
The Effects of NAA on the Tuberous Root Yield and Quality of Rehmannia glutinosa and Its Regulatory Mechanism by Transcriptome and Metabolome Profiling

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Determination of Main R. glutinosa Root Indices
2.3. Determination of Catalpol, Rhmannioside D and Acteoside by HPLC
2.4. Transcriptome Sequencing
2.5. Evaluation of Gene Expression Levels and Data Analyses
2.6. Quantitative Real-Time RT-qPCR
2.7. Screening and Identification of Differential Metabolites
2.8. Data Analysis
3. Results
3.1. Morphological Differences between NTs and CGs and Yield Change
3.2. DAM Screening and Analyses
3.3. Determination of Quality Markers
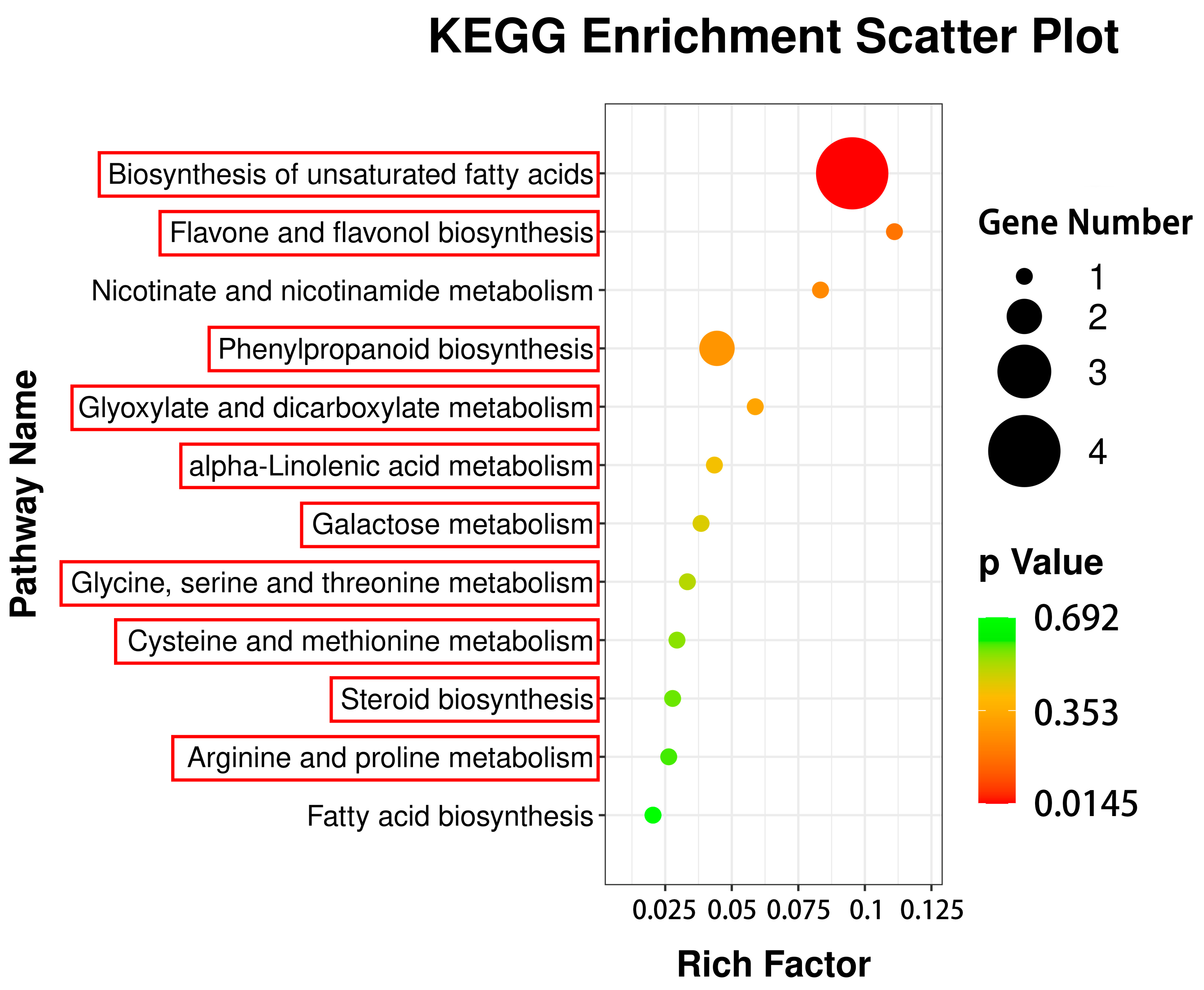
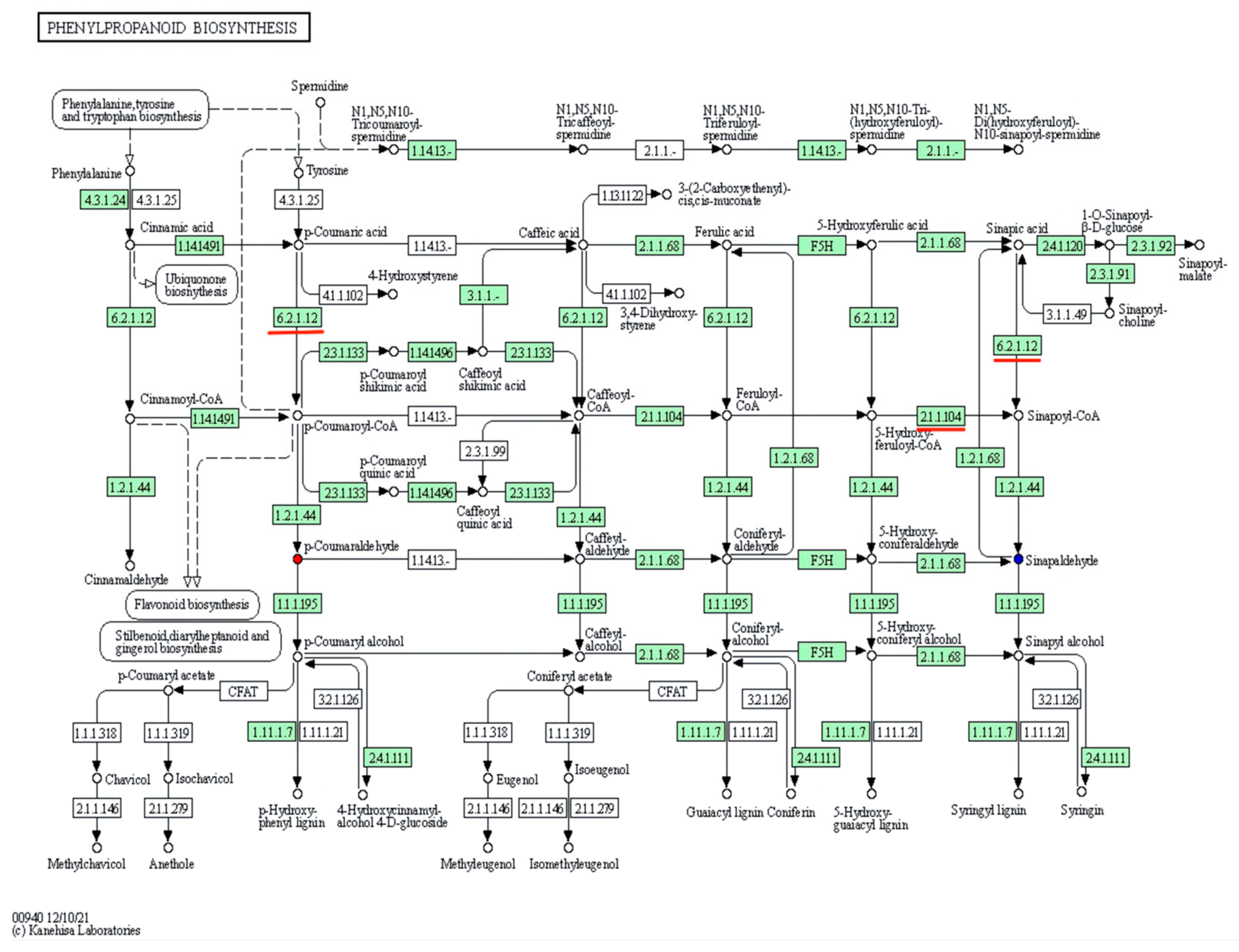
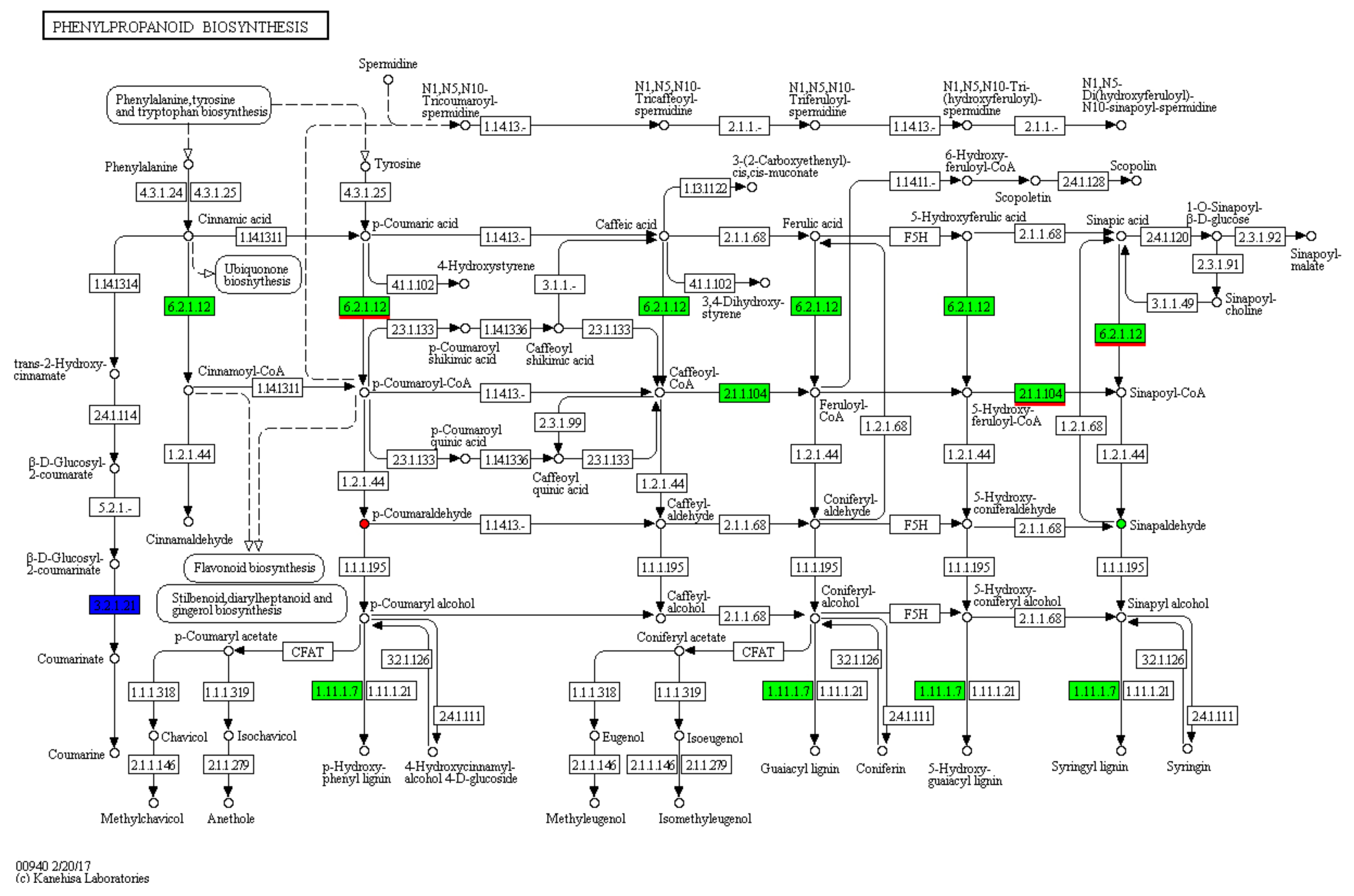
3.4. KEGG Metabolic Pathway Analyses of DAMs
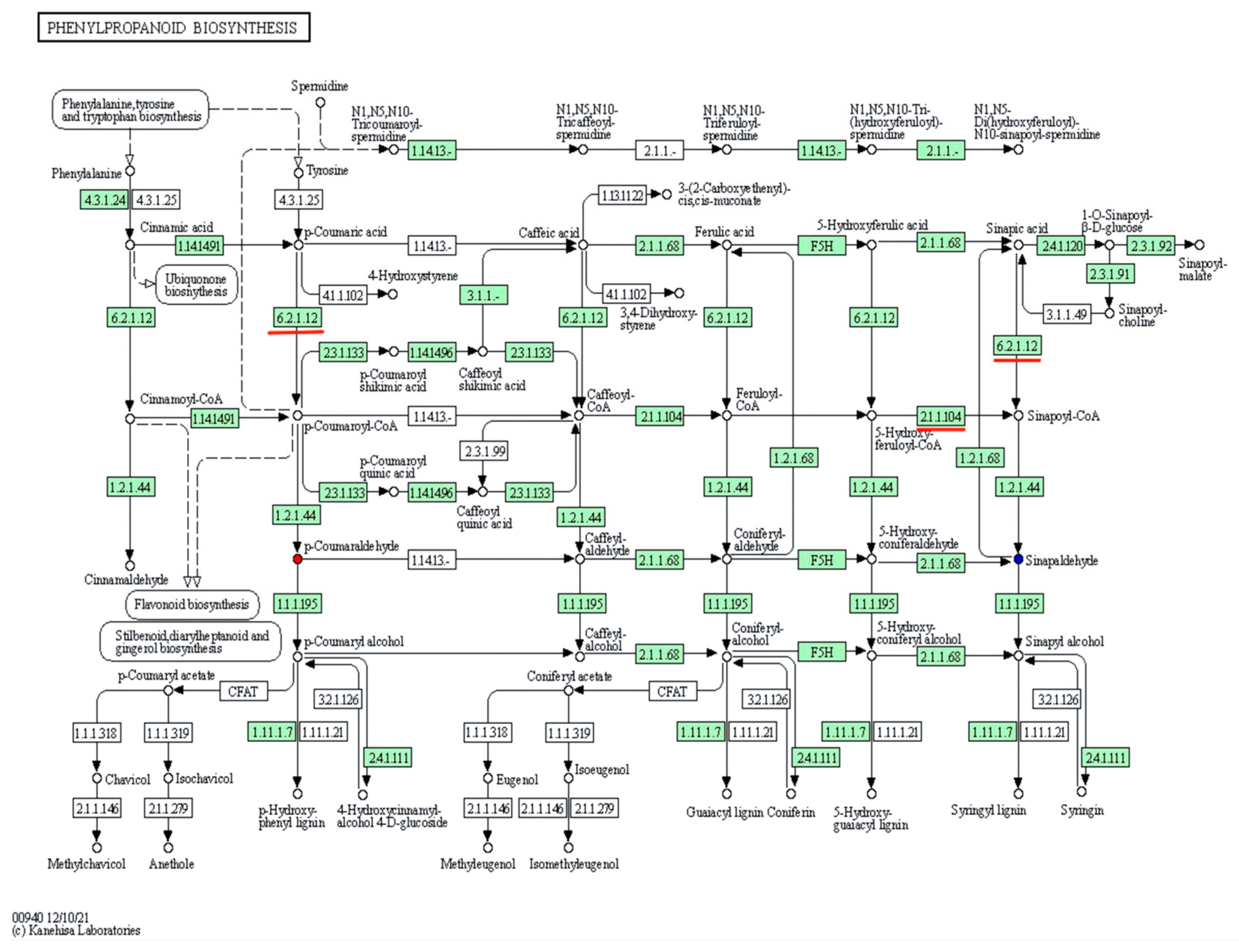
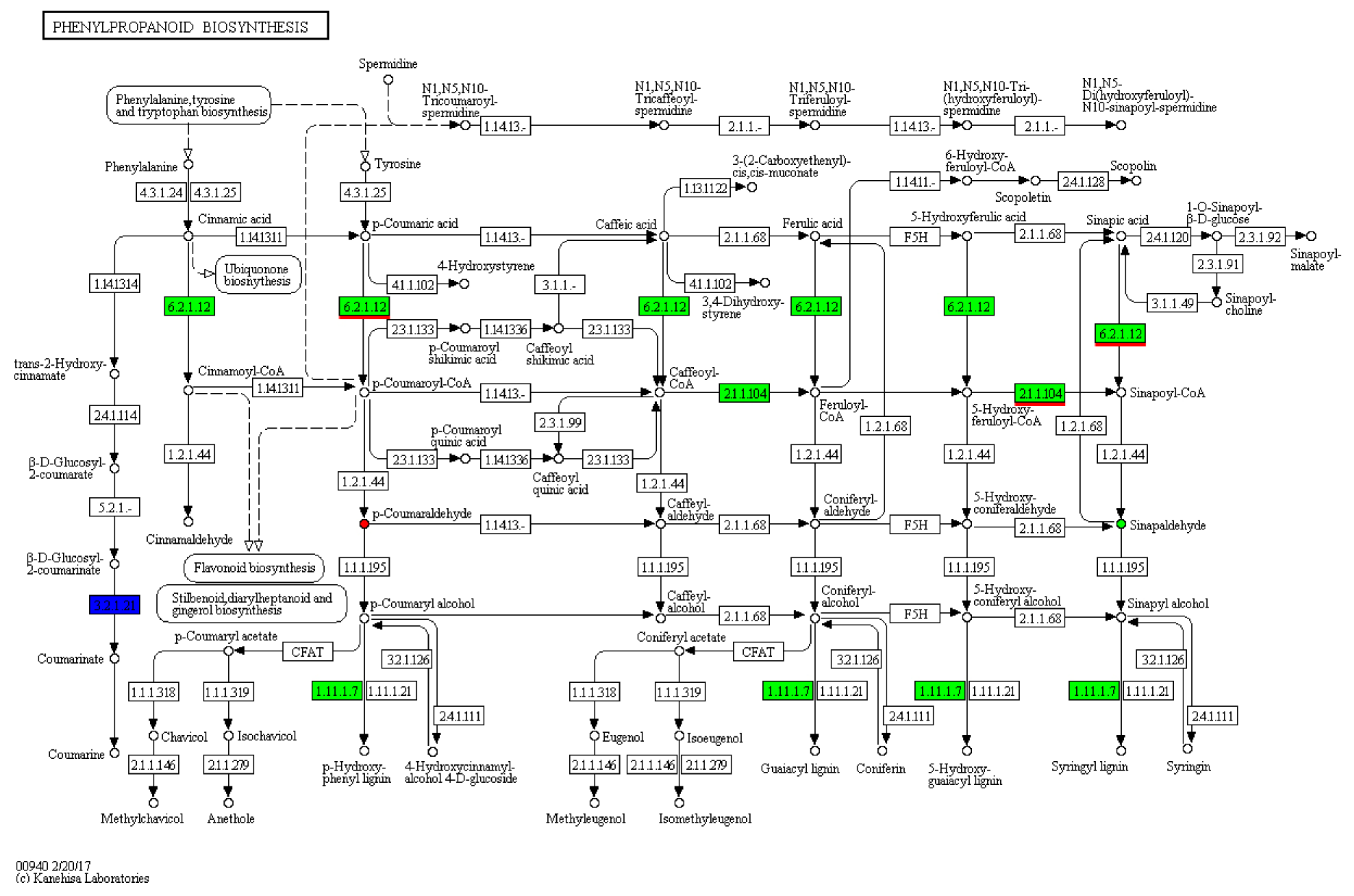
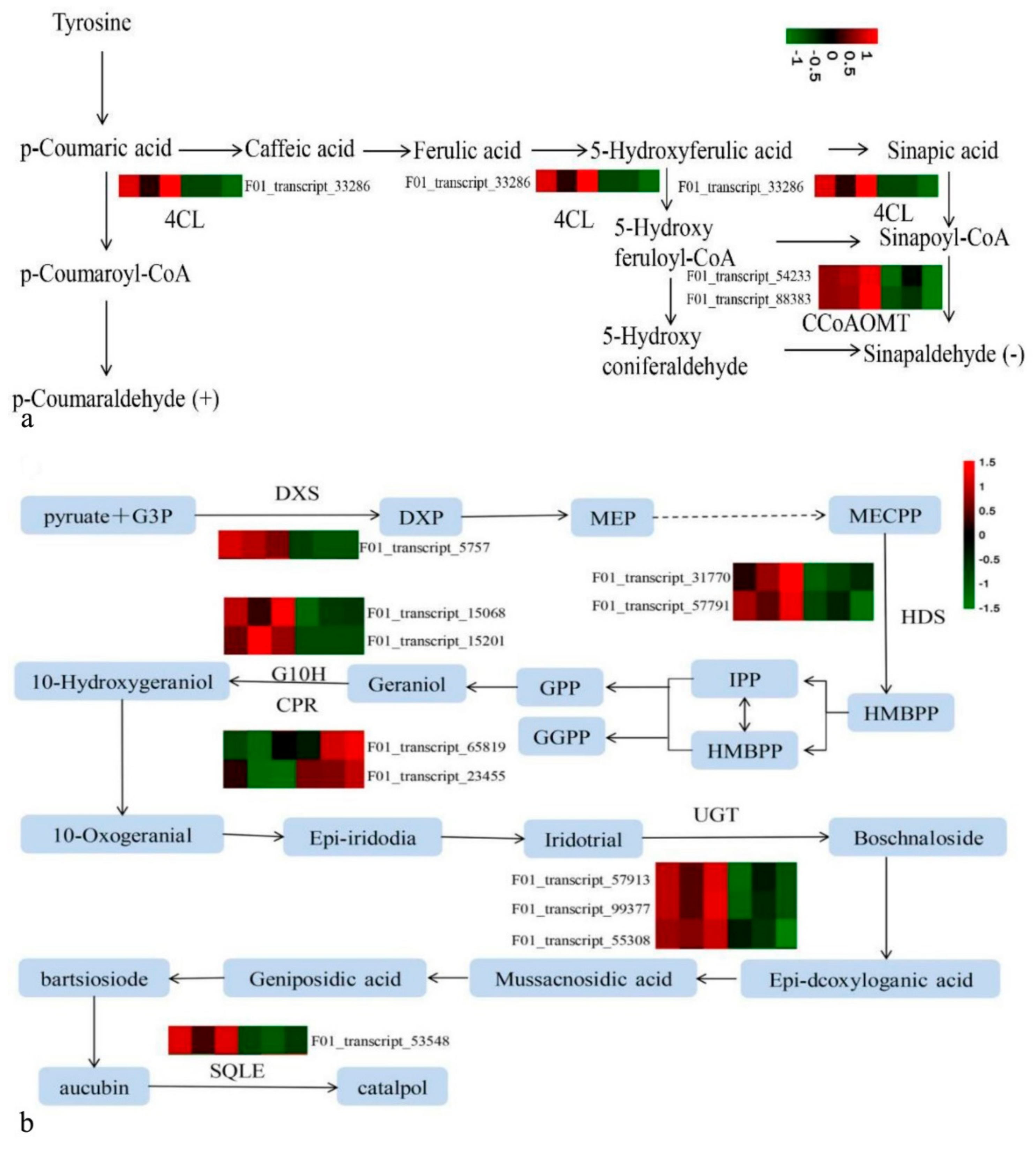
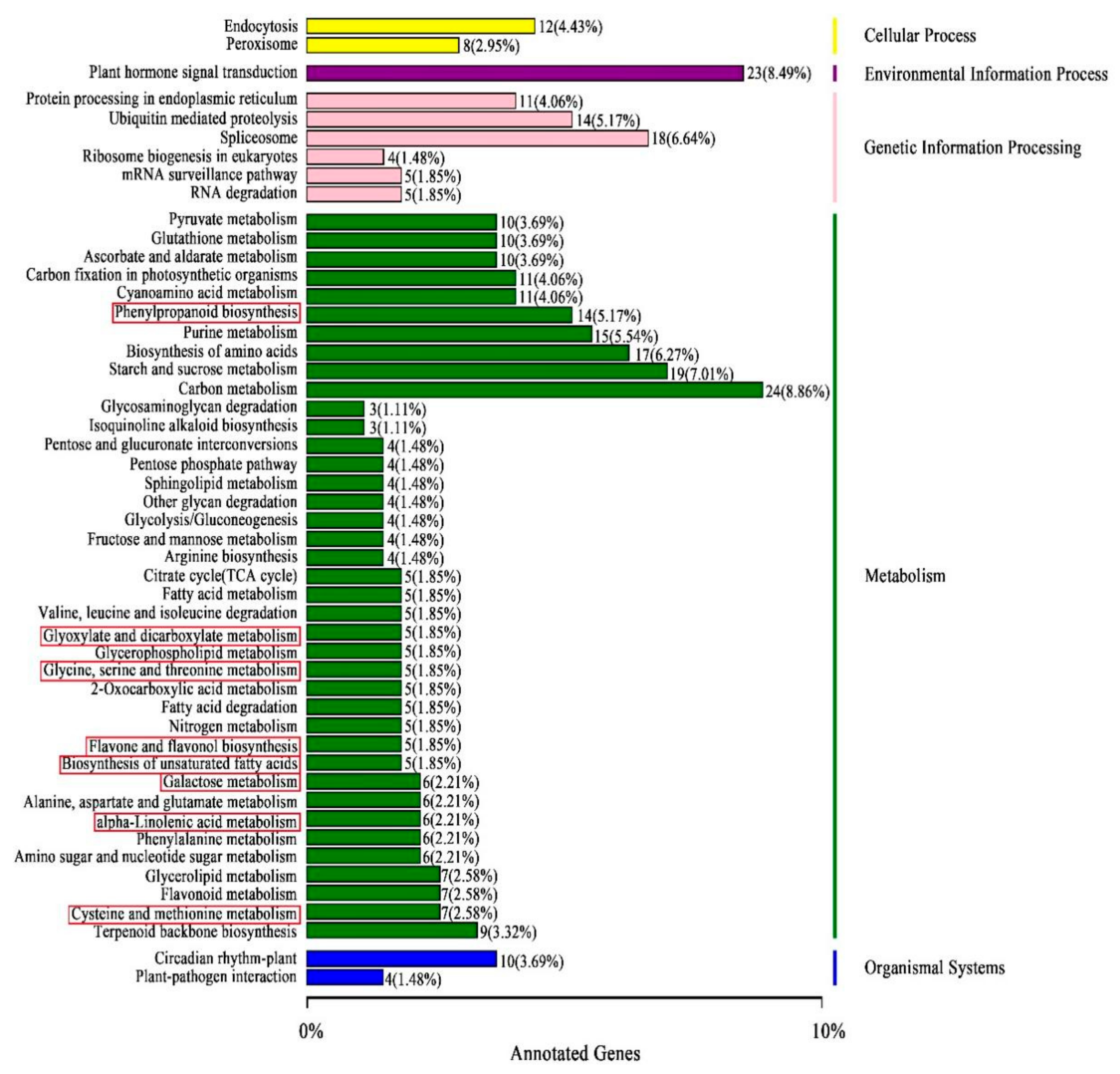
3.5. Identifying DEGs by Transcriptome Sequencing and Their Function Classifications
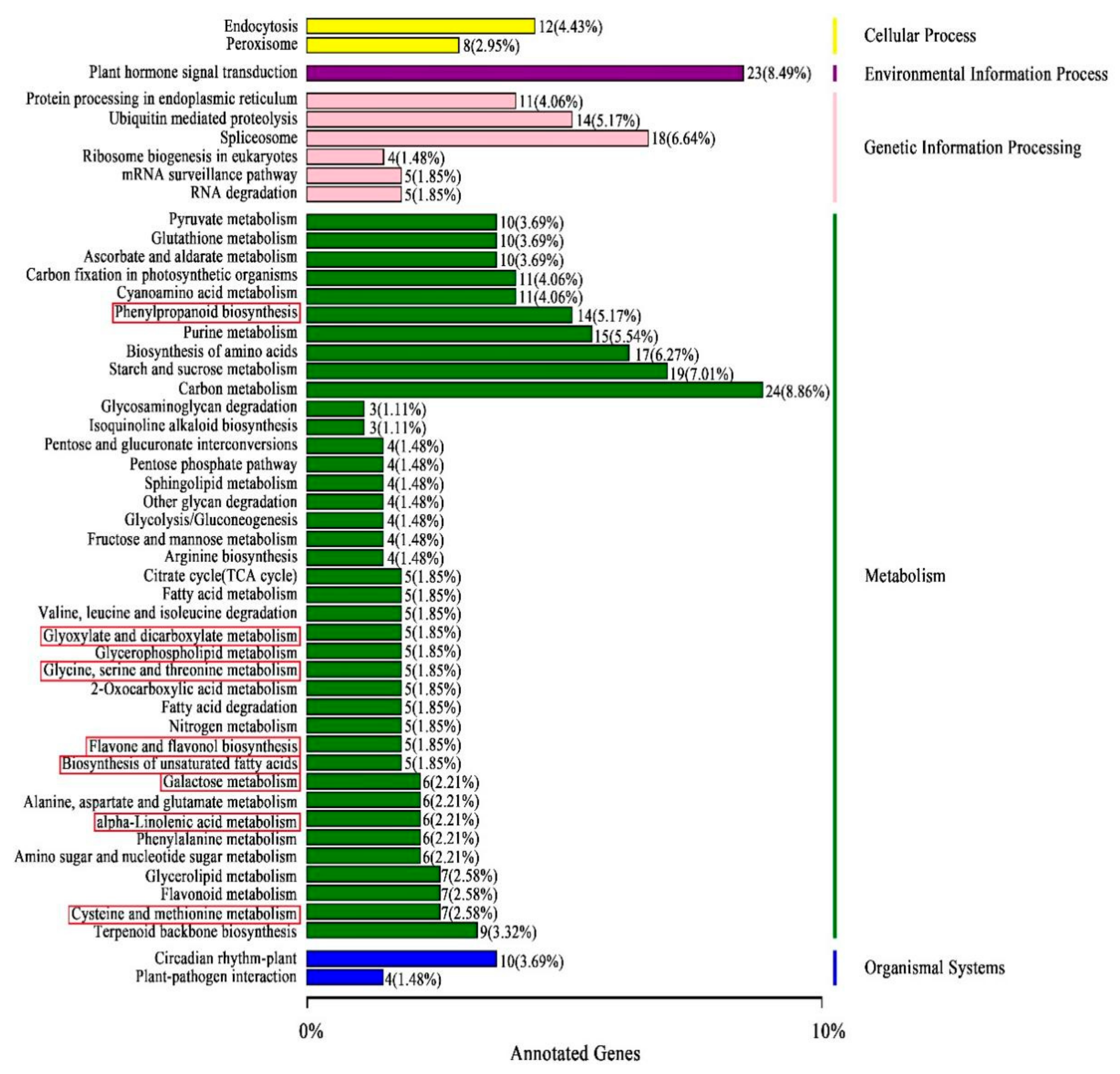
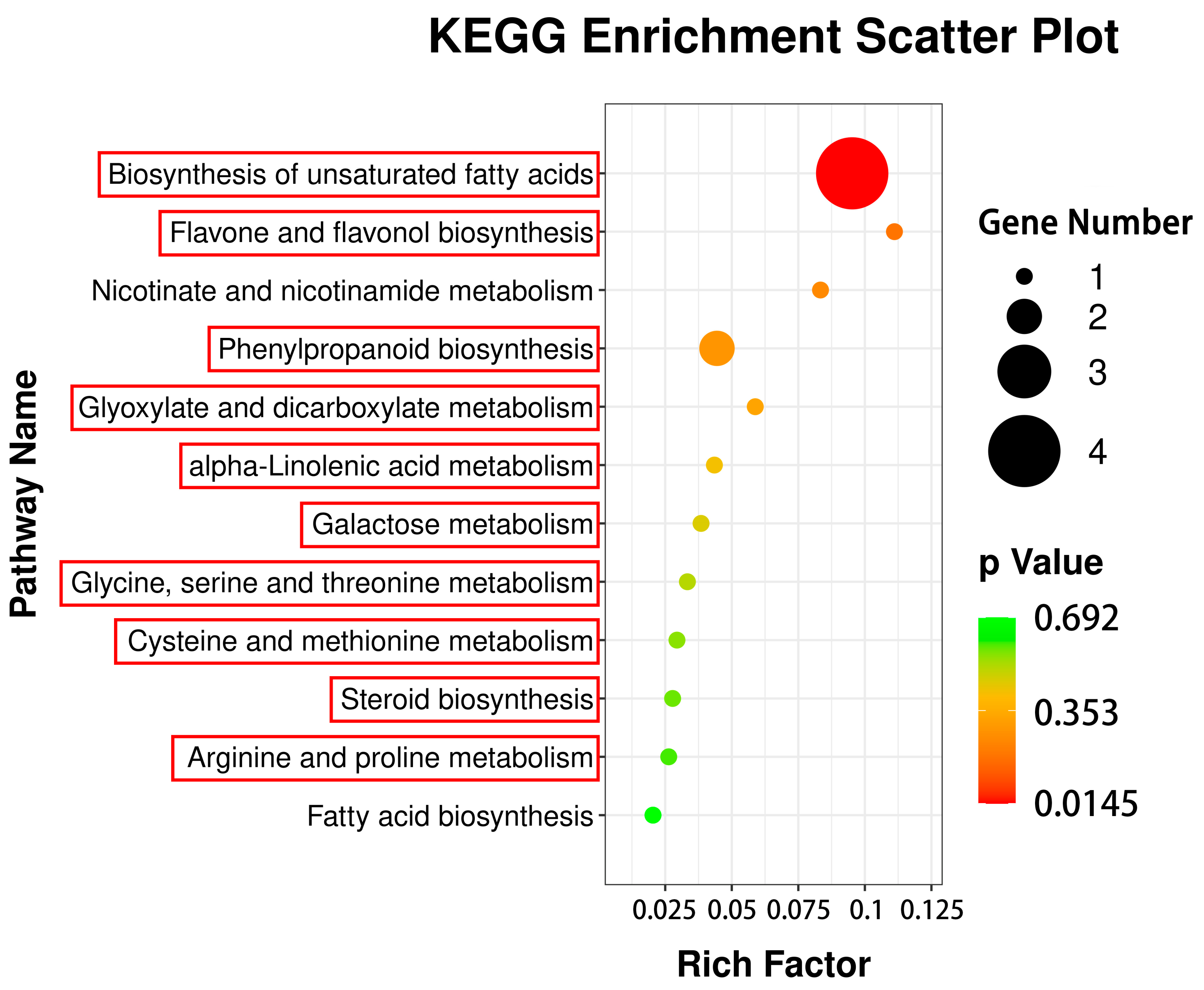
3.6. Correlation Analysis of DEGs to DAMs via Common KEGG Pathways
3.7. Regulation of These DEGs on These DAMs
3.8. Regulation of DAMs by Transcription Factors
3.9. Identification of the DEGs Associated with Yield
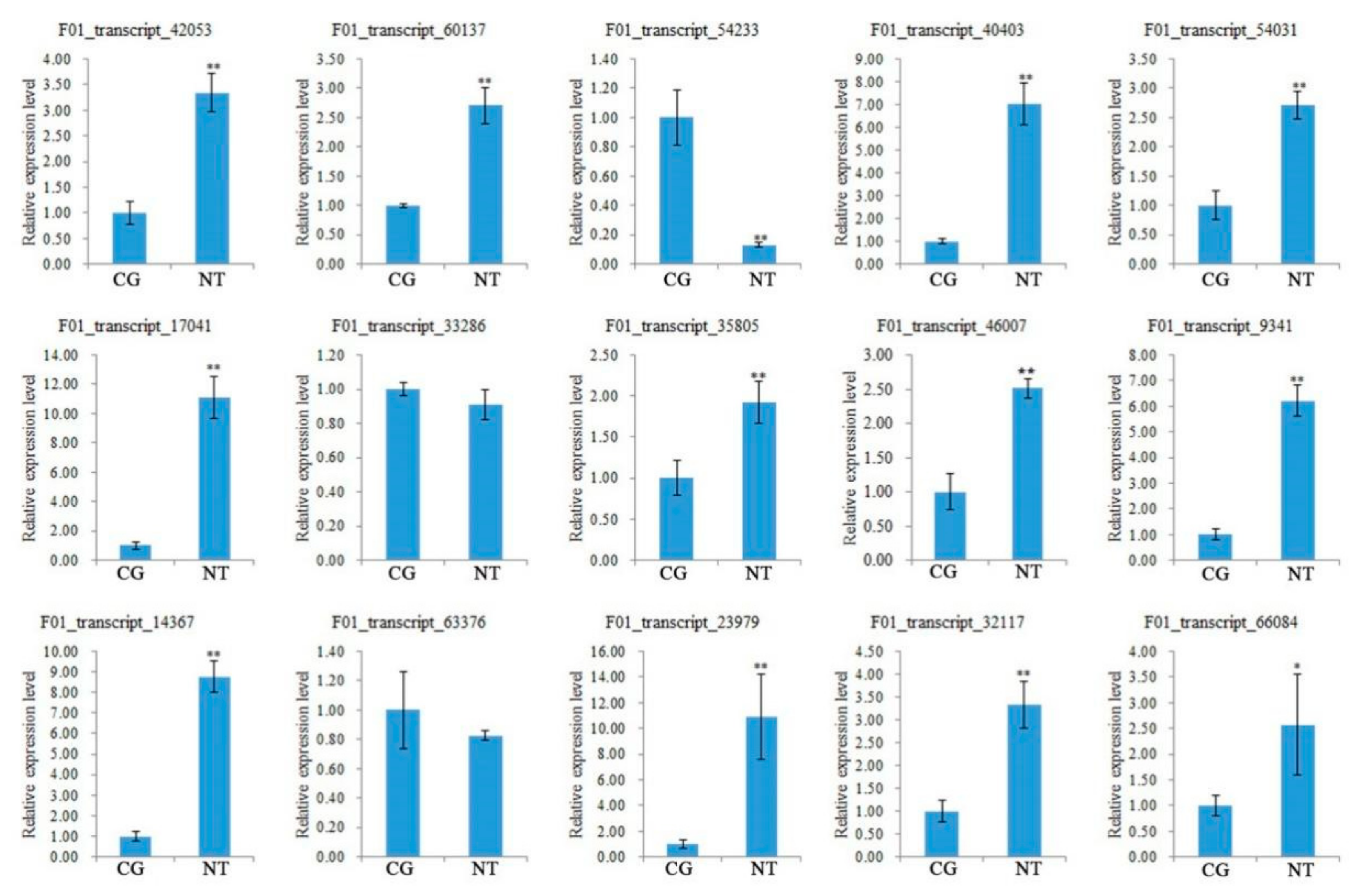
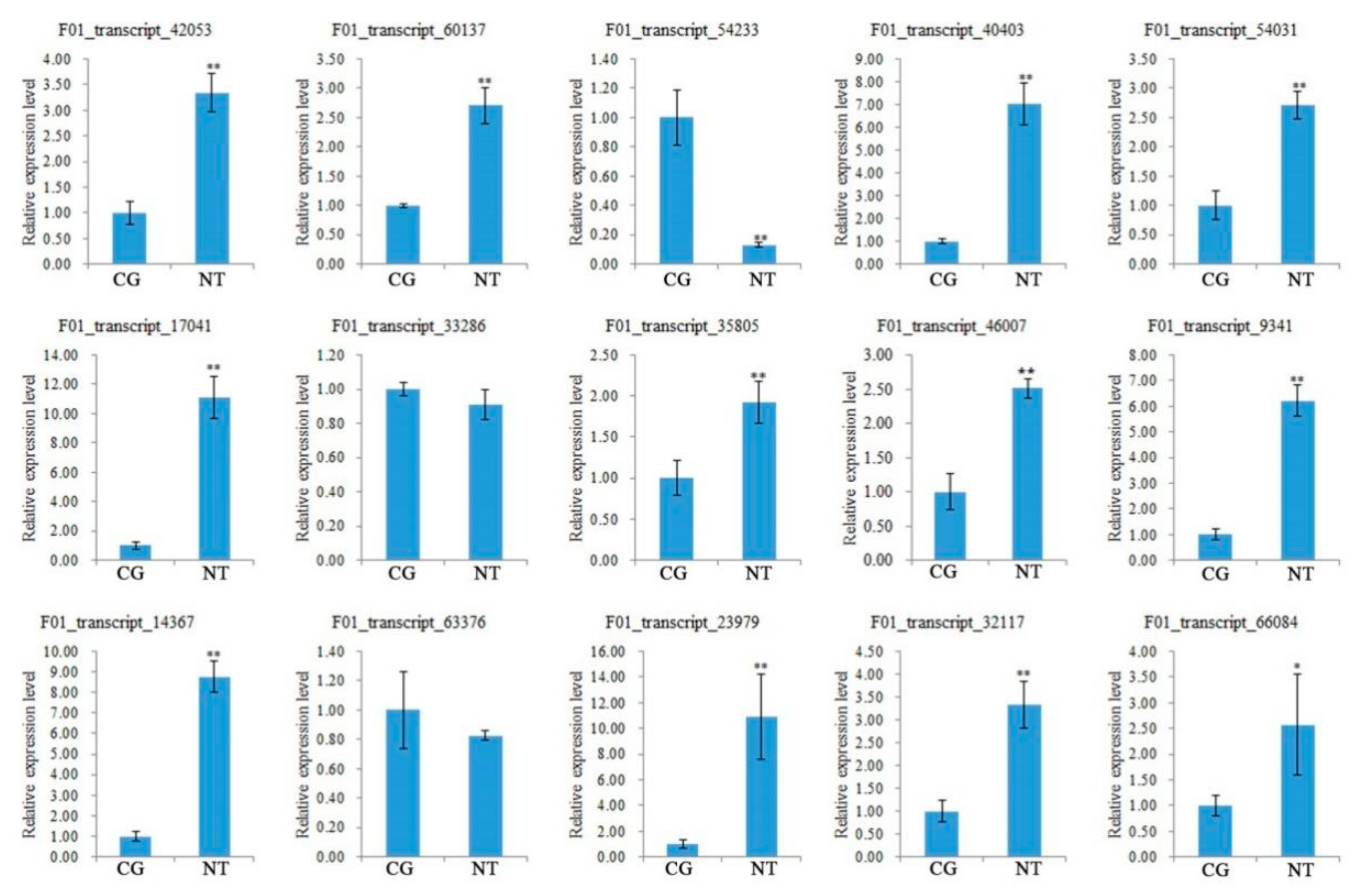
3.10. Verification of DEGs Related to Quality and Yield
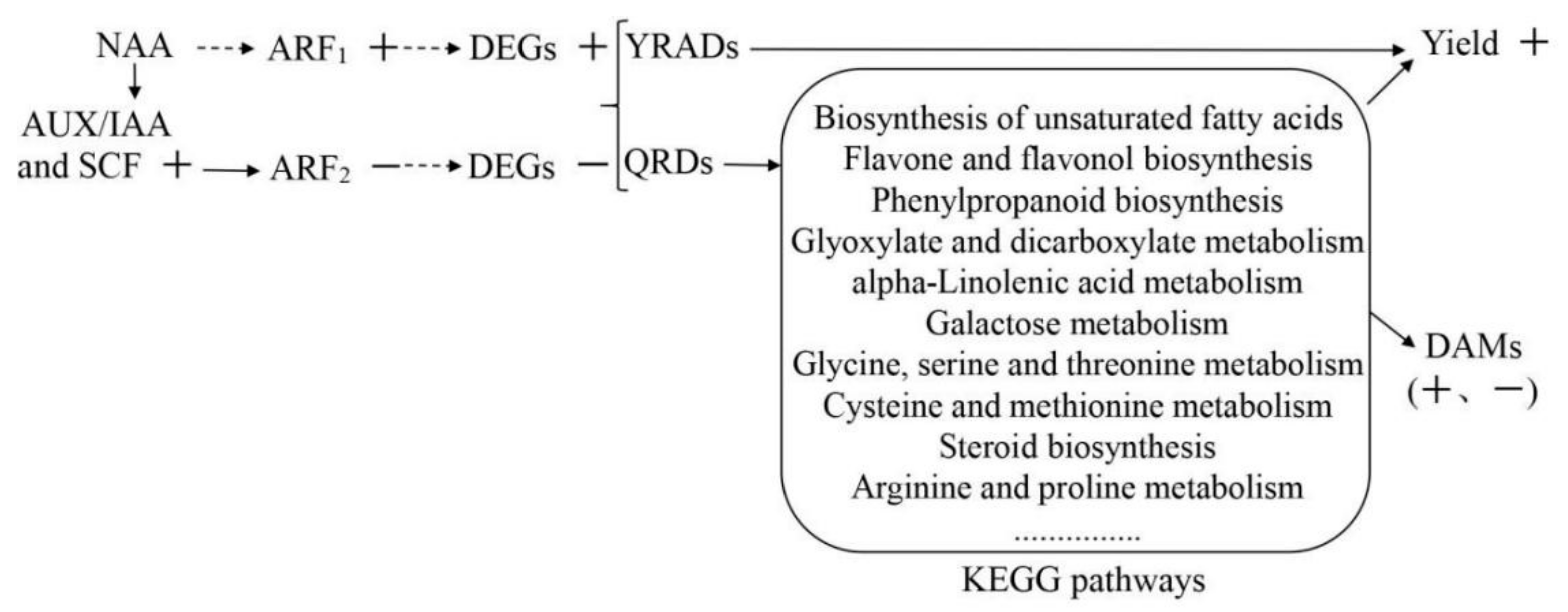
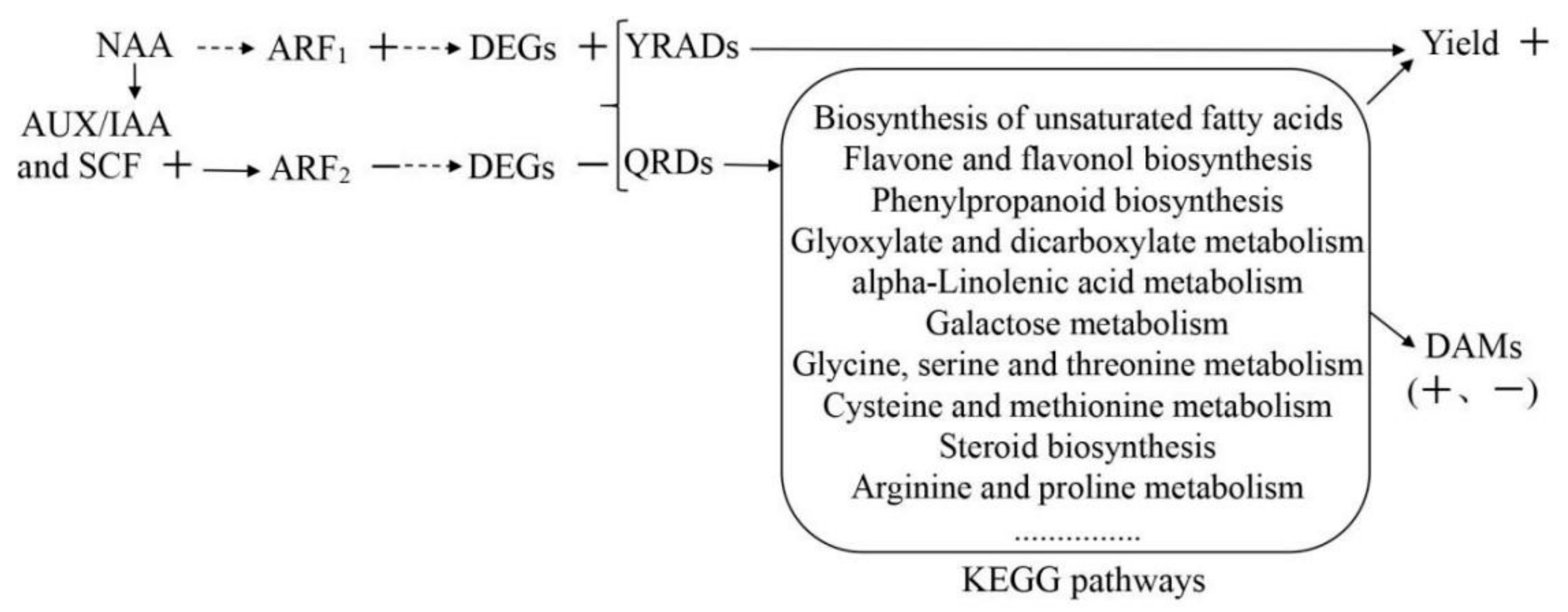
3.11. Identification of Auxin Regulatory Factors and NAA Regulatory Mechanism
4. Discussions
4.1. NAA Could Increase the Yield Related Indices of R. glutinosa Tuberous Roots
4.2. NAA Changed the Quality of R. glutinosa
4.3. Transcriptomics Analysis Validated the Effect of NAA on R. glutinosa Tuberous Root Quality
4.4. Molecular Regulatory Mechanism of NAA Effects on R. glutinosa Quality and Yield
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, Y.Q.; Yang, K.; Zhang, D.D.; Duan, H.Y.; Liu, Y.K.; Guo, M.M. Metabolite accumulation and metabolic network in developing roots of Rehmannia glutinosa Libosch reveals its root developmental mechanism and quality. Sci. Rep. 2018, 8, 14127. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.L.; Wei, X.Y.; Qi, Q.T.; Jia, W.J.; Zhao, M.W.; Wang, H.N.; Zhou, Y.Q.; Duan, H.Y. Study of terpenoid synthesis and prenyltransferase in roots of Rehmannia glutinosa based on iTRAQ quantitative proteomics. Front. Plant Sci. 2021, 12, 693758. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.Q.; Shao, L.Y.; Li, H.M.; Zhu, J.L. Full-length transcriptome analysis and the mining and elucidation of key enzymes of downstream acteoside biosynthesis pathways in Rehmannia glutinosa. J. Plant Physiol. 2022, 58, 197–206. [Google Scholar]
- Li, J.J.; Cheng, T.; Ma, J.X.; He, J.Y.; Dong, S.K. Comparative study on morphological structure and index components of different main cultivars of Rehmannia glutinosa. J. Chin. Med. Mater. 2022, 45, 21–26. [Google Scholar]
- Zhou, Y.; Shao, L.; Zhu, J.; Li, H.; Duan, H. Comparative analysis of tuberous root metabolites between cultivated and wild varieties of Rehmannia glutinosa by widely targeted metabolomics. Sci. Rep. 2021, 11, 11460. [Google Scholar] [CrossRef]
- Cance, C.; Martin-Arevalillo, R.; Boubekeur, K.; Dumas, R. Auxin response factors are keys to the many auxin doors. New Phytol. 2022. [Google Scholar] [CrossRef]
- Qiao, Z.W.; Ji, X.L.; Li, H.L.; Wang, X.; Zhang, C.L.; Wang, X.F.; You, C.X. MdARF8: An auxin response factor involved in hasmonate signaling pathway in Malus domestica. J. Plant Growth Regul. 2022, 1–12. [Google Scholar] [CrossRef]
- Hu, X.; Luo, X.Q.; Zhou, Z.S.; Wang, R.; Hu, Y.Q.; Zhang, G.M.; Hang, G.W. Multi-spectroscopic and molecular simulation approaches to characterize the intercalation binding of 1-naphthaleneacetic acid with Calf Thymus DNA. Front. Toxicol. 2021, 3, 620501. [Google Scholar] [CrossRef]
- Huang, Y. Effects of Root Application Sodium Naphthalene Acetate and Polyaspartic Acid on Growth, Physiological Characteristics and Yield of Cucumber. Master’s Thesis, Chinese Academy of Agricultural Sciences, Beijing, China, 2017. [Google Scholar]
- Sun, J.; Xiong, S.M.; Pi, X.L.; Le, M.W.; Rao, Y.L.; Yan, T.X.; Yan, X.W.; Zhou, H.Y. Effects of foliar spraying naphthylacetic acid at different periods on yield and seed quality of autumn Sesame variety Ganzhi No.13. J. South. Agric. 2018, 49, 1318–1323. [Google Scholar]
- Hussain, I.; Khakwani, A.A.; Bakhsh, I.; Khan, E.A.; Sheheryar. Effect of naphthalene acetic acid (NAA) on grain yield and bioeconomic efficiency of coarse rice (Oryza Sativa L.). Pak. J. Bot. 2021, 53, 2017–2023. [Google Scholar] [CrossRef]
- Tripathi, V.K.; Shukla, P.K. Influence of plant bio-regulators, boric acid and zinc sulphate on yield and fruit characters of Strawberry cv. Chandler. Progress. Hortic. 2010, 42, 186–188. [Google Scholar]
- An, L.; Ma, J.W.; Qin, D.M.; Wang, H.; Yuan, Y.L.; Li, H.L.; Na, R.S.; Wu, X.J. Novel strategy to decipher the regulatory mechanism of 1-naphthaleneacetic acid in strawberry maturation. J. Agric. Food Chem. 2019, 67, 129–1301. [Google Scholar] [CrossRef] [PubMed]
- Rahmat, E.; Okello, D.; Kim, H.; Lee, J.; Chung, Y.; Komakech, R.; Kang, Y. Scale-up production of Rehmannia glutinosa adventitious root biomass in bioreactors and improvement of its acteoside content by elicitation. Ind. Crop. Prod. 2021, 172, 114059. [Google Scholar] [CrossRef]
- Yang, Y.H.; Zhang, Z.Y.; Li, R.F.; Yi, Y.J.; Yang, H.; Wang, C.J.; Wang, Z.S.Q.; Liu, Y.Y. RgC3H involves in the biosynthesis of allelopathic phenolic acids and alters their release amount in Rehmannia glutinosa Roots. Plants 2020, 9, 567. [Google Scholar] [CrossRef]
- Lu, C.H.; Dong, J.; Xu, Y.X. A preliminary report on the application of expansin (CPPU) in the production of Rehmannia glutinosa Libosch. Anhui Agri. Sci. Bull. 2019, 25, 20–27. [Google Scholar]
- Ma, L.G.; Dong, C.M.; Song, C.; Wang, X.L.; Zheng, X.K.; Niu, Y.; Chen, S.L.; Feng, W.S. De novo genome assembly of the potent medicinal plant Rehmannia glutinosa using nanopore technology. Comput Struct Biotec 2021, 19, 3954–3963. [Google Scholar] [CrossRef]
- Cho, E.J.; Nguyen, Q.A.; Lee, Y.G.; Song, Y.; Park, B.J.; Bae, H.J. Enhanced biomass yield of and saccharification in transgenic tobacco over-expressing beta-glucosidase. Biomolecules 2020, 10, 806. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Liu, J.; Wang, Y.; Zhang, X.X.; Shen, Z.G.; Hu, Z.B. Ectopic expression of Vicia sativa caffeoyl-CoA O-methyltransferase (VsCCoAOMT) increases the uptake and tolerance of cadmium in Arabidopsis. Environ. Exp. Bot. 2018, 145, 47–53. [Google Scholar] [CrossRef]
- Yang, J.M.; Chen, F.; Yu, O.; Beachy, R.N. Controlled silencing of 4-coumarate: CoA ligase alters lignocellulose composition without affecting stem growth. Plant Physiol. Bioch. 2011, 49, 103–109. [Google Scholar] [CrossRef]
- Li, X.; Dong, C.M.; Qi, D.M.; Li, M.; Xing, B.; Hou, Y.W. Quality evaluation of fresh Rehmannia glutinosa Libosch from different habitats by HPLC multi-component determination and principal component analysis. Jiangsu Agric. Sci. 2020, 48, 225–229. [Google Scholar]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.G.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.Q.; Wang, X.N.; Wang, W.S.; Duan, H.Y. De novo transcriptome sequencing-based discovery and expression analyses of verbascoside biosynthesis-associated genes in Rehmannia glutinosa tuberous roots. Mol. Breed. 2016, 36, 139. [Google Scholar] [CrossRef]
- Hou, W.H.; Sun, P.; Chen, Q.J.; Li, X.N. Selection of the Reference Genes for Gene Expression Studies in Rehmanniaglutinosa by Real-time Quantitative PCR. Chin. Agric. Sci Bull. 2011, 27, 76–82. [Google Scholar]
- Ponnusamy, K.; Choi, J.N.; Kim, J.; Lee, S.Y.; Lee, C.H. Microbial community and metabolomic comparison of irritable bowel syndrome faeces. J. Med. Microbiol. 2011, 60, 817–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, J.S.; Ryan, U.; Trengove, R.D.; Maker, G.L. Development of an untargeted metabolomics method for the analysis of human faecal samples using Cryptosporidium-infected samples. Mol. Biochem. Parasitol. 2012, 185, 145–150. [Google Scholar] [CrossRef]
- Sangster, T.; Major, H.; Plumb, R.; Wilson, A.J.; Wilson, I.D. A pragmatic and readily implemented quality control strategy for HPLC-MS and GC-MS-based metabonomic analysis. Analyst 2006, 131, 1075–1078. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Chen, W.X.; Chen, H.M.; Zhong, Q.P.; Yun, Y.H.; Chen, W.J. Metabolomics analysis of the deterioration mechanism and storage time limit of tender coconut water during storage. Foods 2020, 9, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Zhang, A.H.; Song, Q.; Fang, H.; Liu, X.Y.; Su, J.; Yang, L.; Yu, M.D.; Wang, X.J. Functional metabolomics discover pentose and glucuronateinter conversion pathways as promising targets for Yang Huang syndrome treatment with Yinchenhao Tang. RSC Adv. 2018, 8, 36831–36839. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Thevenot, E.A.; Roux, A.; Xu, Y.; Ezan, E.; Junot, C. Analysis of the human adult urinary metabolome variations with age, body mass index and gender by implementing a comprehensive workflow for univariate and OPLS statistical analyses. J. Proteome Res. 2015, 14, 3322–3335. [Google Scholar] [CrossRef]
- Trygg, J.; Wold, S. Orthogonal projections to latent structures (O-PLS). J. Chemometr. 2002, 16, 119–128. [Google Scholar] [CrossRef]
- Wang, J.B.; Pu, S.B.; Sun, Y.; Li, Z.F.; Niu, M.; Yan, X.Z.; Zhao, Y.L.; Wang, L.F.; Qin, X.M.; Ma, Z.J.; et al. Metabolomic profiling of autoimmune hepatitis: The diagnostic utility of nuclear magnetic resonance spectroscopy. J. Proteome Res. 2014, 13, 3792–3801. [Google Scholar] [CrossRef] [PubMed]
- Tahir, N.I.; Rozali, N.L.; Rahmah, A.R.S.; Amiruddin, M.D.; Hwa, L.F.; Shaari, K.; Abas, F.; Othman, A.; Parveez, G.K.A.; Ramli, U.S. Metabolome Study of Oil Palm (Elaeis guineensis Jacq.) Planted in Different Environment Conditions. Trop. Plant Biol. 2022, 1–22. [Google Scholar] [CrossRef]
- Sun, P.; Xiao, X.G.; Duan, L.S.; Guo, Y.H.; Qi, J.J.; Liao, D.Q.; Zhao, C.L.; Liu, Y.; Zhou, L.L.; Li, X.E. Dynamic transcriptional profiling provides insights into tuberous root development in Rehmannia glutinosa. Front. Plant Sci. 2015, 6, 396. [Google Scholar] [CrossRef] [Green Version]
- Song, S.K.; Hofhuis, H.; Lee, M.M.; Clark, S.E. Key divisions in the early arabidopsis embryo require POL and PLL1 phosphatases to establish the root stem cell organizer and vascular axis. Del. Cell 2008, 15, 98–109. [Google Scholar] [CrossRef] [Green Version]
- Deng, Q.; Wang, X.; Zhang, D.Z.; Wang, X.M.; Feng, C.Z.; Xu, S.B. BRS1 function in facilitating lateral root emergence in Arabidopsis. Int. J. Mol. Sci. 2017, 18, 1549. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.Y.; Zhou, W.L.; Zhou, F.J.; Shi, L.J.; He, B.; Huang, C.Y. Effects of sprinkling the seedlings with allantoin on the later growth and yield of maize. Genom. Appl. Biol. 2005, 1, 38–42. [Google Scholar]
- Wang, B.; Sang, Y.L.; Song, H.; Gao, X.Q.; Zhang, X.S. Expression of a rice OsARGOS gene in Arabidopsis promotes cell division and expansion and increases organ size. J Genet. Genomics. 2009, 36, 31–40. [Google Scholar] [CrossRef]
- Qi, Y.H.; Wang, S.K.; Shen, C.J. OsARF12, a transcription activator on auxin response gene, regulates root elongation and affects iron accumulation in rice (Oryza sativa). New Phytol. 2012, 193, 109–120. [Google Scholar] [CrossRef]
- Wei, K.; Wang, L.Y.; Wu, L.Y.; Zhang, C.C.; Li, H.L.; Tan, L.Q.; Cao, H.L.; Cheng, H. Transcriptome Analysis of Indole-3-Butyric Acid-Induced Adventitious Root Formation in Nodal Cuttings of Camellia sinensis (L.). PLoS ONE 2014, 9, e107201. [Google Scholar]
- Abuqamar, S.; Ajeb, S.; Sham, A.; Enan, M.R.; Iratni, R. A mutation in the expansin-like A2 gene enhances resistance to necrotrophic fungi and hypersensitivity to abiotic stress in Arabidopsis thaliana. Mol. Plant Pathol. 2013, 14, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Lian, Q.L.; Xin, H.B.; Li, X.X.; Zhong, X.H.; Yin, Y.L.; Yi, M.F. Effect of GhLOX1gene on the corm enlargement in gladiolus hybridus. Acta Hort. Sinica 2012, 39, 1983–1990. [Google Scholar]
- Chen, L.B.; Li, S.Y.; Li, Y.; Yin, J.J.; Chen, X.H.; Li, L.J. Expression of genes related to starch synthesis during rhizome swelling of lotus root (Nelumbonucifera Gaertn). Sci. Agric. Sin. 2012, 45, 3330–3336. [Google Scholar]
- Wang, L.P.; Qin, K.J.; Zhao, L.C.; Yan, Z.G.; Huang, Z.Q.; Tu, D.P. Research progress of plant root expansion. Hubei Agric. Sci. 2020, 59, 5–9. [Google Scholar]
- Grandis, A.; Leite, D.C.C.; Tavares, E.Q.P.; Arenque-Musa, B.C.; Gaiarsa, J.W.; Martins, M.C.M.; De Souza, A.P.; Gomez, L.D.; Fabbri, C.; Mattei, B.; et al. Cell wall hydrolases act in concert during aerenchyma development in sugarcane roots. Ann. Bot. 2019, 124, 1067–1089. [Google Scholar] [CrossRef] [PubMed]
- Gui, J.S.; Shen, J.H.; Li, L.G. Functional characterization of evolutionarily divergent 4-coumarate: Coenzyme A ligases in rice. Plant Physiol. 2011, 157, 574–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.X.; Yang, J.; Zhang, M.; Fan, W.J.; Firon, N.; Pattanaik, S.; Yuan, L.; Zhang, P. Altered phenylpropanoid metabolism in the maize Lc-expressed sweet potato (Ipomoea batatas) affects storage root development. Sci. Rep. 2016, 6, 18645. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, B.; Kondhare, K.R.; Hannapel, D.J.; Banerjee, A.K. Mobile RNAs and proteins: Prospects in storage organ development of tuber and root crops. Plant Sci. 2019, 284, 73–81. [Google Scholar] [CrossRef]
- Xie, Y.; Ying, J.L.; Tang, M.J.; Wang, Y.; Xu, L.; Liu, M.; Liu, L. Genome-wide identification of AUX/IAA in radish and functional characterization of RsIAA33 gene during taproot thickening. Gene 2021, 795, 145782. [Google Scholar] [CrossRef]
- Parveen, S.; Iqbal, R.M.; Akram, M.; Iqbal, F.; Tahir, M.; Rafay, M. Improvement of growth and productivity of cotton (Gossypium hirsutum L.) through foliar applications of naphthalene acetic acid. Semin.-Cienc. Agrar. 2017, 38, 561–570. [Google Scholar] [CrossRef]
- Wang, B.; Zhou, X.; Xu, F.; Gao, J.W. Ectopic expression of a Chinese cabbage BrARGOS gene in Arabidopsis increases organ size. Transgenic Res. 2010, 19, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.H.; Wu, W.L. Effects of NAA on fruit development and quality traits of blue beery. South China Fruits 2021, 50, 147–152. [Google Scholar]
- He, L.; Ren, Z.Y.; Wang, Y.; Fu, Y.Q.; Li, Y.; Meng, N.; Pan, Q.H. Variation of growth-to-ripening time interval induced by abscisic acid and synthetic auxin affecting transcriptome and flavor compounds in cabernet sauvignon grape berry. Plants 2020, 9, 630. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Bhat, R.; Kumar, A.; Dar, K.R.; Ali, M.T. Chemical thinning improves the fruit size and quality in silver king nectarine. Indian J. Hortic. 2020, 77, 389–393. [Google Scholar] [CrossRef]
- Kaur, N.; Bons, H.K. Influence of different plant growth regulators on yield and quality of sapota (Manilkara achras) cv. Kalipatti. Indian J. Agr. Sci. 2019, 89, 842–845. [Google Scholar]
- Neil, J.S.; Matthew, K.A.; Laura, G.W.; Dayton, C.B.; Jorge, L.; Yang, X.J.; Matthew, R.T. Translating auxin responses into ovules, seeds and yield: Insight from Arabidopsis and the cereals. J. Integr. Plant Biol. 2019, 61, 310–336. [Google Scholar]
- Wang, S.X.; Shi, F.Y.; Dong, X.X.; Li, Y.X.; Zhang, Z.H.; Li, H. Genome-wide identification and expression analysis of auxin response factor (ARF) gene family in strawberry (Fragariavesca). J. IntegrAgr. 2019, 18, 1587–1603. [Google Scholar]
- Xia, X.B.; Xu, P.; Xia, R.; Ren, M. Research advances of iridoids glycosides biosynthesis in Rehmannia glutinosa. Biotechnology 2018, 28, 508–512. [Google Scholar]
- The State Food and Drug Administration; The Ministry of Agriculture and Rural Areas; The State Forestry and Herbal Administration; The State Administration of Traditional Chinese Medicine. Standard for Production and Management of Traditional to Chinese Medicine. Announcement. No. 22. 2022. Available online: https://baijiahao.baidu.com/s?id=1727526180433126717&wfr=spider&for=pc (accessed on 24 March 2022).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Root Mass Per Plant/g | Root Number Per Plant | The Longest Root Length/cm | The Thickest Root Diameter/cm | Mean Root Diameter Per Plant/cm |
|---|---|---|---|---|---|
| CK | 212.02 ± 70.35 a | 6.00 ± 1.21 b | 18.87 ± 4.57 c | 3.40 ± 0.71 d | 2.32 ± 0.17 e |
| NT | 256.84 ± 78.66 a′ | 12.00 ± 2.08 b′ | 17.58 ± 0.24 c′ | 3.20 ± 0.24 d′ | 2.52 ± 0.34 e′ |
| Sample | Catalpol/% | Rehmannioside D/% | Acteoside/% |
|---|---|---|---|
| CK | 0.7633 ± 0.0002 a | 0.2067 ± 0.0000 b | 0.0620 ± 0.0006 c |
| NT | 1.9867 ± 0.0003 a′ | 0.2436 ± 0.0000 b′ | 0.0717 ± 0.0000 c′ |
| TF Name | R1 | Enzyme Name | R2 | Pathway | DAM | R3 |
|---|---|---|---|---|---|---|
| EIN3 | up | ASP5, LIP1, CMT2, CYSD2 | up | d, h, j | ① | up |
| EIN3 | up | PED, ACX1, CYP75B2, CCoAOMT, CHI,RG, RFS *, RFS6 *, SHM2, CS, ACO, MET | down | a, b, c, d, e, f, g, i | ② | down |
| agl, CMT2, CYSD2 | up | |||||
| EIN3 | up | CYP75B2, TAT, RFS *, LPD1, GS1-1, SQE1 | down | b, d, e, f, g, h, j | ③ | up |
| ASP5, CMT2, CYSD2 | up | |||||
| EIN3 | up | ACO | down | h | ④ | down |
| EIN3 | down | PED, ACX1, CYP75B2, CcoAOMT, 4CL, TAT, unknow, RG, BGLU12 *, PER21, GOLS1, RFS *, RFS6 *, SHM2, CS, GS1-1, HD, PGDH2, SQE1, AOC3, SAMDC *, MET | down | a, b, c, d, e, f, g, h, i, j | ⑤ | up |
| BGLU1 * | up | |||||
| EIN3 | down | PED, ACX1, CYP75B2, CCoAOMT *, CHI, TAT, RG, BGLU12 *, RFS *, RFS6 *, SHM2, CS, ACO, HD, PGDH2, MET | down | a, b, c, d, e, f, g, i, j | ⑥ | down |
| ASP5, CMT2, CYSD2 | up | |||||
| EIN3 | down | PED, ACX1, CYP75B2, CcoAOMT *, CHI, 4CL *, unknow, RG, BGLU12 *, PER21, GOLS1, RFS *, RFS6 *, SHM2, CS, ACO, HD, PGDH2, SQE1, AOC3, SAMDC *, MET | down | a, b, c, d, e, f, g, h, i, j | ⑦ | down |
| CYSD2 | up | |||||
| BZR1 | up | ASP5, LIP1, CMT2, CYSD2 | up | d, h, j | ① | up |
| BZR1 | up | ACO | down | h | ④ | down |
| BZR1 | up | PED, ACX1, CYP75B2, CCoAOMT *, CHI, RG, RFS *, RFS6 *, SHM2, CS, ACO, MET | down | a, b, c, d, e, f, g, i | ② | down |
| agl, CMT2, CYSD2 | up | |||||
| BZR1 | up | CYP75B2, TAT, RFS *, LPD1, GS1-1, SQE1 | down | b, d, e, f, g, h, j | ③ | up |
| ASP5, CMT2, CYSD2 | up | |||||
| Aux/IAA | up | ASP5, LIP1, CMT2, CYSD2 | up | d, h, j | ① | up |
| Aux/IAA | up | CYP75B2, TAT, RFS *, LPD1, GS1-1, SQE1 | down | b, d, e, f, g, h, j | ③ | up |
| ASP5, CMT2, CYSD2 | up | |||||
| ASP5, CMT2, CYSD2 | up |
| Gene Description | Gene ID | Yield Related Factors | References |
|---|---|---|---|
| beta-glucosidase | F01_transcript_45042, F01_transcript_14367, F01_transcript_57187, F01_transcript_57503, F01_transcript_79209, F01_transcript_83451, F01_transcript_86382, F01_transcript_88550, F01_transcript_45952, F01_transcript_48883 | biomass | [18] |
| 4-coumarate--CoA ligase | F01_transcript_33286 | length | [20] |
| caffeoyl-CoA O-methyltransferase | F01_transcript_54233, F01_transcript_88383 | biomass | [19] |
| protein phosphatase 2C | F01_transcript_10164, F01_transcript_60137, F01_transcript_40403 | branching root numbers | [36] |
| brassinosteroid resistant 1/2 | F01_transcript_54031, F01_transcript_17041, F01_transcript_16804 | branching root numbers | [37] |
| auxin responsive GH3 gene family | F01_transcript_99345 | branching root numbers | [41] |
| Auxin-regulated gene involved in organ size | F01_transcript_35805, F01_transcript_46007 | width (thickening) | [39] |
| Expansin-like A1 | F01_transcript_22738 | width (thickening) | [42] |
| Lipoxygenase | F01_transcript_63376, F01_transcript_80652 | width (thickening) | [43] |
| MADS-box | F01_transcript_48636 | width (thickening) | [44] |
| Allantoinase | F01_transcript_32117, F01_transcript_15391, F01_transcript_66084, F01_transcript_14633, F01_transcript_48348, F01_transcript_98250 | branching root numbers | [38] |
| Probable starch synthase 4 | F01_transcript_2028 | biomass | [35] |
| Galactinol-sucrose galactosyltransferase | F01_transcript_5582, F01_transcript_97134, F01_transcript_6840 | biomass | [35] |
| Auxin response factor | F01_transcript_46036, F01_transcript_79445 | length | [40] |
| ID | Gene | Regulation | CG-Mean (FPKM) | NT-Mean (FPKM) |
|---|---|---|---|---|
| F01_transcript_4608 | TIR1 | normal | 9.54 | 9.85 |
| F01_transcript_5273 | normal | 0.13 | 0.18 | |
| F01_transcript_5421 | normal | 10.59 | 9.12 | |
| F01_transcript_83028 | normal | 7.32 | 6.19 | |
| F01_transcript_86411 | normal | 0.42 | 0.46 | |
| F01_transcript_89970 | normal | 12.59 | 10.31 | |
| F01_transcript_73082 | normal | 7.56 | 6.82 | |
| F01_transcript_18581 | SCF | up | 14.96 | 23.54 |
| F01_transcript_19200 | up | 7.27 | 19.75 | |
| F01_transcript_23979 | up | 86.73 | 176.44 | |
| F01_transcript_41747 | up | 17.40 | 29.12 | |
| F01_transcript_47554 | up | 2.86 | 5.73 | |
| F01_transcript_49230 | up | 12.32 | 26.83 | |
| F01_transcript_4394 | up | 10.93 | 17.55 | |
| F01_transcript_4877 | up | 9.03 | 15.48 | |
| F01_transcript_82073 | up | 1.95 | 4.66 | |
| F01_transcript_83142 | up | 2.59 | 5.14 | |
| F01_transcript_83618 | up | 0.79 | 2.11 | |
| F01_transcript_91955 | up | 12.04 | 18.84 | |
| F01_transcript_79445 | ARF1 | up | 1.41 | 3.73 |
| F01_transcript_46036 | ARF2 | down | 7.73 | 2.64 |
| F01_transcript_21517 | AUX/IAA | up | 14.40 | 26.55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Zhu, J.; Li, H.; Ma, J.; Chen, P.; Zhou, Y. The Effects of NAA on the Tuberous Root Yield and Quality of Rehmannia glutinosa and Its Regulatory Mechanism by Transcriptome and Metabolome Profiling. Curr. Issues Mol. Biol. 2022, 44, 3291-3311. https://doi.org/10.3390/cimb44080227
Li J, Zhu J, Li H, Ma J, Chen P, Zhou Y. The Effects of NAA on the Tuberous Root Yield and Quality of Rehmannia glutinosa and Its Regulatory Mechanism by Transcriptome and Metabolome Profiling. Current Issues in Molecular Biology. 2022; 44(8):3291-3311. https://doi.org/10.3390/cimb44080227
Chicago/Turabian StyleLi, Jianjun, Jialin Zhu, Huimin Li, Jingxiao Ma, Peilei Chen, and Yanqing Zhou. 2022. "The Effects of NAA on the Tuberous Root Yield and Quality of Rehmannia glutinosa and Its Regulatory Mechanism by Transcriptome and Metabolome Profiling" Current Issues in Molecular Biology 44, no. 8: 3291-3311. https://doi.org/10.3390/cimb44080227
APA StyleLi, J., Zhu, J., Li, H., Ma, J., Chen, P., & Zhou, Y. (2022). The Effects of NAA on the Tuberous Root Yield and Quality of Rehmannia glutinosa and Its Regulatory Mechanism by Transcriptome and Metabolome Profiling. Current Issues in Molecular Biology, 44(8), 3291-3311. https://doi.org/10.3390/cimb44080227