
Targeted Delivery of Chimeric Antigen Receptor into T Cells via CRISPR-Mediated Homology-Directed Repair with a Dual-AAV6 Transduction System



Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Guide RNAs and Plasmids
2.3. Analysis of Double-Strand Break Efficiency
2.4. Viral Vector Production
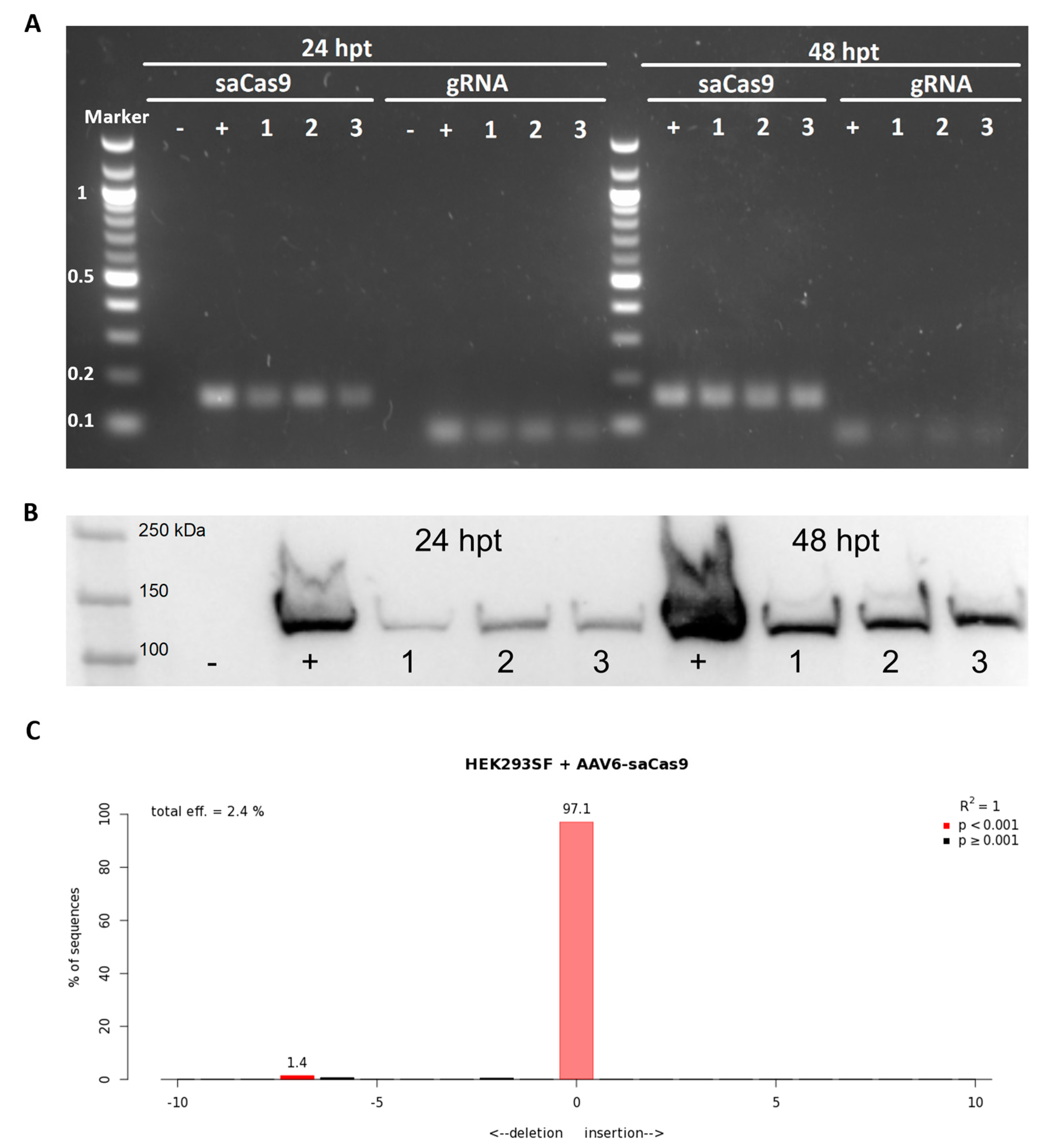
2.5. Analysis of AAV6-Delivered CRISPR/Cas9
2.6. Dual-AAV Transduction of Jurkat Cells
2.7. Genomic Analysis of Modified Jurkat Cells
2.8. Analysis of CRISPR/Cas9 Off-Targets
3. Results
3.1. Guide RNA Design, Cloning, and Selection
3.2. Evaluation of Viral Delivery in HEK293SF Cells
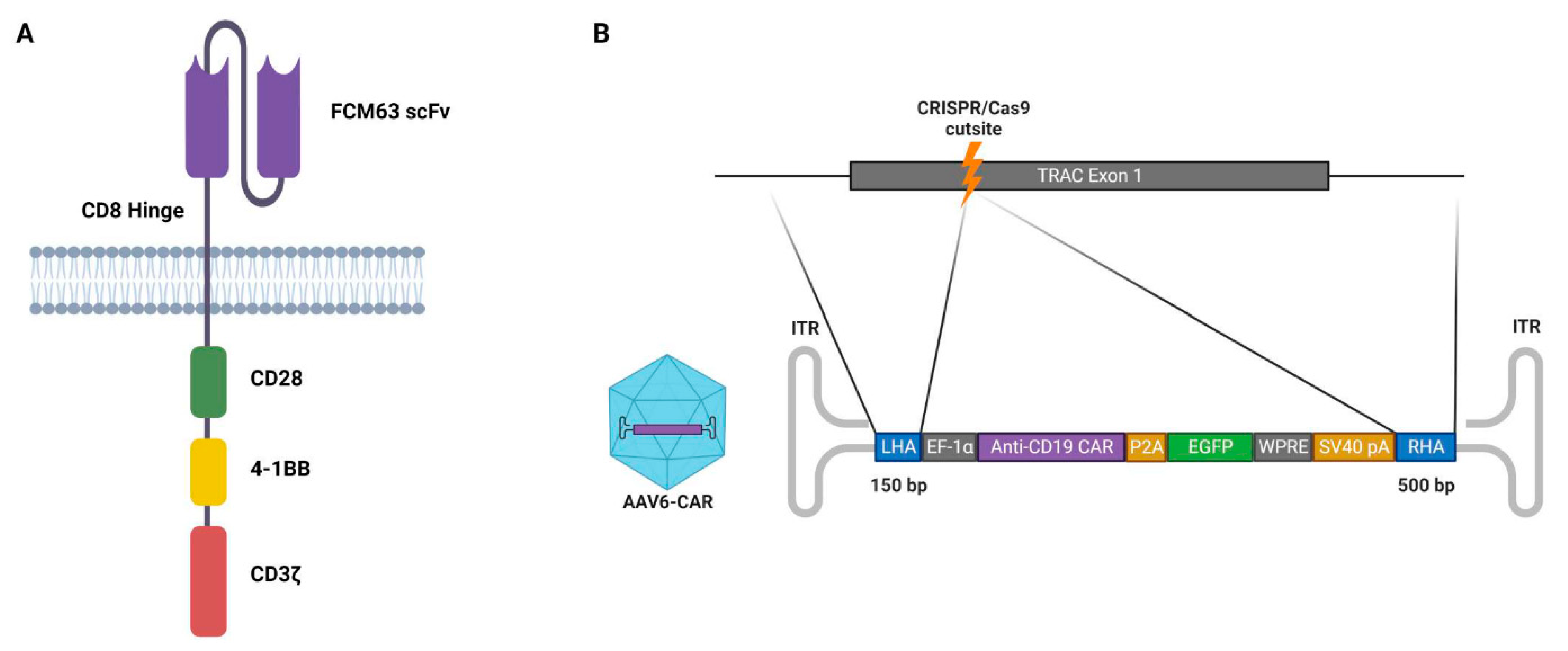
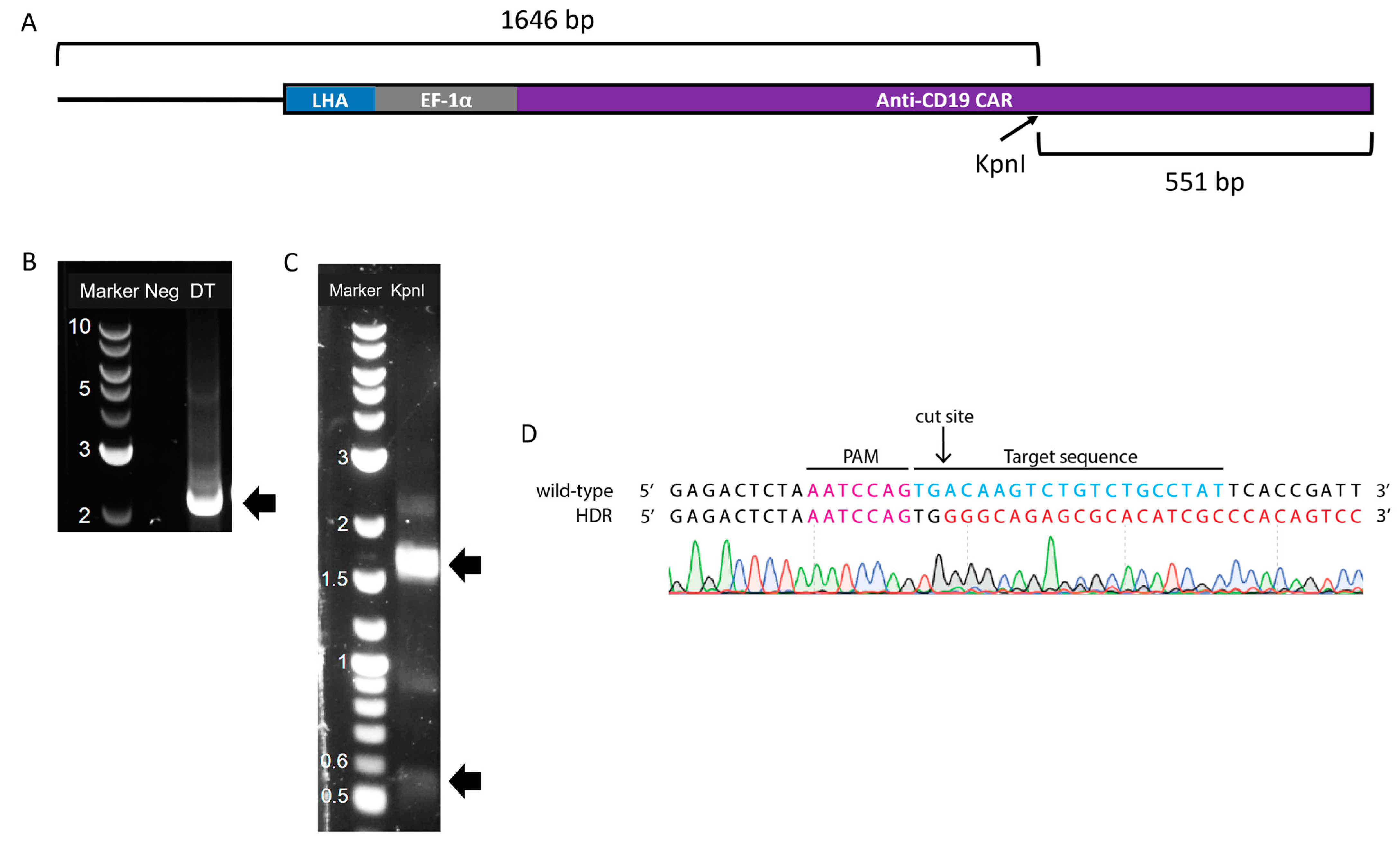
3.3. Design of the DNA Template for Homology-Directed Repair
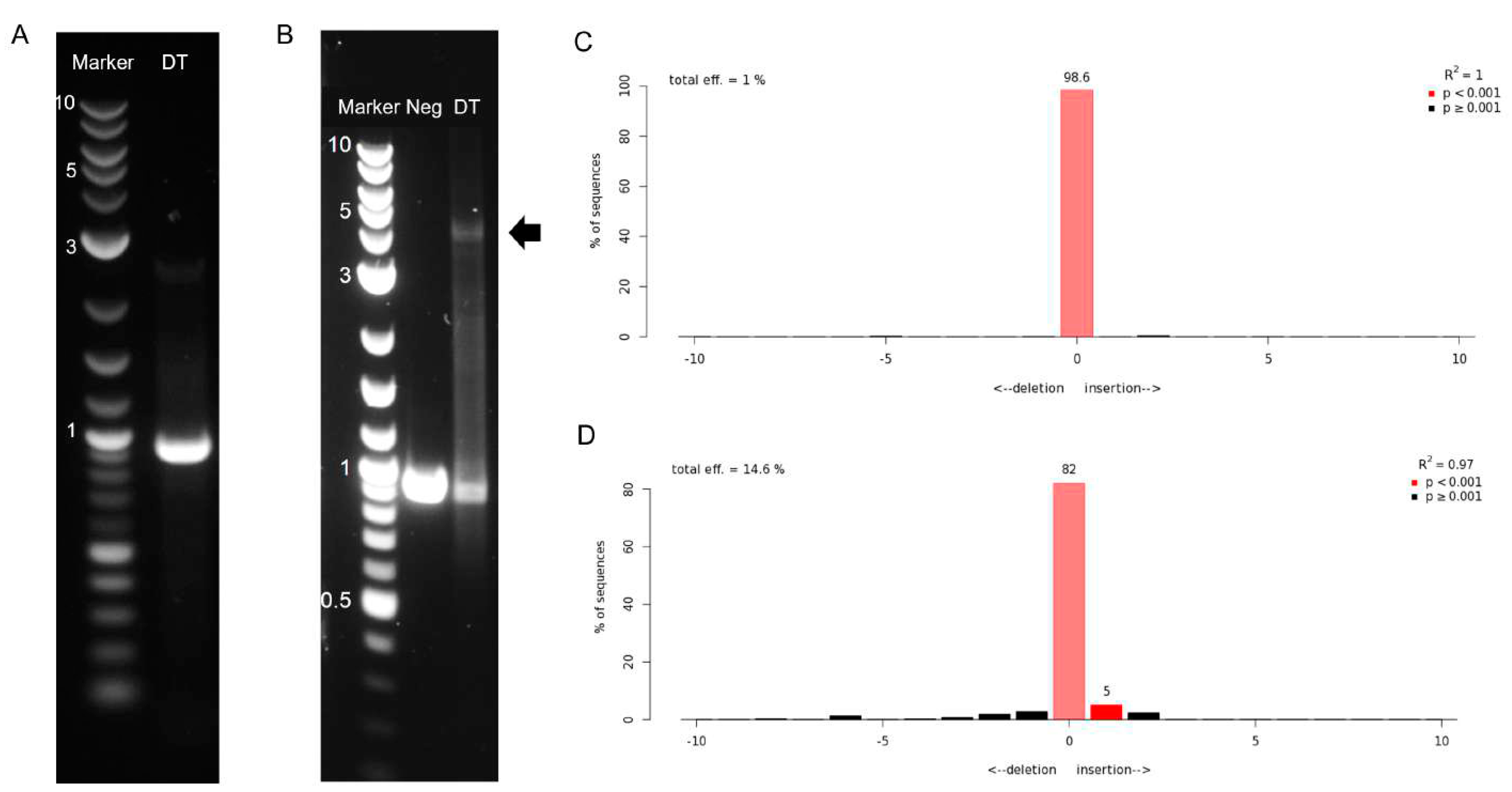
3.4. Production of Adeno-Associated Viral Vectors and Site-Specific Modification of Jurkat Cells via Dual-AAV Transduction
3.5. Off-Target Evaluation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lu, J.; Jiang, G. The journey of CAR-T therapy in hematological malignancies. Mol. Cancer 2022, 21, 194. [Google Scholar] [CrossRef] [PubMed]
- Finney, H.M.; Lawson, A.D.G.; Bebbington, C.R.; Weir, A.N.C.; Christopher, R.; Weir, A.N.C.; Bebbington, C.R.; Weir, A.N.C. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J. Immunol. 1998, 161, 2791–2797. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Santos, E.; Nikhamin, Y.; Yeh, R.; Matsushita, M.; La Perle, K.; Quintás-Cardama, A.; Larson, S.M.; Sadelain, M. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin. Cancer Res. 2007, 13, 5426–5435. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Cheng, J.; Mu, W.; Zhou, J.; Zhu, L. Advances in Universal CAR-T Cell Therapy. Front. Immunol. 2021, 12, 744823. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.K.; Lerman, B.J.; Barnes, J.I.; Boursiquot, B.C.; Tan, Y.J.; Robinson, A.Q.L.; Davis, K.L.; Owens, D.K.; Goldhaber-Fiebert, J.D. Cost Effectiveness of Chimeric Antigen Receptor T-Cell Therapy in Relapsed or Refractory Pediatric B-Cell Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2018, 36, 3192–3202. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, D.A.; Shutova, M.V.; Johnston, N.R.; Smith, O.P.; Fedorin, V.V.; Kukushkin, Y.S.; van der Loo, J.C.M.; Johnstone, E.C. The clinical landscape for AAV gene therapies. Nat. Rev. Drug Discov. 2021, 20, 173–174. [Google Scholar] [CrossRef]
- McCarty, D.M.; Young, S.M., Jr.; Samulski, R.J. Integration of adeno-associated virus (AAV) and recombinant AAV vectors. Annu. Rev. Genet. 2004, 38, 819–845. [Google Scholar] [CrossRef]
- Dhungel, B.P.; Bailey, C.G.; Rasko, J.E.J. Journey to the Center of the Cell: Tracing the Path of AAV Transduction. Trends Mol. Med. 2021, 27, 172–184. [Google Scholar] [CrossRef]
- Russell, D.W.; Hirata, R.K. Human gene targeting by viral vectors. Nat. Genet. 1998, 18, 325–330. [Google Scholar] [CrossRef]
- Vasileva, A.; Jessberger, R. Precise hit: Adeno-associated virus in gene targeting. Nat. Rev. Microbiol. 2005, 3, 837–847. [Google Scholar] [CrossRef]
- Khan, I.F.; Hirata, R.K.; Russell, D.W. AAV-mediated gene targeting methods for human cells. Nat. Protoc. 2011, 6, 482. [Google Scholar] [CrossRef]
- Hirsch, M.L. Adeno-associated virus inverted terminal repeats stimulate gene editing. Gene Ther. 2015, 22, 190–195. [Google Scholar] [CrossRef]
- Barzel, A.; Paulk, N.K.; Shi, Y.; Huang, Y.; Chu, K.; Zhang, F.; Valdmanis, P.N.; Spector, L.P.; Porteus, M.H.; Gaensler, K.M.; et al. Promoterless gene targeting without nucleases ameliorates haemophilia B in mice. Nature 2015, 517, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Gaj, T.; Epstein, B.E.; Schaffer, D.V. Genome Engineering Using Adeno-associated Virus: Basic and Clinical Research Applications. Mol. Ther. 2016, 24, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Porteus, M.H.; Cathomen, T.; Weitzman, M.D.; Baltimore, D. Efficient gene targeting mediated by adeno-associated virus and DNA double-strand breaks. Mol. Cell. Biol. 2003, 23, 3558–3565. [Google Scholar] [CrossRef]
- Miller, D.G.; Petek, L.M.; Russell, D.W. Adeno-associated virus vectors integrate at chromosome breakage sites. Nat. Genet. 2004, 36, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Dever, D.P.; Bak, R.O.; Reinisch, A.; Camarena, J.; Washington, G.; Nicolas, C.E.; Pavel-Dinu, M.; Saxena, N.; Wilkens, A.B.; Mantri, S.; et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature 2016, 539, 384–389. [Google Scholar] [CrossRef]
- Dabiri, H.; Safarzadeh Kozani, P.; Habibi Anbouhi, M.; Mirzaee Godarzee, M.; Haddadi, M.H.; Basiri, M.; Ziaei, V.; Sadeghizadeh, M.; Hajizadeh Saffar, E. Site-specific transgene integration in chimeric antigen receptor (CAR) T cell therapies. Biomark. Res. 2023, 11, 67. [Google Scholar] [CrossRef]
- Moço, P.D.; Aharony, N.; Kamen, A. Adeno-Associated Viral Vectors for Homology-Directed Generation of CAR-T Cells. Biotechnol. J. 2020, 15, 1900286. [Google Scholar] [CrossRef]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gonen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef]
- Guo, Y.; Tong, C.; Su, L.; Zhang, W.; Jia, H.; Liu, Y.; Yang, Q.; Wu, Z.; Wang, Y.; Han, W. CRISPR/Cas9 genome-edited universal CAR T cells in patients with relapsed/refractory lymphoma. Blood Adv. 2022, 6, 2695–2699. [Google Scholar] [CrossRef] [PubMed]
- McGarrity, G.J.; Hoyah, G.; Winemiller, A.; Andre, K.; Stein, D.; Blick, G.; Greenberg, R.N.; Kinder, C.; Zolopa, A.; Binder-Scholl, G.; et al. Patient monitoring and follow-up in lentiviral clinical trials. J. Gene. Med. 2013, 15, 78–82. [Google Scholar] [CrossRef]
- Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef]
- Mitchell, R.S.; Beitzel, B.F.; Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004, 2, E234. [Google Scholar] [CrossRef] [PubMed]
- Themis, M.; Waddington, S.N.; Schmidt, M.; von Kalle, C.; Wang, Y.; Al-Allaf, F.; Gregory, L.G.; Nivsarkar, M.; Themis, M.; Holder, M.V.; et al. Oncogenesis following delivery of a nonprimate lentiviral gene therapy vector to fetal and neonatal mice. Mol. Ther. 2005, 12, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Investig. 2008, 118, 3143–3150. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142. [Google Scholar] [CrossRef]
- Schlimgen, R.; Howard, J.; Wooley, D.; Thompson, M.; Baden, L.R.; Yang, O.O.; Christiani, D.C.; Mostoslavsky, G.; Diamond, D.V.; Duane, E.G.; et al. Risks Associated With Lentiviral Vector Exposures and Prevention Strategies. J. Occup. Environ. Med. 2016, 58, 1159–1166. [Google Scholar] [CrossRef]
- Aiuti, A.; Biasco, L.; Scaramuzza, S.; Ferrua, F.; Cicalese, M.P.; Baricordi, C.; Dionisio, F.; Calabria, A.; Giannelli, S.; Castiello, M.C.; et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science 2013, 341, 1233151. [Google Scholar] [CrossRef]
- Ellis, J. Silencing and variegation of gammaretrovirus and lentivirus vectors. Hum. Gene Ther. 2005, 16, 1241–1246. [Google Scholar] [CrossRef]
- Torikai, H.; Reik, A.; Liu, P.Q.; Zhou, Y.; Zhang, L.; Maiti, S.; Huls, H.; Miller, J.C.; Kebriaei, P.; Rabinovich, B.; et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood 2012, 119, 5697–5705. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Cheng, C.; Cheng, A.W.; Zhang, X.; Li, N.; Xia, C.; Wei, X.; Liu, X.; Wang, H. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. 2017, 27, 154–157. [Google Scholar] [CrossRef]
- Anwer, F.; Shaukat, A.A.; Zahid, U.; Husnain, M.; McBride, A.; Persky, D.; Lim, M.; Hasan, N.; Riaz, I.B. Donor origin CAR T cells: Graft versus malignancy effect without GVHD, a systematic review. Immunotherapy 2017, 9, 123–130. [Google Scholar] [CrossRef]
- Dimitri, A.; Herbst, F.; Fraietta, J.A. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol. Cancer 2022, 21, 78. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Zhang, X.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget 2017, 8, 17002–17011. [Google Scholar] [CrossRef]
- Dai, X.; Park, J.J.; Du, Y.; Kim, H.R.; Wang, G.; Errami, Y.; Chen, S. One-step generation of modular CAR-T cells with AAV-Cpf1. Nat. Methods 2019, 16, 247–254. [Google Scholar] [CrossRef]
- Gomes-Silva, D.; Srinivasan, M.; Sharma, S.; Lee, C.M.; Wagner, D.L.; Davis, T.H.; Rouce, R.H.; Bao, G.; Brenner, M.K.; Mamonkin, M. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood 2017, 130, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Borot, F.; Wang, H.; Ma, Y.; Jafarov, T.; Raza, A.; Ali, A.M.; Mukherjee, S. Gene-edited stem cells enable CD33-directed immune therapy for myeloid malignancies. Proc. Natl. Acad. Sci. USA 2019, 116, 11978–11987. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.M.; Ikeda, K.; Cromer, M.K.; Uchida, N.; Nishimura, T.; Romano, R.; Tong, A.J.; Lemgart, V.T.; Camarena, J.; Pavel-Dinu, M.; et al. Highly Efficient and Marker-free Genome Editing of Human Pluripotent Stem Cells by CRISPR-Cas9 RNP and AAV6 Donor-Mediated Homologous Recombination. Cell Stem Cell 2019, 24, 821–828. [Google Scholar] [CrossRef]
- Fu, Y.-W.; Dai, X.-Y.; Wang, W.-T.; Yang, Z.-X.; Zhao, J.-J.; Zhang, J.-P.; Wen, W.; Zhang, F.; Oberg, K.C.; Zhang, L.; et al. Dynamics and competition of CRISPR–Cas9 ribonucleoproteins and AAV donor-mediated NHEJ, MMEJ and HDR editing. Nucleic Acids Res. 2021, 49, 969–985. [Google Scholar] [CrossRef]
- Ellis, B.L.; Hirsch, M.L.; Barker, J.C.; Connelly, J.P.; Steininger, R.J., 3rd; Porteus, M.H. A survey of ex vivo/in vitro transduction efficiency of mammalian primary cells and cell lines with Nine natural adeno-associated virus (AAV1-9) and one engineered adeno-associated virus serotype. Virol. J. 2013, 10, 74. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Exline, C.M.; DeClercq, J.J.; Llewellyn, G.N.; Hayward, S.B.; Li, P.W.-L.; Shivak, D.A.; Surosky, R.T.; Gregory, P.D.; Holmes, M.C.; et al. Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat. Biotechnol. 2015, 33, 1256. [Google Scholar] [CrossRef] [PubMed]
- De Ravin, S.S.; Reik, A.; Liu, P.-Q.; Li, L.; Wu, X.; Su, L.; Raley, C.; Theobald, N.; Choi, U.; Song, A.H.; et al. Targeted gene addition in human CD34+ hematopoietic cells for correction of X-linked chronic granulomatous disease. Nat. Biotechnol. 2016, 34, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; DeClercq, J.J.; Hayward, S.B.; Li, P.W.; Shivak, D.A.; Gregory, P.D.; Lee, G.; Holmes, M.C. Highly efficient homology-driven genome editing in human T cells by combining zinc-finger nuclease mRNA and AAV6 donor delivery. Nucleic Acids Res. 2015, 44, e30. [Google Scholar] [CrossRef] [PubMed]
- Sather, B.D.; Romano Ibarra, G.S.; Sommer, K.; Curinga, G.; Hale, M.; Khan, I.F.; Singh, S.; Song, Y.; Gwiazda, K.; Sahni, J.; et al. Efficient modification of CCR5 in primary human hematopoietic cells using a megaTAL nuclease and AAV donor template. Sci. Transl. Med. 2015, 7, 307ra156. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, D.T.; Antony, J.; Martin, A.J.; Moser, R.J.; Hekele, A.; Wetzel, K.J.; Brown, A.E.; Triggiano, M.A.; Hux, J.A.; Pham, C.D.; et al. Integration of a CD19 CAR into the TCR Alpha Chain Locus Streamlines Production of Allogeneic Gene-Edited CAR T Cells. Mol. Ther. 2017, 25, 949–961. [Google Scholar] [CrossRef]
- Hale, M.; Lee, B.; Honaker, Y.; Leung, W.H.; Grier, A.E.; Jacobs, H.M.; Sommer, K.; Sahni, J.; Jackson, S.W.; Scharenberg, A.M.; et al. Homology-Directed Recombination for Enhanced Engineering of Chimeric Antigen Receptor T Cells. Mol. Ther. Methods Clin. Dev. 2017, 4, 192–203. [Google Scholar] [CrossRef]
- Naeimi Kararoudi, M.; Likhite, S.; Elmas, E.; Yamamoto, K.; Schwartz, M.; Sorathia, K.; de Souza Fernandes Pereira, M.; Sezgin, Y.; Devine, R.D.; Lyberger, J.M.; et al. Optimization and validation of CAR transduction into human primary NK cells using CRISPR and AAV. Cell Rep. Methods 2022, 2, 100236. [Google Scholar] [CrossRef]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef]
- Bloemberg, D.; Nguyen, T.; Maclean, S.; Zafer, A.; Gadoury, C.; Gurnani, K.; Chattopadhyay, A.; Ash, J.; Lippens, J.; Harcus, D.; et al. A High-Throughput Method for Characterizing Novel Chimeric Antigen Receptors in Jurkat Cells. Mol. Ther. Methods Clin. Dev. 2020, 16, 238–254. [Google Scholar] [CrossRef]
- Brinkman, E.K.; Chen, T.; Amendola, M.; van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.R.H.; Bernier, A.; Moço, P.D.; Schrag, J.; Chahal, P.S.; Kamen, A. Development of a Scalable and Robust Anion-Exchange Chromatographic Method for Enriched Recombinant Adeno-Associated Virus Preparations in Genome Containing Vector Capsids of Serotypes- 5, 6, 8, and 9. Mol. Ther. Methods Clin. Dev. 2021, 21, 341–356. [Google Scholar] [CrossRef]
- Cradick, T.J.; Qiu, P.; Lee, C.M.; Fine, E.J.; Bao, G. COSMID: A Web-based Tool for Identifying and Validating CRISPR/Cas Off-target Sites. Mol. Ther.-Nucleic Acids 2014, 3, e214. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Samulski, R.J.; Hirsch, M.L. Adeno-Associated Virus Vector Mobilization, Risk Versus Reality. Hum. Gene Ther. 2020, 31, 1054–1067. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Warrington, K.H.; Hearing, P.; Hughes, J.; Muzyczka, N. Adenovirus-Facilitated Nuclear Translocation of Adeno-Associated Virus Type 2. J. Virol. 2002, 76, 11505–11517. [Google Scholar] [CrossRef] [PubMed]
- Fisher, K.J.; Gao, G.P.; Weitzman, M.D.; DeMatteo, R.; Burda, J.F.; Wilson, J.M. Transduction with recombinant adeno-associated virus for gene therapy is limited by leading-strand synthesis. J. Virol. 1996, 70, 520. [Google Scholar] [CrossRef]
- Swiech, L.; Heidenreich, M.; Banerjee, A.; Habib, N.; Li, Y.; Trombetta, J.; Sur, M.; Zhang, F. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol. 2015, 33, 102–106. [Google Scholar] [CrossRef]
- Chew, W.L.; Tabebordbar, M.; Cheng, J.K.W.; Mali, P.; Wu, E.Y.; Ng, A.H.M.; Zhu, K.; Wagers, A.J.; Church, G.M. A multifunctional AAV–CRISPR–Cas9 and its host response. Nat. Methods 2016, 13, 868. [Google Scholar] [CrossRef]
- Pan, X.; Philippen, L.; Lahiri, S.K.; Lee, C.; Park, S.H.; Word, T.A.; Li, N.; Jarrett, K.E.; Gupta, R.; Reynolds, J.O.; et al. In Vivo Ryr2 Editing Corrects Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. Res. 2018, 123, 953–963. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, B.; Xie, H.; Ren, Q.; Liu, X.; Li, F.; Lv, X.; He, X.; Cheng, C.; Deng, R.; et al. Efficient Human Genome Editing Using SaCas9 Ribonucleoprotein Complexes. Biotechnol. J. 2019, 14, 1800689. [Google Scholar] [CrossRef]
- Weng, S.; Gao, F.; Wang, J.; Li, X.; Chu, B.; Wang, J.; Yang, G. Improvement of muscular atrophy by AAV–SaCas9-mediated myostatin gene editing in aged mice. Cancer Gene Ther. 2020, 27, 960–975. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Tang, L.; He, X.; Liu, X.; Zhou, C.; Liu, J.; Ge, X.; Li, J.; Liu, C.; Zhao, J.; et al. SaCas9 Requires 5′-NNGRRT-3′ PAM for Sufficient Cleavage and Possesses Higher Cleavage Activity than SpCas9 or FnCpf1 in Human Cells. Biotechnol. J. 2018, 13, e1700561. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Stanford, W.; de Solis, C.; Aradhana; Abraham, N.D.; Dao, T.J.; Thaseen, S.; Sairavi, A.; Gonzalez, C.U.; Ploski, J.E. The Development of an AAV-Based CRISPR SaCas9 Genome Editing System That Can Be Delivered to Neurons in vivo and Regulated via Doxycycline and Cre-Recombinase. Front. Mol. Neurosci. 2018, 11, 413. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, L.; Bell, P.; McMenamin, D.; He, Z.; White, J.; Yu, H.; Xu, C.; Morizono, H.; Musunuru, K.; et al. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat. Biotechnol. 2016, 34, 334–338. [Google Scholar] [CrossRef]
- Hirata, R.K.; Russell, D.W. Design and packaging of adeno-associated virus gene targeting vectors. J. Virol. 2000, 74, 4612–4620. [Google Scholar] [CrossRef]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat. Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef]
- Chen, G.; Abdeen, A.A.; Wang, Y.; Shahi, P.K.; Robertson, S.; Xie, R.; Suzuki, M.; Pattnaik, B.R.; Saha, K.; Gong, S. A biodegradable nanocapsule delivers a Cas9 ribonucleoprotein complex for in vivo genome editing. Nat. Nanotechnol. 2019, 14, 974–980. [Google Scholar] [CrossRef]
- Wan, T.; Chen, Y.; Pan, Q.; Xu, X.; Kang, Y.; Gao, X.; Huang, F.; Wu, C.; Ping, Y. Genome editing of mutant KRAS through supramolecular polymer-mediated delivery of Cas9 ribonucleoprotein for colorectal cancer therapy. J. Control. Release 2020, 322, 236–247. [Google Scholar] [CrossRef]
- Lostale-Seijo, I.; Louzao, I.; Juanes, M.; Montenegro, J. Peptide/Cas9 nanostructures for ribonucleoprotein cell membrane transport and gene edition. Chem. Sci. 2017, 8, 7923–7931. [Google Scholar] [CrossRef]
- Krishnamurthy, S.; Wohlford-Lenane, C.; Kandimalla, S.; Sartre, G.; Meyerholz, D.K.; Theberge, V.; Hallee, S.; Duperre, A.M.; Del’Guidice, T.; Lepetit-Stoffaes, J.P.; et al. Engineered amphiphilic peptides enable delivery of proteins and CRISPR-associated nucleases to airway epithelia. Nat. Commun. 2019, 10, 4906. [Google Scholar] [CrossRef]
- Lee, K.; Conboy, M.; Park, H.M.; Jiang, F.; Kim, H.J.; Dewitt, M.A.; Mackley, V.A.; Chang, K.; Rao, A.; Skinner, C.; et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng. 2017, 1, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Behr, M.; Zhou, J.; Xu, B.; Zhang, H. In vivo delivery of CRISPR-Cas9 therapeutics: Progress and challenges. Acta Pharm. Sin. B 2021, 11, 2150–2171. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.H.; Suh, Y. In vivo genome editing in animals using AAV-CRISPR system: Applications to translational research of human disease. F1000Res 2017, 6, 2153. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, W.; Huang, B.; Xu, S.; Li, Y.; Zhu, L.; Yiqiao, H.; Wu, Z.; Wu, X. AAV-mediated in vivo CAR gene therapy for targeting human T-cell leukemia. Blood Cancer J. 2021, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, J.; Mikuni, T.; Yasuda, R. Virus-Mediated Genome Editing via Homology-Directed Repair in Mitotic and Postmitotic Cells in Mammalian Brain. Neuron 2017, 96, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Dudek, A.M.; Porteus, M.H. Answered and Unanswered Questions in Early-Stage Viral Vector Transduction Biology and Innate Primary Cell Toxicity for Ex-Vivo Gene Editing. Front. Immunol. 2021, 12, 660302. [Google Scholar] [CrossRef]
- Ghaffari, S.; Torabi-Rahvar, M.; Omidkhoda, A.; Ahmadbeigi, N. Impact of various culture conditions on ex vivo expansion of polyclonal T cells for adoptive immunotherapy. APMIS 2019, 127, 737–745. [Google Scholar] [CrossRef]
- de Azevedo, J.T.C.; Mizukami, A.; Moco, P.D.; Malmegrim, K.C.R. Immunophenotypic Analysis of CAR-T Cells. In Chimeric Antigen Receptor T Cells; Swiech, K., Malmegrim, K.C.R., Picanco-Castro, V., Eds.; Methods in Molecular Biology; Humana: New York, NY, USA, 2020; pp. 195–201. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| gRNA Sequence | Efficiency (%) |
|---|---|
| #1 ataggcagacagacttgtca | 21.7 |
| #2 gtctctcagctggtacacgg | 12.1 |
| #3 tacacggcagggtcagggtt | 2 |
| Vector | Step | Total VG | % Step Recovery | % Overall Recovery |
|---|---|---|---|---|
| AAV6-SaCas9 | Harvest | 4.34 × 1012 | 100 | 100 |
| Purification | 1.91 × 1012 | 44.08 | - | |
| Concentration | 1.76 × 1012 | 91.75 | 40.47 | |
| AAV6-CAR | Harvest | 8.65 × 1012 | 100 | 100 |
| Purification | 8.17 × 1012 | 94.40 | - | |
| Concentration | 3.99 × 1012 | 48.89 | 46.15 |
| Name | Target Sequence | Gene | Product | % Indel | |
|---|---|---|---|---|---|
| HEK293SF | Jurkat | ||||
| gRNA #1 | ATAGGCAGACAGACTTGTCACTGGAT | TRAC | T-cell receptor | 21.7 | 14.6 |
| OT1 | AAGTAAGACAGACTTGTCAGTGGGT | LOC112267962 | Long non-coding RNA | 1.5 | - |
| OT3 | ATAGTCACACAGACTGTCACTGAAT | LINC01035 | Long non-coding RNA | 0.1 | 0 |
| OT4 | ATAGGAGACAGGATTGTCAAGGGAT | CACNB2 | Voltage-gated calcium channel | 1.3 | - |
| OT6 | ATAGAGCAGACAGACAGGTCAAAGAAT | - | - | 1.5 | 0 |
| OT8 | ATGGCAGAGAGACTTGCCATTGGAT | LPAL2 | Lipoprotein(a) 2 pseudogene | 1.1 | 0 |
| OT10 | GTAGTCAGACAGACTTGTATTGGAT | GTDC1 | Glycosyltransferase-like | 0.2 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moço, P.D.; Farnós, O.; Sharon, D.; Kamen, A.A. Targeted Delivery of Chimeric Antigen Receptor into T Cells via CRISPR-Mediated Homology-Directed Repair with a Dual-AAV6 Transduction System. Curr. Issues Mol. Biol. 2023, 45, 7705-7720. https://doi.org/10.3390/cimb45100486
Moço PD, Farnós O, Sharon D, Kamen AA. Targeted Delivery of Chimeric Antigen Receptor into T Cells via CRISPR-Mediated Homology-Directed Repair with a Dual-AAV6 Transduction System. Current Issues in Molecular Biology. 2023; 45(10):7705-7720. https://doi.org/10.3390/cimb45100486
Chicago/Turabian StyleMoço, Pablo D., Omar Farnós, David Sharon, and Amine A. Kamen. 2023. "Targeted Delivery of Chimeric Antigen Receptor into T Cells via CRISPR-Mediated Homology-Directed Repair with a Dual-AAV6 Transduction System" Current Issues in Molecular Biology 45, no. 10: 7705-7720. https://doi.org/10.3390/cimb45100486
APA StyleMoço, P. D., Farnós, O., Sharon, D., & Kamen, A. A. (2023). Targeted Delivery of Chimeric Antigen Receptor into T Cells via CRISPR-Mediated Homology-Directed Repair with a Dual-AAV6 Transduction System. Current Issues in Molecular Biology, 45(10), 7705-7720. https://doi.org/10.3390/cimb45100486