Abstract
Applied to investigate specific sequences, nucleic acid detection assays can help identify novel bacterial and viral infections. Most up-to-date systems combine isothermal amplification with Cas-mediated detection. They surpass standard PCR methods in detection time and sensitivity, which is crucial for rapid diagnostics. The first part of this review covers the variety of isothermal amplification methods and describes their reaction mechanisms. Isothermal amplification enables fast multiplication of a target nucleic acid sequence without expensive laboratory equipment. However, researchers aim for more reliable results, which cannot be achieved solely by amplification because it is also a source of non-specific products. This motivated the development of Cas-based assays that use Cas9, Cas12, or Cas13 proteins to detect nucleic acids and their fragments in biological specimens with high specificity. Isothermal amplification yields a high enough concentration of target nucleic acids for the specific signal to be detected via Cas protein activity. The second part of the review discusses combinations of different Cas-mediated reactions and isothermal amplification methods and presents signal detection techniques adopted in each assay. Understanding the features of Cas-based assays could inform the choice of an optimal protocol to detect different nucleic acids.
1. Introduction
Pathogen detection methods usually target viral or bacterial proteins or nucleic acids. A nucleic acid detection system is a multistep assay that includes (1) extraction of nucleic acids from a biospecimen; (2) isothermal amplification of a nucleic acid fragment to reach the detection threshold; (3) detection of nucleic acids and experimental data analysis. Classic polymerase chain reaction (PCR) [1,2] and real-time PCR [3] currently give way to advanced systems. These systems combine highly specific Cas-based protein complexes with an isothermal amplification step that allows for nucleic acid analysis under constant temperature. Originally, the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas-system is an immune gene cluster that protects bacteria from invading pathogens [4]. Via gene recombination, the protective mechanism inserts a foreign nucleic acid sequence in the gene cluster, downstream to “direct repeats” that are recognized by a Cas protein. During translation, Cas proteins form a complex with their guide RNAs (gRNA) that are complementary to the specific fragments of nucleic acids. If designed artificially, modern CRISPR/Cas systems can incorporate diverse Cas proteins and specific gRNAs. Cas proteins specifically detect various nucleic acids (ss/ds DNA or RNA) and, thus, facilitate in vivo diagnostics. Another advantage of Cas proteins is their tolerance to contaminants, which supports reactions without preliminary purification of nucleic acids and reduces diagnostic time. A wide range of nucleic acid detection platforms varying in Cas protein types and methods of isothermal amplification has been developed. The most prominent systems are Cas9-based, such as CRISPR/Cas9-triggered isothermal exponential amplification reaction (CAS-EXPAR) [5], NASBA–CRISPR cleavage (NASBACC) [6], CRISPR–Cas9-triggered nicking endonuclease-mediated strand displacement amplification method (CRISDA) [7], Cas9-mediated lateral flow nucleic acids assay (CASLFA) [8], or RCA-assisted CRISPR/Cas9 cleavage (RACE) [9]. Cas12 and Cas14 are used in DNA endonuclease-targeted CRISPR trans reporter (DETECTR) [10], one-hour low-cost multipurpose highly efficient system (HOLMES) [11], and HOLMES v.2 [12]), while Cas13 is employed in specific high-sensitivity enzymatic reporter unLOCKing (SHERLOCK) [13], SHERLOCK v.2 [14], combinatorial arrayed reactions for multiplexed evaluation of nucleic acids (CARMEN) [15], SHERLOCK-based profiling of in vitro transcription (SPRINT) [16], and SHERLOCK and HUDSON integration to navigate epidemics (SHINE) [17]. This review covers molecular mechanisms underlying various CRISPR/Cas-based nucleic acid detection systems.
2. Sample Preparation Methods for the Downstream Nucleic Acid Detection
Nucleic acid detection in clinical specimens requires the inactivation of Rnases-specific RNA-cleaving enzymes that are abundant in biofluids. The heating unextracted diagnostic samples to obliterate nucleases (HUDSON) method [18] implies chemical inactivation of RNase by heating clinical specimens to 95 °C. In combination with SHERLOCK (SHINE), this approach allows for detecting up to 5–10 viral RNA copies in less than an hour (Figure 1) [17].
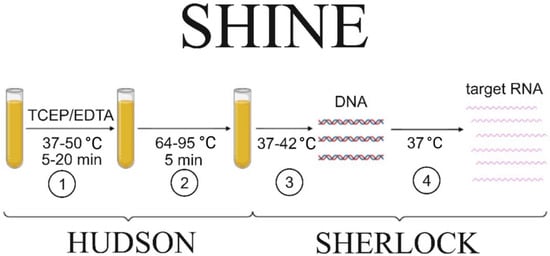
Figure 1.
Scheme of viral particle detection in biological fluids using the SHINE protocol. The main steps are (1) nuclease inactivation to prevent nucleic acid degradation; (2) viral particle inactivation, facilitating the nucleic acid release from the particle; (3) RT–RPA step to amplify a sufficient amount of DNA; (4) target RNA production for the specific Cas13a-mediated detection. The steps that are included either in HUDSON or SHERLOCK protocols are indicated by the curly braces.
3. Methods of Isothermal Amplification
The next step of nucleic acid detection requires amplification of the target sequence. Currently, isothermal methods are being developed and used for rapid and field-deployable amplification.
Nucleic acid sequence-based amplification (NASBA) [19], recombinase polymerase amplification (RPA) [13], RT–RPA (the same as previous but combined with reverse transcription) [13], and loop-mediated isothermal amplification (LAMP) [20] are the most well-known and widely used methods among modern approaches to isothermal amplification. Standard NASBA (Figure 2) is a multi-enzyme reaction involving AMV reverse transcriptase, T7 RNA polymerase, and RNase H [19]. The method achieves 108–109-fold amplification of a target 100–200 nucleotide RNA product in 120 min reaction time [19]. The NASBA isothermal amplification reaction detects both bacterial [21,22,23,24] and viral [25,26,27,28] pathogens. Wu et al. [29] analyzed reaction conditions, such as primer concentration, reaction temperature, and reaction time. Their optimized protocol informed a new isothermal NASBA sequencing-based high-throughput test (INSIGHT method), which provides multiplexed pathogen detection through massively parallel next-generation sequencing. The NASBA reaction is, however, limited in its specificity, which is typical for multi-enzyme reactions. Hence, it is necessary to design specific probes and primers for the target amplicons [30,31,32], as well as to include a preliminary step for annealing at 65 °C.
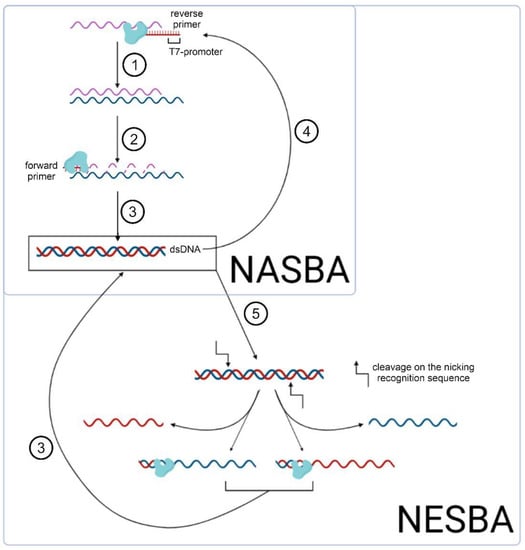
Figure 2.
Scheme of isothermal amplification NASBA and its modification NESBA, including the following steps: (1) reverse transcription of the initial RNA fragment by the AMV reverse transcriptase (shown in the figure); (2) RNase H-mediated cleavage of these RNAs; (3) synthesis of dsDNA as a matrix for (4) subsequent in vitro transcription; (5) nicking of the dsDNA with its subsequent amplification.
In contrast, nicking and extension chain reaction system-based amplification (NESBA) refers to a more sensitive variation of NASBA (Figure 2). Ju et al. demonstrated that NESBA detects RNA fragments, encoding E and N genes of SARS-CoV-2, at a concentration as low as 0.5 copies/ul [33]. Here, the forward T7 primer is extended to form multiple copies of the DNA sequence that enter the T7 RNA polymerase-catalyzed transcription. Thus, compared to the original NASBA method, the NESBA reaction yields a significantly amplified signal, while achieving excellent sensitivity (100%) and specificity (100%).
The RPA reaction, including its combination with Cas proteins, is used extensively for clinical diagnostics in a laboratory setting [34]. The RPA mechanism exploits the activity of homologous recombination proteins. T4 UvsX protein (recombinase) and T4 UvsY (loading factor) bind to the primers, enabling the search for homologous sequences in the template DNA [34]. The complex of primers and proteins creates a D-loop structure in double-stranded DNA, and unwinding DNA strands are stabilized by the single-strand binding (SSB) proteins T4gp32 [34] (Figure 3).
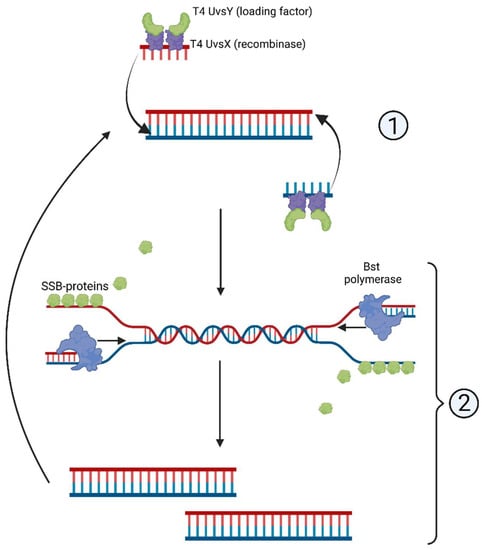
Figure 3.
Scheme of recombinase polymerase amplification, RPA: (1) T4 UvsY- and T4 UvsX-mediated complementary strand recognition and primer annealing; (2) D-loop formation and synthesis of the complementary strand.
A distinct advantage of RPA lies in entirely isothermal amplification: it proceeds without any preliminary steps that would require elevated temperatures. RPA primers are typically longer than those designed for other isothermal methods [34]. Shorter primers, although used in some works, reportedly decrease RPA reaction rate and sensitivity [35,36]. Inhibitors such as hemoglobin (20 g/L), heparin (0.5 U), and urea (1.25%) are shown to not interfere with the reaction significantly [35,36]. Moreover, higher concentrations of hemoglobin (50 g/L), ethanol (4% v/v), or urea (up to 5%) reduce the reaction rate only slightly [37,38,39]. These data favor RPA over detergent-sensitive methods such as NASBA.
To detect a trace quantity of RNA in clinical specimens, RPA is coupled with Cas13-based interference (SHERLOCK, SHERLOCK v.2, CARMEN). Similarly, RT–RPA and LAMP are combined with Cas12-based DNA interference, constituting the SHERLOCK and SHERLOCK v.2 or DETECTR assays, respectively. LAMP is widely used in clinical diagnostics alongside RPA. This type of isothermal amplification is based on the synthesis of concatemeric amplicons from a 100–250 nucleotide DNA sequence, which is facilitated by the Bst DNA polymerase derived from Bacillus subtilis. Two or three pairs of primers are used specifically to amplify the target product [16,36]. To visualize the accumulation of the LAMP product in the visible light range (‘naked-eye visualization’), SYBR Green I is introduced to the reaction mix as a fluorescent dye with a high affinity for double-stranded DNA [40,41]. Drawbacks of LAMP include a high reaction temperature (60–65 °C), complex primer design, and a high probability of producing non-specific amplicons. Additionally, some non-specific products are amplified in the absence of the template because of the interaction between different primer pairs [42]. Many detection methods are currently available that utilize LAMP in a combination with Cas12 and Cas13 proteins that demonstrate specific activity. Additionally, a system named Cas14–DETECTR, which combines LAMP with Cas14 interference, was developed in 2018 [43].
Some detection assays rely on other approaches to isothermal amplification, such as strand displacement amplification (SDA) [9] or rolling circle amplification (RCA) [10]. In SDA, DNA polymerase that possesses strand displacement activity amplifies the DNA template. RCA enables the synthesis of multiple tandem copies from a single DNA strand, resulting in the amplification of a specific concatemeric product. The cross-priming amplification method (CPA) is similarly based on producing a concatemeric amplicon. It allows for amplifying a target sequence from four copies/μL of genomic DNA of Mycobacterium tuberculosis with neither preliminary heating nor nickase in the reaction [44]. However, a clear shortcoming of using the CPA test in the field is that it requires maintaining a high temperature (63–68 °C) to amplify a specific sequence. No reaction product is detected at temperatures below 58 °C [44].
To obtain a specific target product using the above-mentioned isothermal amplification methods, several (typically three) pairs of primers must be used, which considerably complicates the design of an assay. This necessitated the development of other isothermal amplification systems that would require only a pair of primers or just a single primer to achieve the same result in a comparable or even shorter amount of time. For example, polymerase spiral reaction (PSR) utilizing Bst polymerase has already been proven as a reliable detection method to visualize the reaction product by chemiluminescence [45]. Ji et al. applied this method to successfully detect DNA of the porcine circovirus (PSV-3) in clinical specimens. The detection limit reached about 100 copies/μL and there was no cross-reactivity with other circoviruses [45], indicating the high sensitivity and specificity of the method. PSR was found compatible with naked-eye detection based on a mixture of phenol red and cresol red dyes. The color of the mixture changes from pink to yellow if specimens contain the target DNA [45]. In another work [46], SYBR Green I is chosen for visualization in the visible range. PSR reaction mechanism leverages longer primers to obtain a product that contains complementary sequences and, hence, closes into a helix that is subsequently extended [47].
The signal-mediated amplification of RNA technology (SMART) method employs a pair of “probes”, which are complementary to both each other and a specific sequence within the DNA or RNA template (Figure 4). Here, a T-like three-way junction (3WJ) structure is formed. It is then recognized by the Bst polymerase, which extends the ssDNA fragment to form dsDNA that incorporates a promotor of the T7-phage RNA polymerase [48]. Hall et al. were able to specifically detect the mRNA of cyanophages, targeting cyanobacteria Synechococcus sp., even 10 h after transfection [49]. A specific signal was also obtained for similar genes in cyanophages. SMART required neither extraction nor purification of genomic DNA since the detection was performed in cell lysates. The data obtained suggest the high specificity of the SMART system as well as its tolerance to the detergents present in the reaction mix.
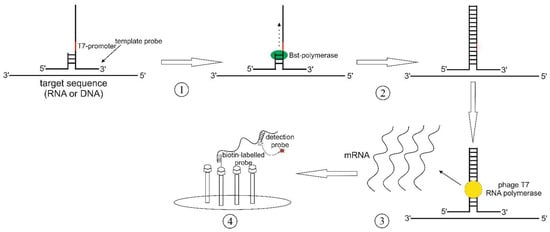
Figure 4.
SMART scheme: (1) 3-way junction (3WJ) formation; (2) synthesis of the second strand by the Bst DNA polymerase; (3) synthesis of the specific mRNA; (4) detection of the synthesized RNA fragment by a specific detection probe that is annealed to a biotin-labeled probe.
Unusual (UIMA) and linear-target (LIMA) isothermal multimerization and amplification systems exploit strand-displacing polymerases that possess reverse transcriptase activity but lack 5′–3′-exonuclease activity. An example of such a polymerase is the Bst polymerase used in LAMP isothermal amplification. Due to its activity, the reaction products comprise long sequences of a multiplied repeated target DNA fragment. Yet, these reactions require a single pair of primers or, as in the case of UIMA, only one primer. The specificity of one-primer reactions has been proven in experiments where the reaction mix contained either no FAM-labeled template or no FAM-labeled primer. No product amplification has been observed on the real-time fluorescence-tracked electropherogram, indicating the absence of contamination [50]. A single primer is sufficient owing to unique reaction conditions. Under high concentrations of Mg2+ (8–10 mM), the primer–template complex tends to form loop structures [50], in which the mismatches are extended by the aforementioned polymerases to form dsDNA. During amplification, the product is extended by one segment, and complementary sequences from different fragments are further extended at the annealing step. Isothermal amplification enables a 106–1010-fold increase in the concentration of the target nucleic acid fragment in clinical specimens over a relatively short time (5 min to 1 h) and at a constant temperature (25–65 °C). Hence, it allows a sample containing as few as 1 to 10 copies of viral RNA to be processed for detection quickly and with minimal use of equipment.
4. Application of Genome-Editing Proteins in Nucleic Acid Detection Assays
A more specific, sensitive, and rapid pathogen identification can be achieved by integrating the above-mentioned types of isothermal amplification with Cas-mediated detection methods. Over the last five years, plenty of new detection systems utilizing Cas proteins have been introduced. We attempt to systemize their parameters and possible application in Table 1. Cas proteins can help detect the majority of viral and bacterial pathogens in extremely low concentrations (see Table 1).

Table 1.
Nucleic acid detection systems based on the activity of Cas proteins.
As can be seen from Table 1, the choice of a Cas-based detection system for a particular application depends on the type of nucleic acid and, consequently, on the detection object.
Due to the enzymatic cleavage activity of the Cas9 protein, Cas-based assays efficiently detect DNA/microRNA when used in combination with various isothermal amplification techniques and different methods of capturing specific signals (Figure 5).
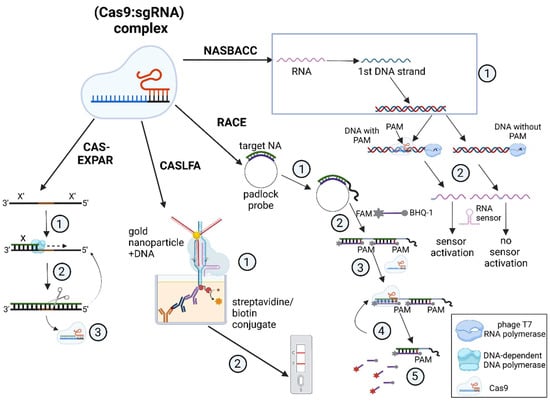
Figure 5.
Nucleic acid detection methods using the Cas9 protein. Cas–EXPAR: (1) dsDNA synthesis combined with (2) its cleavage by a restriction enzyme; (3) Cas9-mediated interference. CASLFA: (1) formation of a complex conjugate due to the recognition of [gold nanoparticle: DNA] complex by Cas9 complex that is immobilized on the streptavidin/biotin conjugate; (2) lateral flow detection; RACE: (1) recognition of target nucleic acid by a padlock probe and (2) synthesis of the specific product by RCA; (3) FAM-labeled probe hybridization; (4) Cas9-mediated [probe: template] recognition and cleavage leading to (5) the emission of a fluorescent signal; NASBACC: (1) simplified outline of NASBA that produces dsDNA; (2) T7 RNA polymerase recognizes dsDNA sequences that, via PAM-specific cleavage, leads to the formation of two kinds of products: the truncated product mediates sensor activation, while the full-length one does not.
For instance, in the CAS–EXPAR system, the Cas9 protein introduces a single-strand break, or nick, in a native DNA molecule and the nicked fragment is annealed to a DNA template. It is then extended by a DNA-dependent DNA polymerase in a strand-displacement reaction, and specific nicking endonucleases cleave the ssDNA fragment, leading to exponential accumulation of the cleaved product [7]. The detection limit for CAS–EXPAR reaches 0.82 aM, as determined by fluorescence analysis [7,57].
CASLFA simplifies the detection through immunochemical analysis in a lateral flow assay that is based on aurum nanoparticles (AuNP). Two molecular mechanisms can lead to the test line appearing on a test strip: either (1) the AuNP–DNA conjugate binds with an immobilized biotinylated non-target DNA strand (NTS), which is mediated by the interaction of the Cas9∙sgRNA complex with the target DNA strand (TS); or (2) the AuNP–DNA conjugate hybridizes with a stem-loop region sequence of the sgRNA when the complex binds anchored DNA [10]. The two ways can be mediated by both native Cas9 protein and its mutated variant dCas9 (deactivated Cas9) that lacks nuclease activity.
Alternatively, the RACE system is based on the RCA amplification method. It allows for the fluorescent detection of miRNAs that are only 19 to 23 nucleotides long, including oncogenic miRNAs. Detection is achieved by hybridizing a probe to a sequence amplified from the template miRNA, which is followed by the cleavage of this double-stranded region by CRISPR/Cas9 [11].
Finally, there are Cas9-based systems such as NASBACC that can assist in recognizing various SNPs in viral RNA [8]. The reaction mechanism depends on a specific toehold switch sensor that recognizes an RNA sequence produced via the NASBA reaction. In this case, the toehold switch can only sense full-sized transcripts that are amplified solely from DNA that is not cut by the Cas9–sgRNA complex owing to the absence of the PAM sequence. The downsides of this method are the long reaction time (3 h of NASBA followed by 30 min of Cas9-based detection) and the lack of a combined protocol for one-pot detection, which can result in contamination and false positives.
Unlike Cas9-based detection systems, proteins Cas12, Cas13, and Cas14 possess non-specific cleaving activity that is triggered by the cleavage of a target sequence. Therefore, if genome-editing proteins of the Cas12–Cas14 family are used to detect nucleic acids after their isothermal amplification, the process consists of two stages: (1) specific cleavage of a target DNA or RNA; (2) cleavage of RNA/DNA probes followed by fluorescent detection (Figure 6).
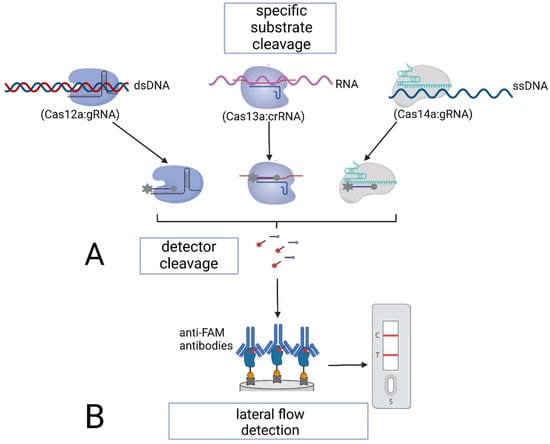
Figure 6.
Specific nucleic acid detection using Cas12–Cas14 proteins: (A) via subsequent fluorescent detection; (B) via lateral flow detection.
Once the target sequence is cleaved, Cas12–Cas14 proteins develop non-specific, collateral cleavage activity. Thus, if the target RNA or DNA is present in the sample, Cas proteins cleave RNA/DNA probes—short (15 to 25 nucleotides) artificial RNA or DNA sequences carrying a fluorescent dye and a quencher. The dissociation of the dye and the quencher after cleavage triggers the fluorescence of the solution [13]. The quantitative fluorescent detection in this case is performed with a fluorescence reader within a relatively short time (less than 1 h).
Gootenberg et al. adapted the SHERLOCK platform for the detection of nucleic acids and developed SHERLOCK v.2 by introducing immunochromatographic analysis that relies on Cas13 and an auxiliary CRISPR-associated enzyme Csm6 [14]. SHERLOCK v2 utilizes an immunochromatographic lateral-flow detection method based on a specific activity of FAM-labeled RNA. These FAM reporters bind anti-FAM antibodies, which are conjugated with gold nanoparticles and anchored on a test strip, thus, preventing the conjugate from recognizing the bacterial protein A. When a FAM reporter is cleaved, the above-mentioned triple conjugates are formed and visualized as the sample (test) line in addition to the control line on a test strip. This modification of SHERLOCK provides even higher sensitivity due to a higher reaction rate, and the reagents are resistant to lyophilization, thus, making field-ready assays possible. Additionally, Joung et al. improved the method by combining isothermal amplification with Cas13-based detection in a one-pot reaction (SHERLOCK testing in one-pot) [51]. Positive samples can be detected within 15 to 45 min, while the specificity and sensitivity of the detection reach 98.5% and 93.1%, respectively.
Combinatorial arrayed reactions for multiplexed evaluation of nucleic acids (CARMEN) suggested by Ackermann et al. involve the Cas13a protein. This assay provides the conditions for multiplexed detection of nucleic acids in a single reaction with attomolar sensitivity. It has been applied to analyze a panel of more than 1000 samples containing 169 previously described viral RNAs. High throughput is achieved via an advanced signal detection technique: four commercially available fluorophores are combined to create highly specific color codes. Clinically, CARMEN allows for multiplexed detection of various viral pathogens in a biofluid sample. In particular, this method is proven to differentiate influenza virus serotypes (H1–H16, N1–N9) [13].
Chuan et al. optimized Cas12/Cas13-mediated detection by introducing a colorimetric approach that reliably detects traces of target nucleic acids (up to 10 copies) in clinical specimens within 3–5 min [52]. The colorimetric assay enables naked-eye visualization and involves complementary binding of linker DNA/RNA with specific probes conjugated with gold nanoparticles. Just as with SHERLOCK v.2, this method does not require any specialized equipment to analyze the results of an experiment.
Lopez-Valls et al. further optimized and modified this approach [53]. Their CRISPR/CAS-based colorimetric nucleic acid detection (CASCADE) method can detect picomolar concentrations of RNA without a preliminary isothermal amplification stage. If coupled with RPA or NASBA, its sensitivity covers the 3 fM and 40 aM ranges, respectively. Moreover, unlike the method described by Chuan et al. [52], CASCADE does not require centrifugation to achieve a qualitative change in the color of the reaction mix. This becomes possible due to extending the length of gold nanoparticle-conjugated oligonucleotides to 33 nucleotides, as well as increasing both the concentration of nanocomplexes and detection time.
An interesting approach to increasing the specificity of viral RNA detection in solutions is presented by Wang et al. [54]. They describe a CRISPR/Cas13-based CASCADE viral RNA assay for SARS-CoV-2 RNA detection. The assay is based on specific cleavage of SARS-CoV-2 RNA fragments and collateral cleavage of a hybrid nucleic acid pre-primer at its UU site to release an inactive phosphorylated primer. The primer is activated by dephosphorylation. Then, the primer binds the DNA sequence carrying T7-promoter and L-broccoli reporter gene and initiates chain extension to dsDNA mediated by the Klenow fragment without 5′–3′ exonuclease activity (exo-). This DNA serves as a template for transcription amplification by RNA polymerase, producing broccoli (RNA aptamers) that bind to the fluorescent dye DFHBI-1. The resulting complex emits a fluorescent signal. This method is highly sensitive (0.06 fM detection limit), but the presence of many enzymes in the assay mixture may trigger unwanted reactions.
Detection systems coupled with isothermal amplification are also available for the Cas14a protein that cleaves ssDNA sequences independent of PAM [55,58]. The Cas14–SDA method is a combination of SDA and specific Cas14a-mediated cleavage and it detects cholangiocarcinoma-associated miR-21 in concentrations as low as 680 fM [55]. The signal acquisition mechanism begins with hybridizing microRNA with a DNA template that contains the miRNA recognition site, a nicking (cleavage) site, and the Cas14a trans cleavage activation site. Bst polymerase recognizes the microRNA template complex and extends the sequences that are then cleaved by an endonuclease. Similarly to the Cas13a-mediated system, Cas14a is activated by amplified sequences, termed activators, when they are cleaved from long single-stranded nucleic acids (DNA in this case). Then, Cas14a cleaves the reporter FAM-labeled ssDNA oligonucleotides, and the fluorescent signal is emitted.
Thus, the systems based on isothermal amplification coupled with Cas-mediated interference reliably detect trace amounts of nucleic acids in biological specimens. These assays outperform alternative methods, such as PCR and real-time PCR, in detection speed, sensitivity, and ease of use. In addition to the concentration of the target DNA or RNA fragment, the most advanced detection systems also identify contaminants in the reaction, which reduces the time dedicated to optimizing reaction conditions.
5. Other Detection Methods Involving Cas Proteins
Although canonical systems that combine isothermal amplification and Cas-based detection are widely used in clinical and research studies, other approaches have been recently developed. Fozouni et al. exclude the isothermal amplification step and instead target multiple crRNAs at different sequences within a full-sized SARS-CoV-2 RNA [56]. Out of twelve crRNA molecules that target different genomic RNA regions of coronavirus, two crRNAs are chosen for the final assay since they generate the strongest fluorescent signal when they form a ribonucleoprotein complex with the Cas13a protein. This assay achieves 100 copies/μL sensitivity, and the detection time is 30 min, which allows the method to compete with isothermal-amplification-based assays in terms of usability and convenience. The specificity of this method enables differentiating SARS-CoV-2 RNA from RNA of other infectious agents, including MERS-CoV and influenza viruses A and B, which contribute the most to seasonal viral infections.
Finally, Iwasaki et al. demonstrate the feasibility of detecting low-molecular-weight compounds in solutions using a method termed SPRINT [16]. In its essence, SPRINT employs the activity of ‘riboswitches’—ligand-specific RNA sequences that recognize small compounds in solution (nucleotides, inorganic ions, etc.), thereby changing the secondary structure of RNA and making the target region available for Cas13a–crRNA-mediated cleavage. The cleavage triggers collateral RNase activity of Cas13a, as described above, and, hence, a specific fluorescent signal is generated by the cleaved reporter RNA oligonucleotides.
6. Conclusions
Cas proteins are increasingly gaining popularity as tools for the diagnostics of viral and bacterial infections due to their ability to recognize nucleic acids with high sensitivity and specificity. The collateral cleavage activity of Cas proteins is used to generate detection signals of various origins, such as fluorescent [13], immunochemical [14], or even colorimetric signal that allows for naked-eye visualization of the positive signal [53]. Furthermore, the broad adoption of isothermal amplification enables combining it with the detection techniques based on genomic-editing proteins. Isothermal amplification yields high concentrations of target nucleic acid fragments with no sophisticated specialized equipment required. It is also possible to carry out these combined reactions beyond the laboratory setting, which is also facilitated by the lyophilization stability of many components of these assays [13]. In addition, it is worth noting that methods that identify target nucleic acid sequences without their preliminary amplification are now actively being developed [56] to further simplify and accelerate the detection of pathogens in clinical and research settings.
Author Contributions
Conceptualization, D.N.A. and G.A.S.; investigation, D.N.A.; writing—original draft preparation, D.N.A.; writing—review and editing, G.A.S.; supervision, G.A.S.; project administration, G.A.S. All authors have read and agreed to the published version of the manuscript.
Funding
The work was funded by the Ministry of Science and Higher Education of the Russian Federation (Agreement No. 075-15-2021-1085).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Saiki, R.K.; Scharf, S.; Faloona, F.; Mullis, K.B.; Horn, G.T.; Erlich, H.A.; Arnheim, N. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 1985, 230, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Saiki, R.K.; Gelfand, D.H.; Stoffel, S.; Scharf, S.J.; Higuchi, R.; Horn, G.T.; Mullis, K.B.; Erlich, H.A. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 1988, 239, 488–491. [Google Scholar] [CrossRef] [PubMed]
- Kubista, M.; Andrade, J.M.; Bengtsson, M.; Forootan, A.; Jonák, J.; Lind, K.; Sindelka, R.; Sjöback, R.; Sjögreen, B.; Strömbom, L.; et al. The real-time polymerase chain reaction. Mol. Aspects Med. 2006, 27, 95–125. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Huang, M.; Zhou, X.; Wang, H.; Xing, D. Clustered Regularly Interspaced Short Palindromic Repeats/Cas9 Triggered Isothermal Amplification for Site-Specific Nucleic Acid Detection. Anal. Chem. 2018, 90, 2193–2200. [Google Scholar] [CrossRef]
- Pardee, K.; Green, A.A.; Takahashi, M.K.; Braff, D.; Lambert, G.; Lee, J.W.; Ferrante, T.; Ma, D.; Donghia, N.; Fan, M.; et al. Rapid, Low-Cost Detection of Zika Virus Using Programmable Biomolecular Components. Cell 2016, 165, 1255–1266. [Google Scholar] [CrossRef]
- Zhou, W.; Hu, L.; Ying, L.; Zhao, Z.; Chu, P.K.; Yu, X.F. A CRISPR-Cas9-triggered strand displacement amplification method for ultrasensitive DNA detection. Nat. Commun. 2018, 9, 5012. [Google Scholar] [CrossRef]
- Wang, X.; Xiong, E.; Tian, T.; Cheng, M.; Lin, W.; Wang, H.; Zhang, G.; Sun, J.; Zhou, X. Clustered Regularly Interspaced Short Palindromic Repeats/Cas9-Mediated Lateral Flow Nucleic Acid Assay. ACS Nano 2020, 14, 2497–2508. [Google Scholar] [CrossRef]
- Wang, R.; Zhao, X.; Chen, X.; Qiu, X.; Qing, G.; Zhang, H.; Zhang, L.; Hu, X.; He, Z.; Zhong, D.; et al. Rolling Circular Amplification (RCA)-Assisted CRISPR/Cas9 Cleavage (RACE) for Highly Specific Detection of Multiple Extracellular Vesicle MicroRNAs. Anal. Chem. 2020, 92, 2176–2185. [Google Scholar] [CrossRef]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef]
- Li, S.Y.; Cheng, Q.-X.; Wang, J.-M.; Li, X.-Y.; Zhang, Z.-L.; Gao, S.; Cao, R.-B.; Zhao, G.-P.; Wang, J. CRISPR-Cas12a-assisted nucleic acid detection. Cell Discov. 2018, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, S.; Wu, N.; Wu, J.; Wang, G.; Zhao, G.; Wang, J. HOLMESv2: A CRISPR-Cas12b-assisted platform for nucleic acid detection and DNA methylation quantitation. ACS Synth. Biol. 2019, 8, 2228–2237. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Gootenberg, J.S.; Abudayyeh, O.O.; Kellner, M.J.; Joung, J.; Collins, J.J.; Zhang, F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 2018, 360, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, C.M.; Myhrvold, C.; Thakku, S.G.; Freije, C.A.; Metsky, H.C.; Yang, D.K.; Ye, S.H.; Boehm, C.K.; Kosoko-Thoroddsen, T.-S.F.; Kehe, J.; et al. Massively multiplexed nucleic acid detection with Cas13. Nature 2020, 582, 277–282. [Google Scholar] [CrossRef]
- Iwasaki, R.S.; Batey, R.T. SPRINT: A Cas13a-based platform for detection of small molecules. Nucleic Acids Res. 2020, 48, 101. [Google Scholar] [CrossRef]
- Arizti-Sanz, J.; Freije, C.A.; Stanton, A.C.; Petros, B.A.; Boehm, C.K.; Siddiqui, S.; Shaw, B.M.; Adams, G.; Kosoko-Thoroddsen, T.-S.F.; Kemball, M.E.; et al. Integrated sample inactivation, amplification, and Cas13-based detection of SARS-CoV-2. Nat. Commun. 2020, 1, 5921. [Google Scholar] [CrossRef]
- Myhrvold, C.; Freije, C.A.; Gootenberg, J.S.; Abudayyeh, O.O.; Metsky, H.C.; Durbin, A.F.; Kellner, M.J.; Tan, A.; Paul, L.; Parham, L.; et al. Field-deployable viral diagnostics using CRISPR-Cas13. Science 2018, 360, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Compton, J. Nucleic acid sequence-based amplification. Nature 1991, 350, 91–92. [Google Scholar] [CrossRef] [PubMed]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000, 28, e63. [Google Scholar] [CrossRef]
- Scheler, O.; Kaplinski, L.; Glynn, B.; Palta, P.; Parkel, S.; Toome, K.; Maher, M.; Barry, T.; Remm, M.; Kurg, A. Detection of NASBA amplified bacterial tmRNA molecules on SLICSel designed microarray probe. BMC Biotechnol. 2011, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Zhang, J.Z.; Zhang, M.J. Application of NASBA and RPA in detection of pathogenic bacteria. Zhonghua Liu Xing Bing Xue Za Zhi 2019, 40, 1018–1022. [Google Scholar] [PubMed]
- Zhai, L.; Liu, H.; Li, J.; Lu, Z.; Bie, X. A duplex real-time NASBA assay targeting a serotype-specific gene for rapid detection of viable Salmonella Paratyphi C in retail foods of animal origin. Can. J. Microbiol. 2022, 68, 259–268. [Google Scholar] [CrossRef]
- Cook, N. The use of NASBA for the detection of microbial pathogens in food and environmental samples. J. Microbiol. Methods 2003, 68, 165–174. [Google Scholar] [CrossRef]
- Romano, J.W.; van Gemen, B.; Kievits, T. NASBA: A novel, isothermal detection technology for qualitative and quantitative HIV-1 RNA measurements. Clin. Lab. Med. 1996, 16, 89–103. [Google Scholar] [CrossRef]
- Kia, V.; Tafti, A.; Paryan, M.; Mohammadi-Yeganeh, S. Evaluation of real-time NASBA assay for the detection of SARS-CoV-2 compared with real-time PCR. Ir. J. Med. Sci. 2022, 6, 1–7. [Google Scholar] [CrossRef]
- Lau, L.T.; Fung, Y.W.; Yu, A.C. Detection of animal viruses using nucleic acid sequence-based amplification (NASBA). Dev. Biol. 2006, 126, 7–15. [Google Scholar]
- Lau, L.T.; Reid, S.M.; King, D.P.; Lau, A.M.; Shaw, A.E.; Ferris, N.P.; Yu, A.C. Detection of foot-and-mouth disease virus by nucleic acid sequence-based amplification (NASBA). Vet. Microbiol. 2008, 126, 101–110. [Google Scholar] [CrossRef]
- Wu, Q.; Suo, C.; Brown, T.; Wang, T.; Teichmann, S.A.; Bassett, A.R. INSIGHT: A population-scale COVID-19 testing strategy combining point-of-care diagnosis with centralized high-throughput sequencing. Sci. Adv. 2021, 7, eabe5054. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, N.; Reed, A.; Gerasimova, Y.V.; Kolpashchikov, D.M. Split Dapoxyl Aptamer for Sequence-Selective Analysis of Nucleic Acid Sequence Based Amplification Amplicons. Anal. Chem. 2019, 91, 2667–2671. [Google Scholar] [CrossRef]
- Lamhoujeb, S.; Charest, H.; Fliss, I.; Ngazoa, S.; Jean, J. Real-time molecular beacon NASBA for rapid and sensitive detection of norovirus GII in clinical samples. Can. J. Microbiol. 2009, 55, 1375–1380. [Google Scholar] [CrossRef]
- Morabito, K.; Wiske, C.; Tripathi, A. Engineering Insights for Multiplexed Real-Time Nucleic Acid Sequence-Based Amplification (NASBA): Implications for Design of Point-of-Care Diagnostics. Mol. Diagn. Ther. 2013, 17, 185–192. [Google Scholar] [CrossRef]
- Ju, Y.; Kim, J.; Park, Y.; Lee, C.Y.; Kim, K.; Hong, K.H.; Lee, H.; Yong, D.; Park, H.G. Rapid and accurate clinical testing for COVID-19 by nicking and extension chain reaction system-based amplification (NESBA). Biosens. Bioelectron. 2022, 196, 113689. [Google Scholar] [CrossRef]
- Li, J.; Macdonald, J.; von Stetten, F. Review: A comprehensive summary of a decade development of the recombinase polymerase amplification. Analyst 2020, 145, 1950–1960. [Google Scholar] [CrossRef]
- Mayboroda, O.; Gonzalez Benito, A.; Sabaté del Rio, J.; Svobodova, M.; Julich, S.; Tomaso, H.; O’Sullivan, C.K.; Katakis, I. Isothermal solid-phase amplification system for detection of Yersinia pestis. Anal. Bioanal. Chem. 2016, 408, 671–676. [Google Scholar] [CrossRef]
- Martorell, S.; Palanca, S.; Maquieira, Á.; Tortajada-Genaro, L.A. Blocked recombinase polymerase amplification for mutation analysis of PIK3CA gene. Anal. Biochem. 2018, 544, 49–56. [Google Scholar] [CrossRef]
- Kersting, S.; Rausch, V.; Bier, F.F.; von Nickisch-Rosenegk, M. Rapid detection of Plasmodium falciparum with isothermal recombinase polymerase amplification and lateral flow analysis. Malar. J. 2014, 13, 99. [Google Scholar] [CrossRef]
- Rosser, A.; Rollinson, D.; Forrest, M.; Webster, B. Isothermal Recombinase Polymerase amplification (RPA) of Schistosoma haematobium DNA and oligochromatographic lateral flow detection. Parasites Vectors 2015, 8, 446. [Google Scholar] [CrossRef]
- Krõlov, K.; Frolova, J.; Tudoran, O.; Suhorutsenko, J.; Lehto, T.; Sibul, H.; Mäger, I.; Laanpere, M.; Tulp, I.; Langel, Ü. Sensitive and rapid detection of Chlamydia trachomatis by recombinase polymerase amplification directly from urine samples. J. Mol. Diagn. 2015, 16, 127–135. [Google Scholar] [CrossRef]
- Lai, M.Y.; Ooi, C.H.; Lau, Y.L. Validation of SYBR green I based closed-tube loop-mediated isothermal amplification (LAMP) assay for diagnosis of knowlesi malaria. Malar. J. 2021, 20, 166. [Google Scholar] [CrossRef]
- Singh, R.; Singh, D.P.; Savargaonkar, D.; Singh, O.P.; Bhatt, R.M.; Valecha, N. Evaluation of SYBR green I based visual loop-mediated isothermal amplification (LAMP) assay for genus and species-specific diagnosis of malaria in P. vivax and P. falciparum endemic regions. J. Vector Borne Dis. 2017, 54, 54–56. [Google Scholar]
- Rolando, J.C.; Jue, E.; Barlow, J.T.; Ismagilov, R. Real-time kinetics and high-resolution melt curves in single-molecule digital LAMP to differentiate and study specific and non-specific amplification. Nucleic Acids Res. 2020, 48, 42. [Google Scholar] [CrossRef]
- Harrington, L.B.; Burstein, D.; Chen, J.S.; Paez-Espino, D.; Ma, E.; Witte, I.P.; Cofsky, J.C.; Kyrpides, N.C.; Banfield, J.F.; Doudna, J.A. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science 2018, 362, 839–842. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Hu, L.; Zhong, H.; Wang, H.; Yusa, S.; Weiss, T.C.; Romaniuk, P.J.; Pickerill, S.; You, Q. Cross priming amplification: Mechanism and optimization for isothermal DNA amplification. Sci. Rep. 2012, 2, 246. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Xu, X.; Wang, X.; Zuo, K.; Li, Z.; Leng, C.; Kan, Y.; Yao, L.; Bi, Y. Novel polymerase spiral reaction assay for the visible molecular detection of porcine circovirus type 3. BMC Vet. Res. 2019, 15, 322. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Dong, D.; Yang, Z.; Zou, D.; Chen, Z.; Yuan, J.; Huang, L. Polymerase Spiral Reaction (PSR): A novel isothermal nucleic acid amplification method. Sci. Rep. 2015, 29, 123–127. [Google Scholar] [CrossRef]
- Gupta, V.; Chakravarti, S.; Chander, V.; Majumder, S.; Bhat, S.A.; Gupta, V.K.; Nandi, S. Polymerase spiral reaction (PSR): A novel, visual isothermal amplification method for detection of canine parvovirus 2 genomic DNA. Arch. Virol. 2017, 162, 1995–2001. [Google Scholar] [CrossRef]
- Wharam, S.D.; Marsh, P.; Lloyd, J.S.; Ray, T.D.; Mock, G.A.; Assenberg, R.; McPhee, J.E.; Brown, P.; Weston, A.; Cardy, D.L.N. Specific detection of DNA and RNA targets using a novel isothermal nucleic acid amplification assay based on the formation of a three-way junction structure. Nucleic Acids Res. 2001, 29, e54. [Google Scholar] [CrossRef]
- Hall, M.J.; Wharam, S.D.; Weston, A.; Cardy, D.L.; Wilson, W.H. Use of signal-mediated amplification of RNA technology (SMART) to detect marine cyanophage DNA. Biotechniques 2002, 32, 604–606, 608–611. [Google Scholar] [CrossRef]
- Wang, G.; Ding, X.; Hu, J.; Wu, W.; Sun, J.; Mu, Y. Unusual isothermal multimerization and amplification by the strand-displacing DNA polymerases with reverse transcription activities. Sci. Rep. 2017, 7, 13928. [Google Scholar] [CrossRef]
- Joung, J.; Ladha, A.; Saito, M.; Kim, N.G.; Woolley, A.E.; Segel, M.; Barretto, R.; Ranu, A.; Macrae, R.; Faure, G.; et al. Detection of SARS-CoV-2 with SHERLOCK One-Pot Testing. N. Engl. J. Med. 2020, 383, 1492–1494. [Google Scholar] [CrossRef]
- Yuan, C.; Tian, T.; Sun, J.; Hu, M.; Wang, X.; Xiong, E.; Cheng, M.; Bao, Y.; Lin, W.; Jiang, J.; et al. Universal and Naked-Eye Gene Detection Platform Based on the Clustered Regularly Interspaced Short Palindromic Repeats/Cas12a/13a System. Anal. Chem. 2020, 92, 4029–4037. [Google Scholar] [CrossRef] [PubMed]
- López-Valls, M.; Escalona-Noguero, C.; Rodríguez-Díaz, C.; Pardo, D.; Castellanos, M.; Milán-Rois, P.; Martínez-Garay, C.; Coloma, R.; Abreu, M.; Cantón, R.; et al. CASCADE: Naked eye-detection of SARS-CoV-2 using Cas13a and gold nanoparticles. Anal. Chim. Acta 2022, 1205, 339749. [Google Scholar] [CrossRef]
- Wang, Y.; Xue, T.; Wang, M.; Ledesma-Amaro, R.; Lu, Y.; Hu, X.; Zhang, T.; Yang, M.; Li, Y.; Xiang, J.; et al. CRISPR-Cas13a cascade-based viral RNA assay for detecting SARS-CoV-2 and its mutations in clinical samples. Sens. Actuators B Chem. 2022, 362, 131765. [Google Scholar] [CrossRef]
- Chi, Z.; Wu, Y.; Chen, L.; Yang, H.; Khan, M.R.; Busquets, R.; Huang, N.; Lin, X.; Deng, R.; Yamg, W.; et al. CRISPR-Cas14a-integrated strand displacement amplification for rapid and isothermal detection of cholangiocarcinoma associated circulating microRNAs. Anal. Chim. Acta 2022, 1205, 339763. [Google Scholar] [CrossRef]
- Fozouni, P.; Son, S.; Díaz de León Derby, M.; Knott, G.J.; Gray, C.N.; D’Ambrosio, M.V.; Zhao, C.; Switz, N.; Kumar, R.; Stephens, S.; et al. Amplification-free detection of SARS-CoV-2 with CRISPR-Cas13a and mobile phone microscopy. Cell 2021, 184, 323–333. [Google Scholar] [CrossRef]
- Jia, F.; Li, X.; Zhang, C.; Tang, X. The expanded development and application of CRISPR system for sensitive nucleotide detection. Protein Cell 2017, 11, 624–629. [Google Scholar] [CrossRef]
- Karvelis, T.; Bigelyte, G.; Young, J.K.; Hou, Z.; Zedaveinyte, R.; Budre, K.; Paulraj, S.; Djukanovic, V.; Gasior, S.; Silanskas, A.; et al. PAM recognition by miniature CRISPR-Cas12f nucleases triggers programmable double-stranded DNA target cleavage. Nucleic Acids Res. 2020, 48, 5016–5023. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).