocytes. The roles of ras and mitogen-activated protein kinases. J. Biol. Chem. 1998, 273, 15249–15256. [Google Scholar] [CrossRef] [PubMed]
- Aydemir-Koksoy, A.; Abramowitz, J.; Allen, J.C. Ouabain-induced signaling and vascular smooth muscle cell proliferation. J. Biol. Chem. 2001, 276, 46605–46611. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Liu, J.; Garlid, K.D.; Shapiro, J.I.; Xie, Z. Involvement of mitogen-activated protein kinases and reactive oxygen species in the inotropic action of ouabain on cardiac myocytes. A potential role for mitochondrial k(atp) channels. Mol. Cell Biochem. 2003, 242, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Cai, T.; Yuan, Z.; Wang, H.; Liu, L.; Haas, M.; Maksimova, E.; Huang, X.Y.; Xie, Z.J. Binding of src to Na+/K+-atpase forms a functional signaling complex. Mol. Biol. Cell 2006, 17, 317–326. [Google Scholar] [CrossRef]
- Morth, J.P.; Pedersen, B.P.; Toustrup-Jensen, M.S.; Sorensen, T.L.; Petersen, J.; Andersen, J.P.; Vilsen, B.; Nissen, P. Crystal structure of the sodium-potassium pump. Nature 2007, 450, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Blanco, G.; Mercer, R.W. Isozymes of the Na-K-atpase: Heterogeneity in structure, diversity in function. Am. J. Physiol. 1998, 275, F633–F650. [Google Scholar] [CrossRef]
- Jimenez, T.; McDermott, J.P.; Sanchez, G.; Blanco, G. Na,K-atpase alpha4 isoform is essential for sperm fertility. Proc. Natl. Acad. Sci. USA 2011, 108, 644–649. [Google Scholar] [CrossRef]
- Berry, R.G.; Despa, S.; Fuller, W.; Bers, D.M.; Shattock, M.J. Differential distribution and regulation of mouse cardiac Na+/K+-atpase alpha1 and alpha2 subunits in t-tubule and surface sarcolemmal membranes. Cardiovasc. Res. 2007, 73, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Sweadner, K.J.; Herrera, V.L.; Amato, S.; Moellmann, A.; Gibbons, D.K.; Repke, K.R. Immunologic identification of Na+,K(+)-atpase isoforms in myocardium. Isoform change in deoxycorticosterone acetate-salt hypertension. Circ. Res. 1994, 74, 669–678. [Google Scholar] [CrossRef]
- Aronsen, J.M.; Swift, F.; Sejersted, O.M. Cardiac sodium transport and excitation-contraction coupling. J. Mol. Cell Cardiol. 2013, 61, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Chow, D.C.; Forte, J.G. Functional significance of the beta-subunit for heterodimeric p-type atpases. J. Exp. Biol. 1995, 198, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Geering, K. Functional roles of Na,K-atpase subunits. Curr. Opin. Nephrol. Hypertens. 2008, 17, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Shattock, M.J.; Ottolia, M.; Bers, D.M.; Blaustein, M.P.; Boguslavskyi, A.; Bossuyt, J.; Bridge, J.H.; Chen-Izu, Y.; Clancy, C.E.; Edwards, A.; et al. Na+/Ca2+ exchange and Na+/K+-atpase in the heart. J. Physiol. 2015, 593, 1361–1382. [Google Scholar] [CrossRef] [PubMed]
- Baloglu, E. Hypoxic stress-dependent regulation of Na,K-atpase in ischemic heart disease. Int. J. Mol. Sci. 2023, 24, 7855. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factor 1 and cardiovascular disease. Annu. Rev. Physiol. 2014, 76, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Wolf, P.L.; Escudero, R.; Deutsch, R.; Jamieson, S.W.; Thistlethwaite, P.A. Early expression of angiogenesis factors in acute myocardial ischemia and infarction. N. Engl. J. Med. 2000, 342, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Jurgensen, J.S.; Rosenberger, C.; Wiesener, M.S.; Warnecke, C.; Horstrup, J.H.; Grafe, M.; Philipp, S.; Griethe, W.; Maxwell, P.H.; Frei, U.; et al. Persistent induction of hif-1alpha and -2alpha in cardiomyocytes and stromal cells of ischemic myocardium. FASEB J. 2004, 18, 1415–1417. [Google Scholar] [CrossRef]
- Sui, X.; Wei, H.; Wang, D. Novel mechanism of cardiac protection by valsartan: Synergetic roles of tgf-beta1 and hif-1alpha in ang ii-mediated fibrosis after myocardial infarction. J. Cell Mol. Med. 2015, 19, 1773–1782. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, J.; Suter, M.; Windak, R.; Krebs, T.; Felley, A.; Montessuit, C.; Tokarska-Schlattner, M.; Aasum, E.; Bogdanova, A.; Perriard, E.; et al. Activation of a hif1alpha-ppargamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 2009, 9, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Takeda, N.; Isagawa, T.; Semba, H.; Nishimura, S.; Morioka, M.S.; Nakagama, Y.; Sato, T.; Soma, K.; Koyama, K.; et al. Macrophage hypoxia signaling regulates cardiac fibrosis via oncostatin m. Nat. Commun. 2019, 10, 2824. [Google Scholar] [CrossRef]
- Tang, H.; Babicheva, A.; McDermott, K.M.; Gu, Y.; Ayon, R.J.; Song, S.; Wang, Z.; Gupta, A.; Zhou, T.; Sun, X.; et al. Endothelial hif-2alpha contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L256–L275. [Google Scholar] [PubMed]
- Bogdanova, A.; Ogunshola, O.O.; Bauer, C.; Nikinmaa, M.; Gassmann, M. Molecular mechanisms of oxygen-induced regulation of Na+/K+ pump. Adv. Exp. Med. Biol. 2003, 536, 231–238. [Google Scholar] [PubMed]
- Schwinger, R.H.; Wang, J.; Frank, K.; Muller-Ehmsen, J.; Brixius, K.; McDonough, A.A.; Erdmann, E. Reduced sodium pump alpha1, alpha3, and beta1-isoform protein levels and Na+,K+-atpase activity but unchanged Na+-Ca2+ exchanger protein levels in human heart failure. Circulation 1999, 99, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Semb, S.O.; Lunde, P.K.; Holt, E.; Tonnessen, T.; Christensen, G.; Sejersted, O.M. Reduced myocardial Na+, K(+)-pump capacity in congestive heart failure following myocardial infarction in rats. J. Mol. Cell Cardiol. 1998, 30, 1311–1328. [Google Scholar] [CrossRef] [PubMed]
- Bossuyt, J.; Ai, X.; Moorman, J.R.; Pogwizd, S.M.; Bers, D.M. Expression and phosphorylation of the na-pump regulatory subunit phospholemman in heart failure. Circ. Res. 2005, 97, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Ishino, K.; Botker, H.E.; Clausen, T.; Hetzer, R.; Sehested, J. Myocardial adenine nucleotides, glycogen, and Na, K-atpase in patients with idiopathic dilated cardiomyopathy requiring mechanical circulatory support. Am. J. Cardiol. 1999, 83, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Pike, M.M.; Luo, C.S.; Clark, M.D.; Kirk, K.A.; Kitakaze, M.; Madden, M.C.; Cragoe, E.J., Jr.; Pohost, G.M. Nmr measurements of Na+ and cellular energy in ischemic rat heart: Role of Na(+)-H+ exchange. Am. J. Physiol. 1993, 265, H2017–H2026. [Google Scholar] [CrossRef] [PubMed]
- Verdonck, F.; Volders, P.G.; Vos, M.A.; Sipido, K.R. Intracellular Na+ and altered Na+ transport mechanisms in cardiac hypertrophy and failure. J. Mol. Cell Cardiol. 2003, 35, 5–25. [Google Scholar] [CrossRef] [PubMed]
- Despa, S.; Islam, M.A.; Weber, C.R.; Pogwizd, S.M.; Bers, D.M. Intracellular Na(+) concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation 2002, 105, 2543–2548. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, K.T. Changes in cellular Ca(2+) and Na(+) regulation during the progression towards heart failure. J. Physiol. 2023, 601, 905–921. [Google Scholar] [CrossRef] [PubMed]
- Swift, F.; Birkeland, J.A.; Tovsrud, N.; Enger, U.H.; Aronsen, J.M.; Louch, W.E.; Sjaastad, I.; Sejersted, O.M. Altered Na+/Ca2+-exchanger activity due to downregulation of Na+/K+-atpase alpha2-isoform in heart failure. Cardiovasc. Res. 2008, 78, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Shamraj, O.I.; Grupp, I.L.; Grupp, G.; Melvin, D.; Gradoux, N.; Kremers, W.; Lingrel, J.B.; De Pover, A. Characterisation of Na/K-atpase, its isoforms, and the inotropic response to ouabain in isolated failing human hearts. Cardiovasc. Res. 1993, 27, 2229–2237. [Google Scholar] [CrossRef] [PubMed]
- Jager, H.; Wozniak, G.; Akinturk, I.H.; Hehrlein, F.W.; Scheiner-Bobis, G. Expression of sodium pump isoforms and other sodium or calcium ion transporters in the heart of hypertensive patients. Biochim. Biophys. Acta 2001, 1513, 149–159. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fedorova, O.V.; Talan, M.I.; Agalakova, N.I.; Lakatta, E.G.; Bagrov, A.Y. Coordinated shifts in Na/K-atpase isoforms and their endogenous ligands during cardiac hypertrophy and failure in nacl-sensitive hypertension. J. Hypertens. 2004, 22, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Book, C.B.; Moore, R.L.; Semanchik, A.; Ng, Y.C. Cardiac hypertrophy alters expression of Na+,K(+)-atpase subunit isoforms at mrna and protein levels in rat myocardium. J. Mol. Cell Cardiol. 1994, 26, 591–600. [Google Scholar] [CrossRef]
- Baloglu, E.; Nonnenmacher, G.; Seleninova, A.; Berg, L.; Velineni, K.; Ermis-Kaya, E.; Mairbaurl, H. The role of hypoxia-induced modulation of alveolar epithelial Na(+)- transport in hypoxemia at high altitude. Pulm. Circ. 2020, 10, 50–58. [Google Scholar] [CrossRef]
- Rao, X.; Huang, X.; Zhou, Z.; Lin, X. An improvement of the 2^(-delta delta ct) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinforma Biomath. 2013, 3, 71–85. [Google Scholar] [PubMed]
- Forbush, B., 3rd. Assay of Na,K-atpase in plasma membrane preparations: Increasing the permeability of membrane vesicles using sodium dodecyl sulfate buffered with bovine serum albumin. Anal. Biochem. 1983, 128, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Takeda, N. The roles of hif-1alpha signaling in cardiovascular diseases. J. Cardiol. 2023, 81, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Loeh, B.; Baloglu, E.; Ke, A.; Bartsch, P.; Mairbaurl, H. Beta2-adrenergic stimulation blunts inhibition of epithelial ion transport by hypoxia of rat alveolar epithelial cells. Cell Physiol. Biochem. 2010, 25, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Wodopia, R.; Ko, H.S.; Billian, J.; Wiesner, R.; Bartsch, P.; Mairbaurl, H. Hypoxia decreases proteins involved in epithelial electrolyte transport in a549 cells and rat lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L1110–L1119. [Google Scholar] [CrossRef]
- Belliard, A.; Sottejeau, Y.; Duan, Q.; Karabin, J.L.; Pierre, S.V. Modulation of cardiac Na+,K+-atpase cell surface abundance by simulated ischemia-reperfusion and ouabain preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H94–H103. [Google Scholar] [CrossRef]
- Petrushanko, I.Y.; Mitkevich, V.; Makarov, A. Molecular mechanisms of the redox regulation of the Na, K-atpase. Biophysics 2020, 65, 711–730. [Google Scholar] [CrossRef]
- Petrushanko, I.Y.; Yakushev, S.; Mitkevich, V.A.; Kamanina, Y.V.; Ziganshin, R.H.; Meng, X.; Anashkina, A.A.; Makhro, A.; Lopina, O.D.; Gassmann, M.; et al. S-glutathionylation of the Na,K-atpase catalytic alpha subunit is a determinant of the enzyme redox sensitivity. J. Biol. Chem. 2012, 287, 32195–32205. [Google Scholar] [CrossRef]
- Kurella, E.G.; Tyulina, O.V.; Boldyrev, A.A. Oxidative resistance of Na/K-atpase. Cell Mol. Neurobiol. 1999, 19, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Figtree, G.A.; Liu, C.C.; Bibert, S.; Hamilton, E.J.; Garcia, A.; White, C.N.; Chia, K.K.; Cornelius, F.; Geering, K.; Rasmussen, H.H. Reversible oxidative modification: A key mechanism of Na+-K+ pump regulation. Circ. Res. 2009, 105, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Bibert, S.; Liu, C.C.; Figtree, G.A.; Garcia, A.; Hamilton, E.J.; Marassi, F.M.; Sweadner, K.J.; Cornelius, F.; Geering, K.; Rasmussen, H.H. Fxyd proteins reverse inhibition of the Na+-K+ pump mediated by glutathionylation of its beta1 subunit. J. Biol. Chem. 2011, 286, 18562–18572. [Google Scholar] [CrossRef]
- Xie, L.; Xiao, K.; Whalen, E.J.; Forrester, M.T.; Freeman, R.S.; Fong, G.; Gygi, S.P.; Lefkowitz, R.J.; Stamler, J.S. Oxygen-regulated beta(2)-adrenergic receptor hydroxylation by egln3 and ubiquitylation by pvhl. Sci. Signal 2009, 2, ra33. [Google Scholar] [CrossRef] [PubMed]
- Recchia, A.G.; De Francesco, E.M.; Vivacqua, A.; Sisci, D.; Panno, M.L.; Ando, S.; Maggiolini, M. The g protein-coupled receptor 30 is up-regulated by hypoxia-inducible factor-1alpha (hif-1alpha) in breast cancer cells and cardiomyocytes. J. Biol. Chem. 2011, 286, 10773–10782. [Google Scholar] [CrossRef]
- Magnani, N.D.; Dada, L.A.; Queisser, M.A.; Brazee, P.L.; Welch, L.C.; Anekalla, K.R.; Zhou, G.; Vagin, O.; Misharin, A.V.; Budinger, G.R.S.; et al. Hif and hoil-1l-mediated pkczeta degradation stabilizes plasma membrane Na,K-atpase to protect against hypoxia-induced lung injury. Proc. Natl. Acad. Sci. USA 2017, 114, E10178–E10186. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, S.V.; Otero, L.; Aristizabal-Pachon, A.F.; Hinostroza, F.; Carvacho, I.; Torres, Y.P. Hypoxic regulation of the large-conductance, calcium and voltage-activated potassium channel, bk. Front. Physiol. 2021, 12, 780206. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gurler, B.; Gencay, G.; Baloglu, E. Hypoxia and HIF-1α Regulate the Activity and Expression of Na,K-ATPase Subunits in H9c2 Cardiomyoblasts. Curr. Issues Mol. Biol. 2023, 45, 8277-8288. https://doi.org/10.3390/cimb45100522
Gurler B, Gencay G, Baloglu E. Hypoxia and HIF-1α Regulate the Activity and Expression of Na,K-ATPase Subunits in H9c2 Cardiomyoblasts. Current Issues in Molecular Biology. 2023; 45(10):8277-8288. https://doi.org/10.3390/cimb45100522
Chicago/Turabian StyleGurler, Beyza, Gizem Gencay, and Emel Baloglu. 2023. "Hypoxia and HIF-1α Regulate the Activity and Expression of Na,K-ATPase Subunits in H9c2 Cardiomyoblasts" Current Issues in Molecular Biology 45, no. 10: 8277-8288. https://doi.org/10.3390/cimb45100522
APA StyleGurler, B., Gencay, G., & Baloglu, E. (2023). Hypoxia and HIF-1α Regulate the Activity and Expression of Na,K-ATPase Subunits in H9c2 Cardiomyoblasts. Current Issues in Molecular Biology, 45(10), 8277-8288. https://doi.org/10.3390/cimb45100522