
Expression of GOT2 Is Epigenetically Regulated by DNA Methylation and Correlates with Immune Infiltrates in Clear-Cell Renal Cell Carcinoma



Abstract
:1. Introduction
2. Materials and Methods
2.1. Differential Expression of GOT2 mRNA and Protein
2.2. Clinical Correlations & Survival Analysis
2.3. GOT2 Methylation Analysis
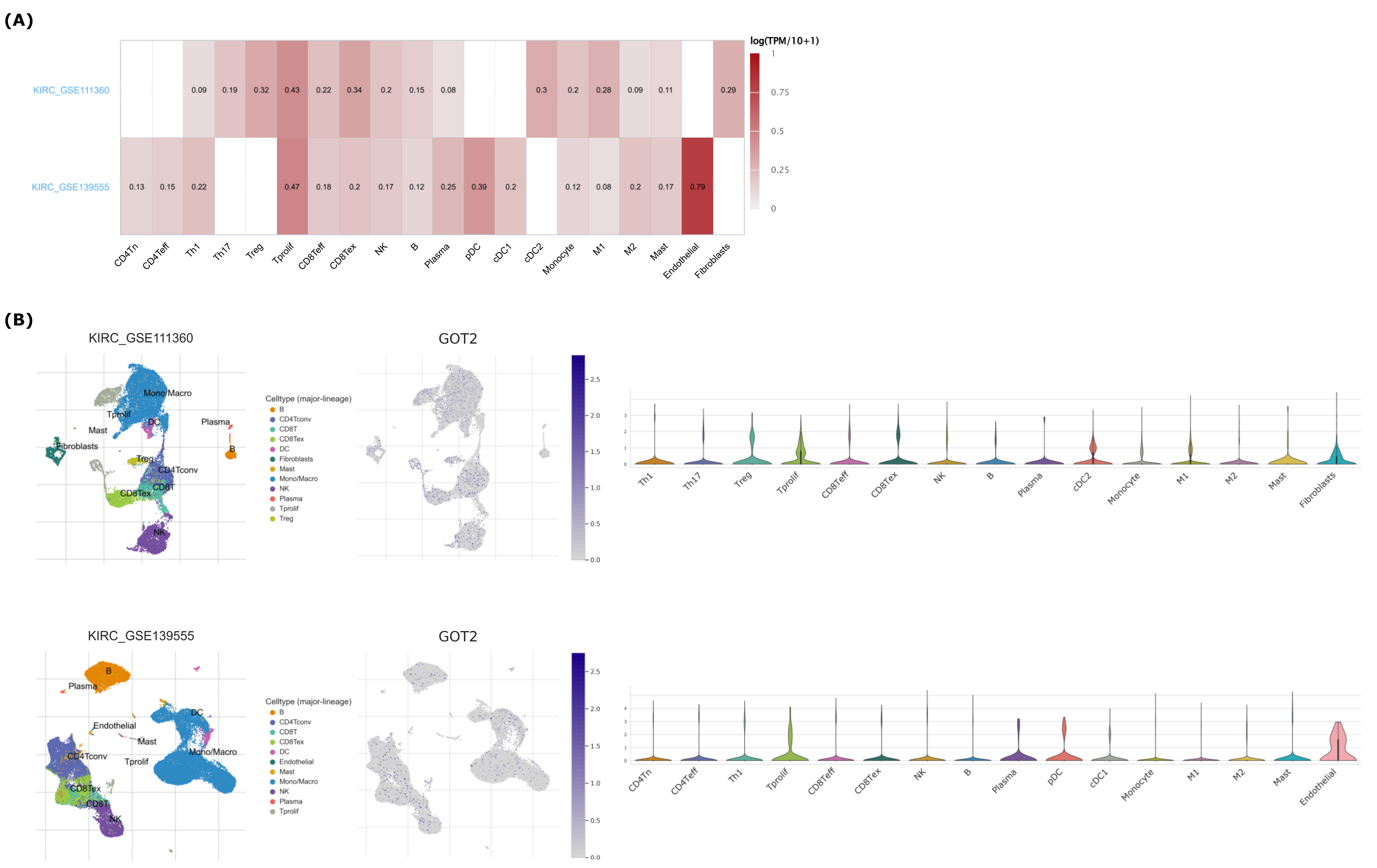
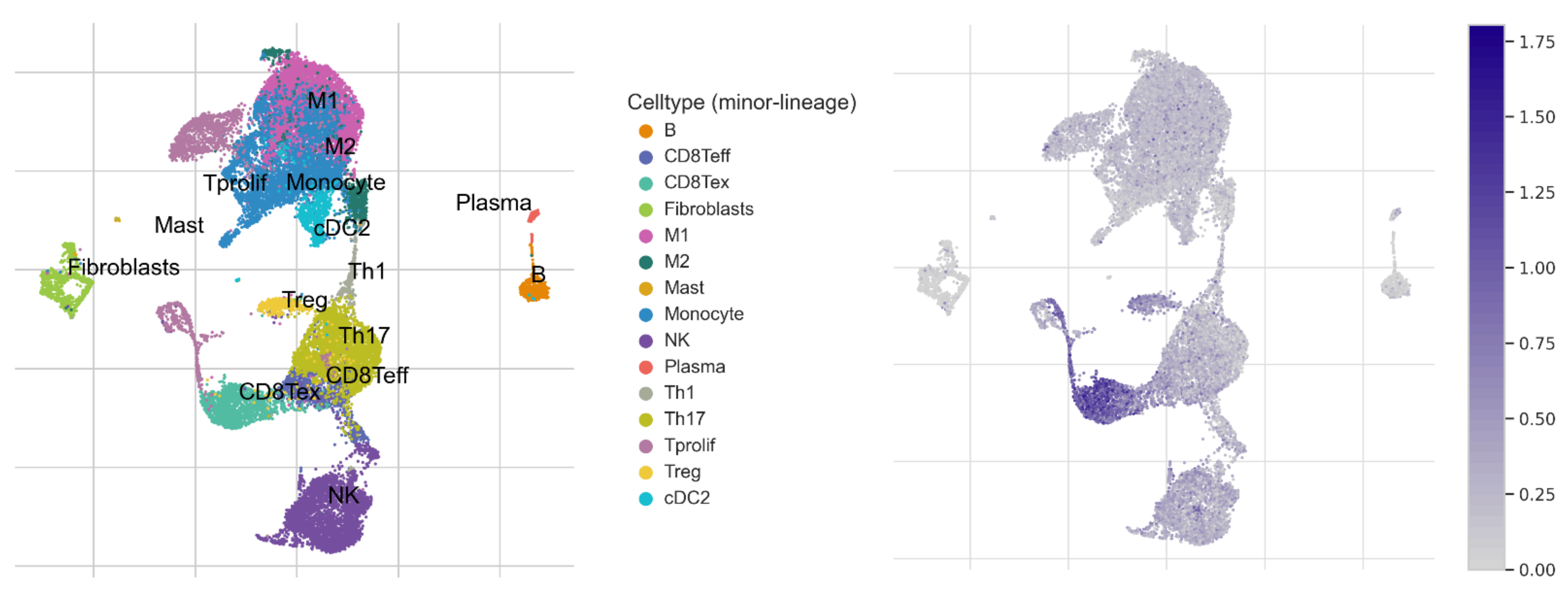
2.4. Analysis of Immune Cell Infiltration
2.5. Association between GOT2 and Tumor Microenvironment Exploration
3. Results
3.1. Expression Level of GOT2 mRNA in Pan-Cancer
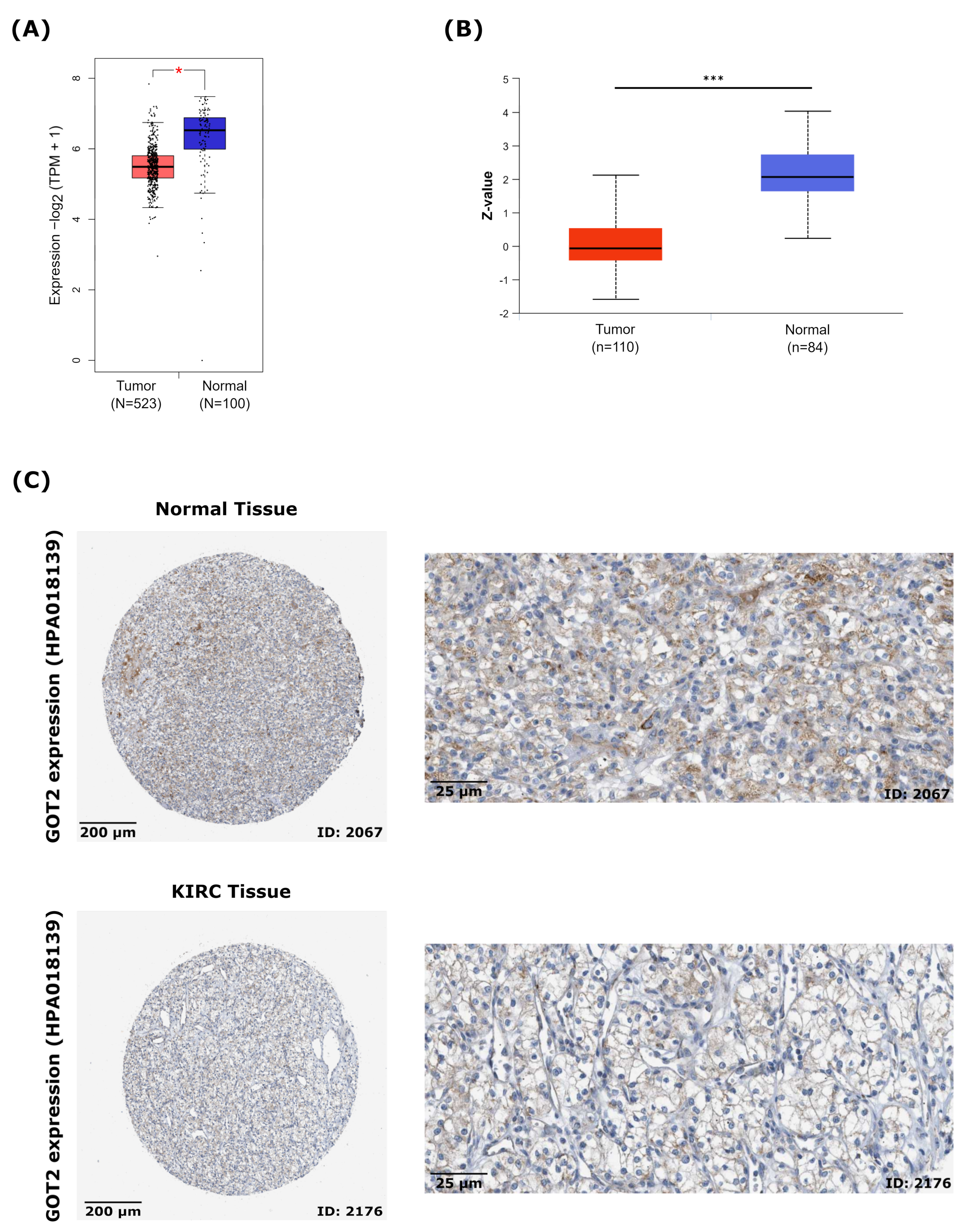
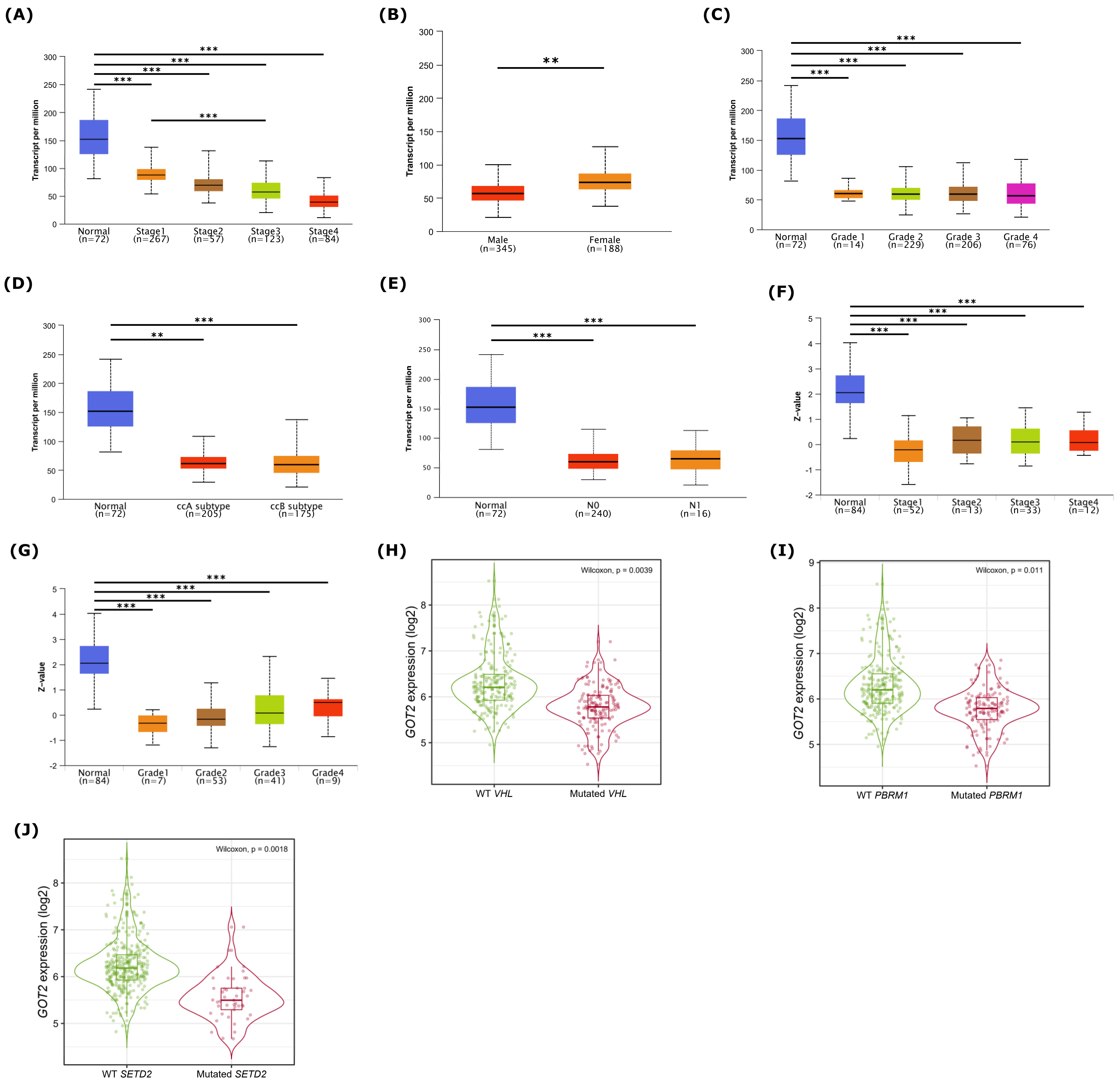
3.2. GOT2 mRNA and Protein Are Downregulated and Correlated with Clinicopathological Parameters in KIRC
3.3. Relationship between GOT2 Expression and Clinical Pathological Parameters of Patients with KIRC
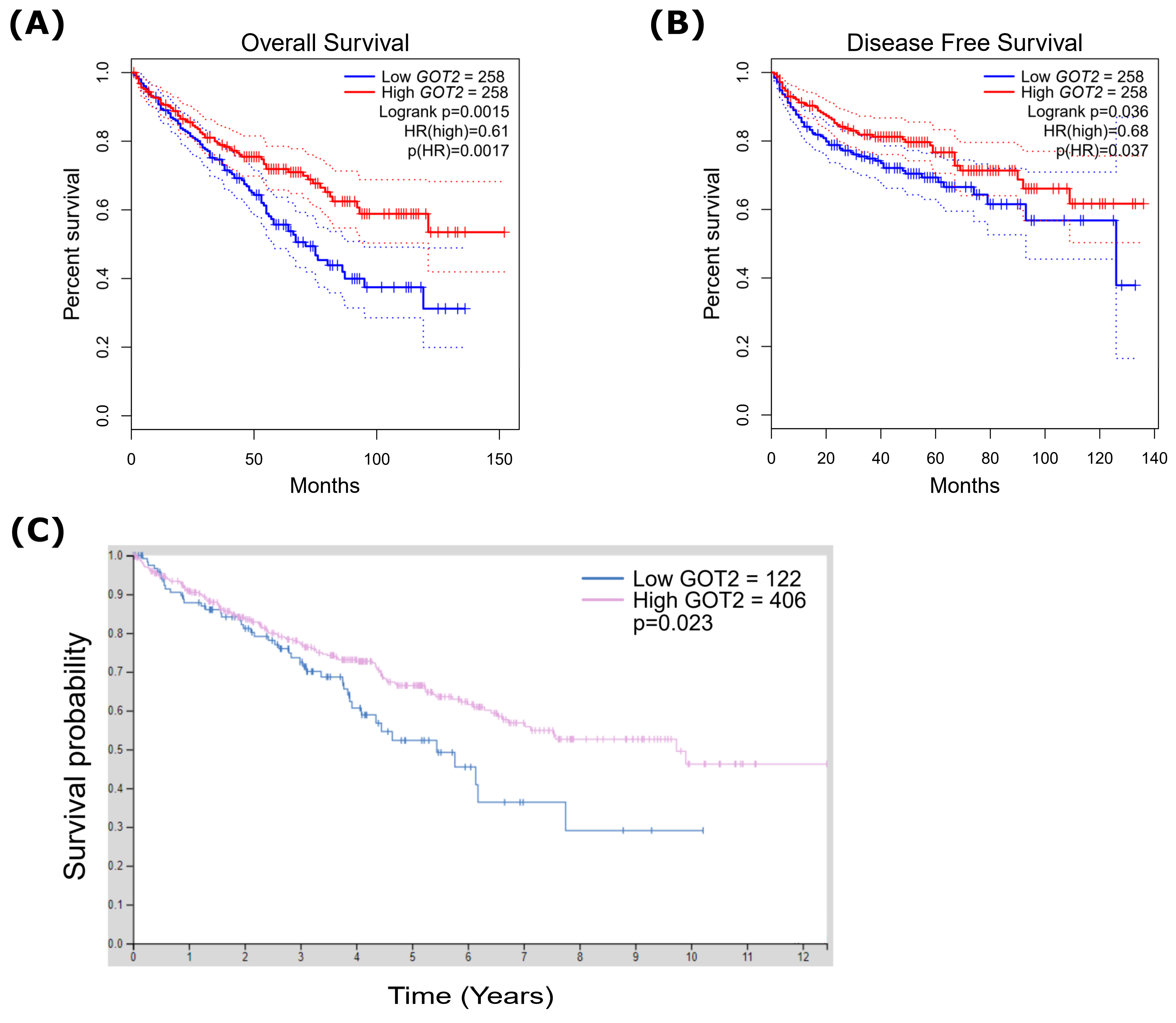
3.4. Low Expression of GOT2 Is Associated with Poor Outcome in KIRC Patients
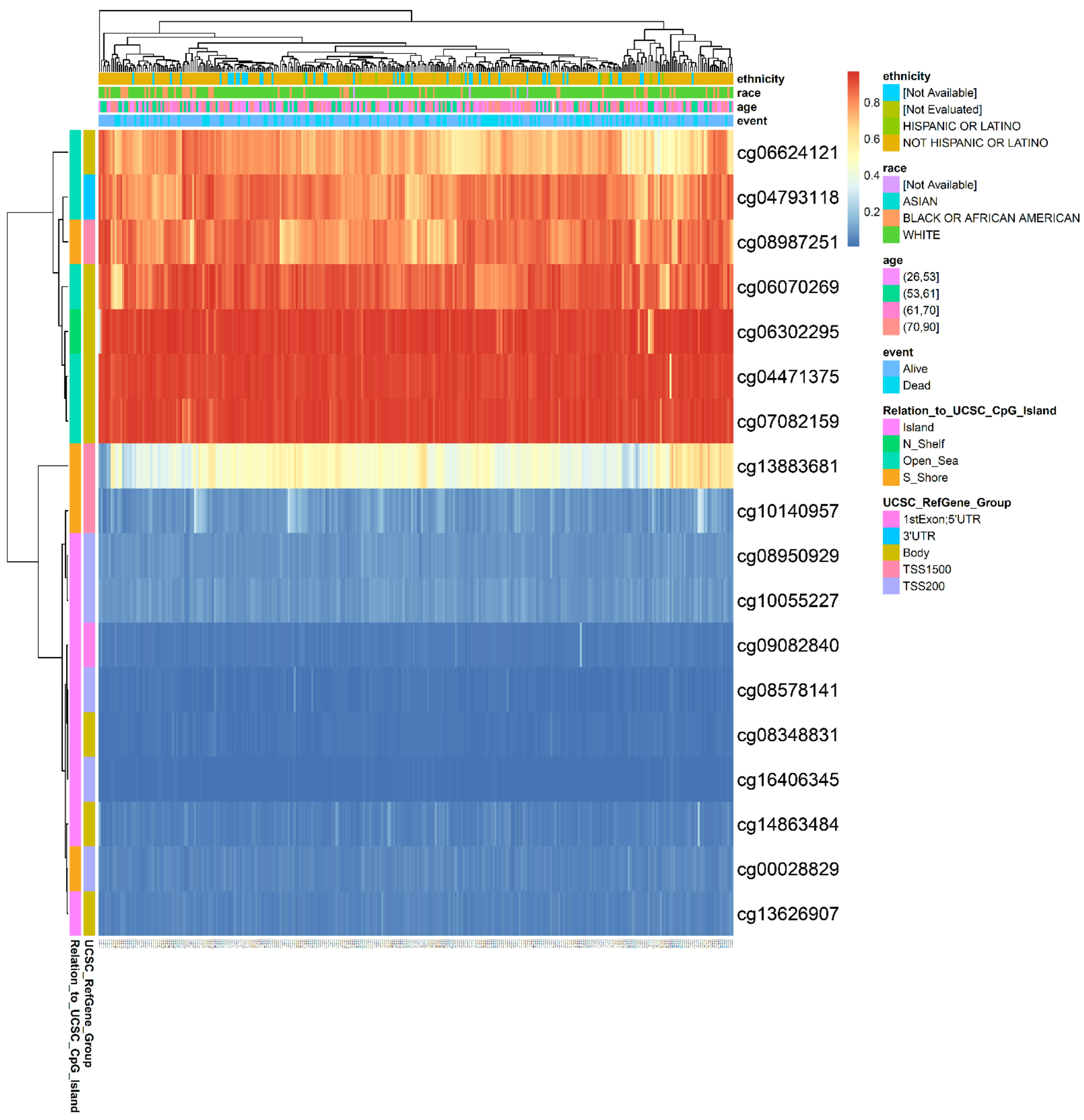
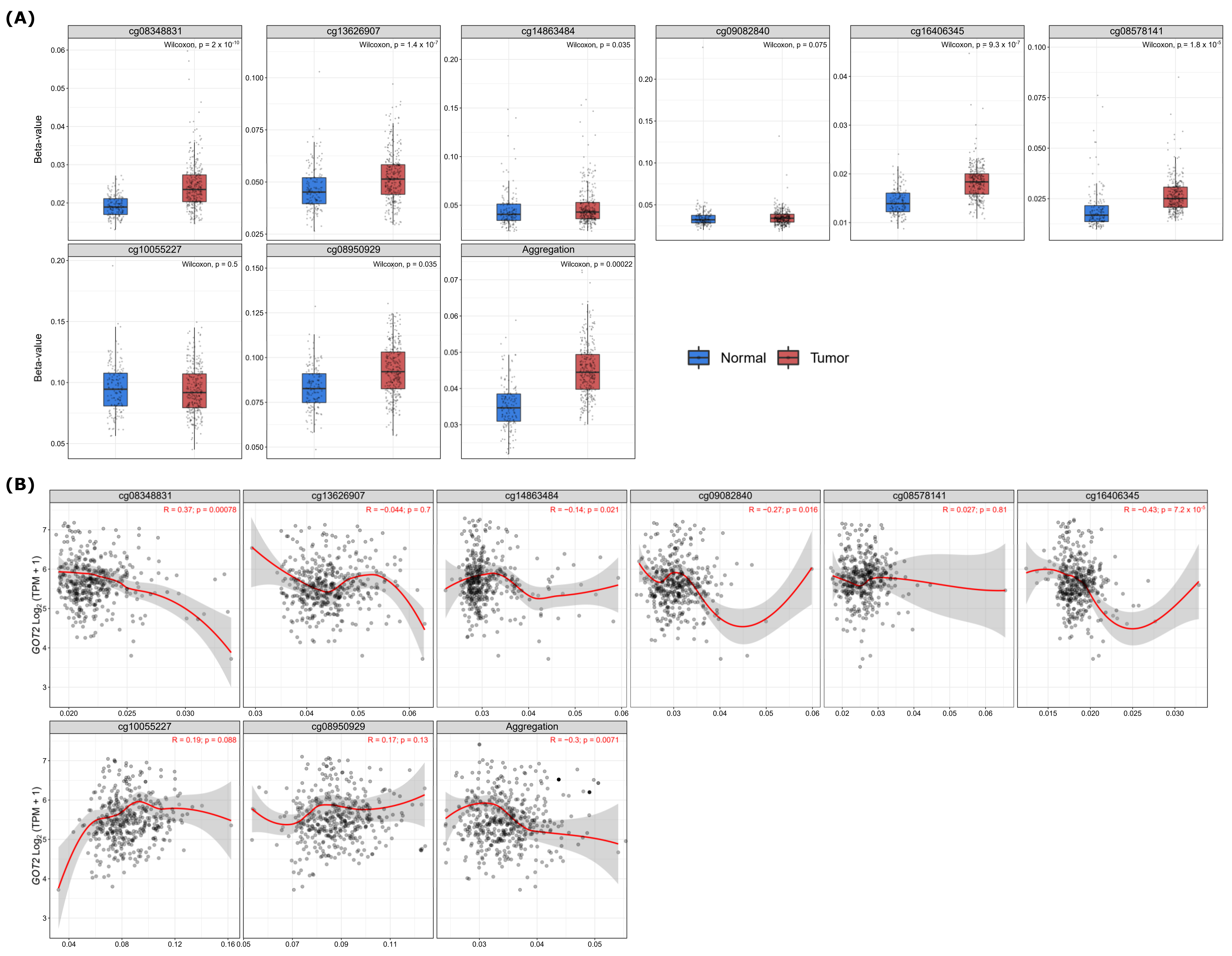
3.5. Hypermethylation of DNA in the Promoter Region Leads to Low Expression of GOT2 in KIRC
3.6. GOT2 Expression Correlates with Immune Cell Infiltration in KIRC
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Beltran, A.; Carrasco, J.C.; Cheng, L.; Scarpelli, M.; Kirkali, Z.; Montironi, R. 2009 update on the classification of renal epithelial tumors in adults. Int. J. Urol. 2009, 16, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Campbell, S.C.; Escudier, B. Renal cell carcinoma. Lancet 2009, 373, 1119–1132. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Gerlinger, M.; Horswell, S.; Larkin, J.; Rowan, A.J.; Salm, M.; Varela, I.; Fisher, R.; McGranahan, N.; Matthews, N.; Santos, C.R.; et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat. Genet. 2014, 46, 225–233. [Google Scholar] [CrossRef]
- Sankin, A.; Hakimi, A.A.; Mikkilineni, N.; Ostrovnaya, I.; Silk, M.T.; Liang, Y.; Mano, R.; Chevinsky, M.; Motzer, R.; Solomon, S.B.; et al. The impact of genetic heterogeneity on biomarker development in kidney cancer assessed by multiregional sampling. Cancer Med. 2014, 3, 1485–1492. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.J.; Chen, D.; Wang, P.I.; Marker, M.; Redzematovic, A.; Chen, Y.-B.; Selcuklu, S.D.; Weinhold, N.; Bouvier, N.; Huberman, K.H.; et al. Genomic Biomarkers of a Randomized Trial Comparing First-line Everolimus and Sunitinib in Patients with Metastatic Renal Cell Carcinoma. Eur. Urol. 2016, 71, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Jonasch, E.; Walker, C.L.; Rathmell, W.K. Clear cell renal cell carcinoma ontogeny and mechanisms of lethality. Nat. Rev. Nephrol. 2020, 17, 245–261. [Google Scholar] [CrossRef]
- Molina, A.; Lin, X.; Korytowsky, B.; Matczak, E.; Lechuga, M.; Wiltshire, R.; Motzer, R. Sunitinib objective response in metastatic renal cell carcinoma: Analysis of 1059 patients treated on clinical trials. Eur. J. Cancer 2013, 50, 351–358. [Google Scholar] [CrossRef]
- Motzer, R.J.; Barrios, C.H.; Kim, T.M.; Falcon, S.; Cosgriff, T.; Harker, W.G.; Srimuninnimit, V.; Pittman, K.; Sabbatini, R.; Rha, S.Y.; et al. Phase II Randomized Trial Comparing Sequential First-Line Everolimus and Second-Line Sunitinib Versus First-Line Sunitinib and Second-Line Everolimus in Patients With Metastatic Renal Cell Carcinoma. J. Clin. Oncol. 2014, 32, 2765–2772. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; Shan, H.; Miao, M.Z.; Jiang, Z.; Meng, Y.; Chen, R.; Zhang, L.; Liu, Y. Identification of the key genes and pathways involved in the tumorigenesis and prognosis of kidney renal clear cell carcinoma. Sci. Rep. 2020, 10, 4271. [Google Scholar] [CrossRef]
- Ragone, R.; Sallustio, F.; Piccinonna, S.; Rutigliano, M.; Vanessa, G.; Palazzo, S.; Lucarelli, G.; Ditonno, P.; Battaglia, M.; Fanizzi, F.P.; et al. Renal Cell Carcinoma: A Study through NMR-Based Metabolomics Combined with Transcriptomics. Diseases 2016, 4, 7. [Google Scholar] [CrossRef]
- Wettersten, H.I.; Aboud, O.A.; Lara, P.N., Jr.; Weiss, R.H. Metabolic reprogramming in clear cell renal cell carcinoma. Nature reviews. Nephrology 2017, 13, 410–419. [Google Scholar]
- Bianchi, C.; Meregalli, C.; Bombelli, S.; Di Stefano, V.; Salerno, F.; Torsello, B.; De Marco, S.; Bovo, G.; Cifola, I.; Mangano, E.; et al. The glucose and lipid metabolism reprogramming is grade-dependent in clear cell renal cell carcinoma primary cultures and is targetable to modulate cell viability and proliferation. Oncotarget 2017, 8, 113502–113515. [Google Scholar] [CrossRef] [Green Version]
- Lucarelli, G.; Loizzo, D.; Franzin, R.; Battaglia, S.; Ferro, M.; Cantiello, F.; Castellano, G.; Bettocchi, C.; Ditonno, P.; Battaglia, M. Metabolomic insights into pathophysiological mechanisms and biomarker discovery in clear cell renal cell carcinoma. Expert Rev. Mol. Diagn. 2019, 19, 397–407. [Google Scholar] [CrossRef]
- Lucarelli, G.; Rutigliano, M.; Sallustio, F.; Ribatti, D.; Giglio, A.; Signorile, M.L.; Grossi, V.; Sanese, P.; Napoli, A.; Maiorano, E.; et al. Integrated multi-omics characterization reveals a distinctive metabolic signature and the role of NDUFA4L2 in promoting angiogenesis, chemoresistance, and mitochondrial dysfunction in clear cell renal cell carcinoma. Aging 2018, 10, 3957–3985. [Google Scholar] [CrossRef]
- Bombelli, S.; Torsello, B.; De Marco, S.; Lucarelli, G.; Cifola, I.; Grasselli, C.; Strada, G.; Bovo, G.; Perego, R.A.; Bianchi, C. 36-kDa Annexin A3 Isoform Negatively Modulates Lipid Storage in Clear Cell Renal Cell Carcinoma Cells. Am. J. Pathol. 2020, 190, 2317–2326. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Dachs, G.U.; Gleadle, J.M.; Nicholls, L.G.; Harris, A.L.; Stratford, I.J.; Hankinson, O.; Pugh, C.W.; Ratcliffe, P.J. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc. Natl. Acad. Sci. USA 1997, 94, 8104–8109. [Google Scholar] [CrossRef] [Green Version]
- Flamme, I.; Krieg, M.; Plate, K.H. Up-Regulation of Vascular Endothelial Growth Factor in Stromal Cells of Hemangioblastomas Is Correlated with Up-Regulation of the Transcription Factor HRF/HIF-2α. Am. J. Pathol. 1998, 153, 25–29. [Google Scholar] [CrossRef]
- Krieg, M.; Haas, R.; Brauch, H.; Acker, T.; Flamme, I.; Plate, K.H. Up-regulation of hypoxia-inducible factors HIF-1α and HIF-2α under normoxic conditions in renal carcinoma cells by von Hippel-Lindau tumor suppressor gene loss of function. Oncogene 2000, 19, 5435–5443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-Montero, C.M.; Rini, B.I.; Finke, J.H. The immunology of renal cell carcinoma. Nat. Rev. Nephrol. 2020, 16, 721–735. [Google Scholar] [CrossRef] [PubMed]
- D’Avella, C.; Abbosh, P.; Pal, S.K.; Geynisman, D.M. Mutations in renal cell carcinoma. Urol. Oncol. Semin. Orig. Investig. 2018, 38, 763–773. [Google Scholar] [CrossRef]
- Pandey, N.; Lanke, V.; Vinod, P.K. Network-based metabolic characterization of renal cell carcinoma. Sci. Rep. 2020, 10, 5955. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Bermudez, J.; Baudrier, L.; La, K.; Zhu, X.G.; Fidelin, J.; Sviderskiy, V.O.; Papagiannakopoulos, T.; Molina, H.; Snuderl, M.; Lewis, C.A.; et al. Publisher Correction: Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat. Cell Biol. 2018, 20, 775–781. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Luengo, A.; Danai, L.V.; Bush, L.N.; Diehl, F.F.; Hosios, A.M.; Lau, A.N.; Elmiligy, S.; Malstrom, S.; Lewis, C.A.; et al. Aspartate is an endogenous metabolic limitation for tumour growth. Nat. Cell Biol. 2018, 20, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, S.; Adler, L.; Yizhak, K.; Sarver, A.; Silberman, A.; Agron, S.; Stettner, N.; Sun, Q.; Brandis, A.; Helbling, D.; et al. Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature 2015, 527, 379–383. [Google Scholar] [CrossRef]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef] [Green Version]
- Van Karnebeek, C.D.M.; Ramos, R.J.; Wen, X.-Y.; Tarailo-Graovac, M.; Gleeson, J.G.; Skrypnyk, C.; Brand-Arzamendi, K.; Karbassi, F.; Issa, M.Y.; van der Lee, R.; et al. Bi-allelic GOT2 Mutations Cause a Treatable Malate-Aspartate Shuttle-Related Encephalopathy. Am. J. Hum. Genet. 2019, 105, 534–548. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Zhou, L.; Shi, Q.; Zhao, Y.; Lin, H.; Zhang, M.; Zhao, S.; Yang, Y.; Ling, Z.-Q.; Guan, K.-L.; et al. SIRT 3-dependent GOT2 acetylation status affects the malate–aspartate NADH shuttle activity and pancreatic tumor growth. EMBO J. 2015, 34, 1110–1125. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.P.; Sabatini, D.M. Cancer Cell Metabolism: Warburg and Beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Hwang, S.; Kim, M.; Seo, S.B.; Lee, J.-H.; Jeong, S.M. Mitochondrial glutamine metabolism via GOT2 supports pancreatic cancer growth through senescence inhibition. Cell Death Dis. 2018, 9, 55. [Google Scholar] [CrossRef]
- Guan, H.; Sun, C.; Gu, Y.; Li, J.; Ji, J.; Zhu, Y. Circular RNA circ_0003028 contributes to tumorigenesis by regulating GOT2 via miR-1298-5p in non-small cell lung cancer. Bioengineered 2021, 12, 2326–2340. [Google Scholar] [CrossRef]
- Jin, M.; Shi, C.; Hua, Q.; Li, T.; Yang, C.; Wu, Y.; Zhao, L.; Yang, H.; Zhang, J.; Hu, C.; et al. High circ-SEC31A expression predicts unfavorable prognoses in non-small cell lung cancer by regulating the miR-520a-5p/GOT-2 axis. Aging 2020, 12, 10381–10397. [Google Scholar] [CrossRef]
- Liu, M.; Liu, X.; Liu, S.; Xiao, F.; Guo, E.; Qin, X.; Wu, L.; Liang, Q.; Liang, Z.; Li, K.; et al. Big Data-Based Identification of Multi-Gene Prognostic Signatures in Liver Cancer. Front. Oncol. 2020, 10, 847. [Google Scholar] [CrossRef]
- Du, F.; Chen, J.; Liu, H.; Cai, Y.; Cao, T.; Han, W.; Yi, X.; Qian, M.; Tian, D.; Nie, Y.; et al. SOX12 promotes colorectal cancer cell proliferation and metastasis by regulating asparagine synthesis. Cell Death Dis. 2019, 10, 239. [Google Scholar] [CrossRef]
- Feist, M.; Schwarzfischer, P.; Heinrich, P.; Sun, X.; Kemper, J.; von Bonin, F.; Perez-Rubio, P.; Taruttis, F.; Rehberg, T.; Dettmer, K.; et al. Cooperative stat/nf-kappab signaling regulates lymphoma metabolic reprogramming and aberrant got2 expression. Nat. Commun. 2018, 9, 1514. [Google Scholar] [CrossRef]
- Minchenko, O.H.; Riabovol, O.O.; Tsymbal, D.O.; Minchenko, D.O.; Ratushna, O.O. Effect of hypoxia on the expression of nuclear genes encoding mitochondrial proteins in U87 glioma cells. Ukr. Biochem. J. 2016, 88, 54–65. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Edwards, N.J.; Oberti, M.; Thangudu, R.R.; Cai, S.; McGarvey, P.B.; Jacob, S.; Madhavan, S.; Ketchum, K.A. The CPTAC Data Portal: A Resource for Cancer Proteomics Research. J. Proteome Res. 2015, 14, 2707–2713. [Google Scholar] [CrossRef]
- Chen, F.; Chandrashekar, D.S.; Varambally, S.; Creighton, C.J. Pan-cancer molecular subtypes revealed by mass-spectrometry-based proteomic characterization of more than 500 human cancers. Nat. Commun. 2019, 10, 5679. [Google Scholar] [CrossRef] [Green Version]
- Monsivais, D.; Vasquez, Y.M.; Chen, F.; Zhang, Y.; Chandrashekar, D.S.; Faver, J.C.; Masand, R.P.; Scheurer, M.E.; Varambally, S.; Matzuk, M.M.; et al. Mass-spectrometry-based proteomic correlates of grade and stage reveal pathways and kinases associated with aggressive human cancers. Oncogene 2021, 40, 2081–2095. [Google Scholar] [CrossRef]
- Uhlén, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, 2507. [Google Scholar] [CrossRef] [Green Version]
- Thul, P.J.; Åkesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Brooks, S.A.; Brannon, A.R.; Parker, J.S.; Fisher, J.C.; Sen, O.; Kattan, M.W.; Hakimi, A.A.; Hsieh, J.J.; Choueiri, T.K.; Tamboli, P.; et al. ClearCode34: A Prognostic Risk Predictor for Localized Clear Cell Renal Cell Carcinoma. Eur. Urol. 2014, 66, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Györffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1809 patients. Breast Cancer Res. Treat. 2009, 123, 725–731. [Google Scholar] [CrossRef] [Green Version]
- Modhukur, V.; Iljasenko, T.; Metsalu, T.; Lokk, K.; Laisk-Podar, T.; Vilo, J. MethSurv: A web tool to perform multivariable survival analysis using DNA methylation data. Epigenomics 2018, 10, 277–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Ge, D.; Lu, C. The SMART App: An interactive web application for comprehensive DNA methylation analysis and visualization. Epigenet. Chromatin 2019, 12, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Tang, Z.; Zhang, W.; Ye, Z.; Liu, F. GEPIA2021: Integrating multiple deconvolution-based analysis into GEPIA. Nucleic Acids Res. 2021, 49, W242–W246. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Wang, J.; Han, Y.; Dong, X.; Ge, J.; Zheng, R.; Shi, X.; Wang, B.; Li, Z.; Ren, P.; et al. TISCH: A comprehensive web resource enabling interactive single-cell transcriptome visualization of tumor microenvironment. Nucleic Acids Res. 2020, 49, D1420–D1430. [Google Scholar] [CrossRef]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.-H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.e16. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.D.; Madireddi, S.; de Almeida, P.E.; Banchereau, R.; Chen, Y.-J.J.; Chitre, A.S.; Chiang, E.Y.; Iftikhar, H.; O’Gorman, W.E.; Au-Yeung, A.; et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 2020, 579, 274–278. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, C.; Xiao, M.; Han, Y.; Zhang, S.; Xu, B. Bioinformatics Analysis of the Prognostic and Biological Significance of ZDHHC-Protein Acyltransferases in Kidney Renal Clear Cell Carcinoma. Front. Oncol. 2020, 10, 565414. [Google Scholar] [CrossRef]
- Ghatalia, P.; Gordetsky, J.; Kuo, F.; Dulaimi, E.; Cai, K.Q.; Devarajan, K.; Bae, S.; Naik, G.; Chan, T.A.; Uzzo, R.; et al. Prognostic impact of immune gene expression signature and tumor infiltrating immune cells in localized clear cell renal cell carcinoma. J. Immunother. Cancer 2019, 7, 139. [Google Scholar] [CrossRef] [Green Version]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [Green Version]
- Lane, A.N.; Fan, T.W.-M. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015, 43, 2466–2485. [Google Scholar] [CrossRef] [Green Version]
- Meléndez-Rodríguez, F.; Urrutia, A.A.; Lorendeau, D.; Rinaldi, G.; Roche, O.; Böğürcü-Seidel, N.; Muelas, M.O.; Ciller, C.M.; Turiel, G.; Bouthelier, A.; et al. HIF1α Suppresses Tumor Cell Proliferation through Inhibition of Aspartate Biosynthesis. Cell Rep. 2019, 26, 2257–2265.e4. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef] [Green Version]
- Lieu, E.L.; Nguyen, T.; Rhyne, S.; Kim, J. Amino acids in cancer. Exp. Mol. Med. 2020, 52, 15–30. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, J.; Wang, S.; Jiang, Q.; Xu, K. Identification and Validation of a Nine-Gene Amino Acid Metabolism-Related Risk Signature in HCC. Front. Cell Dev. Biol. 2021, 9, 731790. [Google Scholar] [CrossRef]
- Hakimi, A.A.; Reznik, E.; Lee, C.-H.; Creighton, C.J.; Brannon, A.R.; Luna, A.; Aksoy, B.A.; Liu, E.M.; Shen, R.; Lee, W.; et al. An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell 2016, 29, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Shroff, E.H.; Eberlin, L.S.; Dang, V.M.; Gouw, A.M.; Gabay, M.; Adam, S.J.; Bellovin, D.I.; Tran, P.T.; Philbrick, W.M.; Garcia-Ocana, A.; et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc. Natl. Acad. Sci. USA 2015, 112, 6539–6544. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef]
- Sandoval, J.; Esteller, M. Cancer epigenomics: Beyond genomics. Curr. Opin. Genet. Dev. 2012, 22, 50–55. [Google Scholar] [CrossRef]
- Das, P.M.; Singal, R. DNA methylation and cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2004, 22, 4632–4642. [Google Scholar] [CrossRef] [Green Version]
- Lasseigne, B.N.; Brooks, J.D. The Role of DNA Methylation in Renal Cell Carcinoma. Mol. Diagn. Ther. 2018, 22, 431–442. [Google Scholar] [CrossRef]
- Mehdi, A.; Riazalhosseini, Y. Epigenome Aberrations: Emerging Driving Factors of the Clear Cell Renal Cell Carcinoma. Int. J. Mol. Sci. 2017, 18, 1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research, N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Borcherding, N.; Vishwakarma, A.; Voigt, A.P.; Bellizzi, A.; Kaplan, J.; Nepple, K.; Salem, A.K.; Jenkins, R.W.; Zakharia, Y.; Zhang, W. Mapping the immune environment in clear cell renal carcinoma by single-cell genomics. Commun. Biol. 2021, 4, 122. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, J.; Kang, W.; Wang, J.; Xiang, Y.; Fu, M.; Gao, H.; Huang, Z. Tumor microenvironment immune subtypes for classification of novel clear cell renal cell carcinoma profiles with prognostic and therapeutic implications. Medicine 2021, 100, e24949. [Google Scholar] [CrossRef] [PubMed]
- Nakano, O.; Sato, M.; Naito, Y.; Suzuki, K.; Orikasa, S.; Aizawa, M.; Suzuki, Y.; Shintaku, I.; Nagura, H.; Ohtani, H. Proliferative activity of intratumoral CD8(+) T-lymphocytes as a prognostic factor in human renal cell carcinoma: Clinicopathologic demonstration of antitumor immunity. Cancer Res. 2001, 61, 5132–5136. [Google Scholar] [PubMed]
- Qi, Y.; Xia, Y.; Lin, Z.; Qu, Y.; Qi, Y.; Chen, Y.; Zhou, Q.; Zeng, H.; Wang, J.; Chang, Y.; et al. Tumor-infiltrating CD39+CD8+ T cells determine poor prognosis and immune evasion in clear cell renal cell carcinoma patients. Cancer Immunol. Immunother. 2020, 69, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, N.; Becht, E.; Pagès, F.; Skliris, G.P.; Verkarre, V.; Vano, Y.A.; Mejean, A.; Saint-Aubert, N.; Lacroix, L.; Natario, I.; et al. Orchestration and Prognostic Significance of Immune Checkpoints in the Microenvironment of Primary and Metastatic Renal Cell Cancer. Clin. Cancer Res. 2015, 21, 3031–3040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, E.; Whiteman, K.; Pai, T.; Hickman, T.; Johnson, T.; Friedman, T.; Parikh, A.; Gilbert, M.; Shen, B.; Weiss, G.J.; et al. Abstract 2184: BOXR1030: A first-in-class CAR T-cell therapy co-expressing GOT2 enhances T-cell metabolic function for the treatment of GPC3-positive solid tumors. Immunology 2020, 80, 2184. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinicopathological Characteristics | n | Hazard Ratio (95% CI) | Logrank p |
|---|---|---|---|
| Stage | |||
| 1 | 265 | 1.67 (0.92–3.03) | 8.94 × 10−2 |
| 2 | 57 | 0.29 (0.06–1.31) | 8.56 × 10−2 |
| 3 | 123 | 0.35 (0.74–2.39) | 3.47 × 10−1 |
| 4 | 82 | 0.56 (0.32–0.97) | 3.50× 10−2 |
| Gender | |||
| Female | 186 | 0.62 (0.36–1.05) | 7.45 × 10−2 |
| Male | 344 | 0.71 (0.47–1.06) | 9.56 × 10−2 |
| Grade | |||
| 1 | 14 | - | - |
| 2 | 227 | 1.51 (0.8–2.84) | 2.04 × 10−1 |
| 3 | 206 | 0.53 (0.33–0.85) | 7.90 ×10−3 |
| 4 | 75 | 0.64 (0.35–1.19) | 1.60 × 10−1 |
| Mutation burden | |||
| High | 168 | 1.34 (0.77–2.34) | 2.94 × 10−1 |
| Low | 164 | 2.28 (1.04–4.99) | 3.49× 10−2 |
| Race | |||
| White | 459 | 0.72 (0.5–1.02) | 6.17× 10−2 |
| Asian | 8 | - | - |
| Black/African American | 56 | 0.39 (0.12–1.29) | 1.10 × 10−1 |
| Hemoglobin result | |||
| Elevated | 5 | 1.73 (0.1076–27.8905) | 6.98 × 10−1 |
| Normal | 184 | 0.70 (0.3784–1.3129) | 2.70 × 10−1 |
| Low | 261 | 1.22 (0.8484–1.7628) | 2.81 × 10−1 |
| Laterality | |||
| Right | 280 | 1.12 (0.7181–1.7561) | 6.11 × 10−1 |
| Left | 248 | 0.71 (0.4721–1.0682) | 1.00 × 10−1 |
| Bilateral | 4 | - | - |
| Serum calcium result | |||
| Elevated | 10 | 0.65 (0.1599–2.6446) | 5.48 × 10−1 |
| Low | 203 | 0.79 (0.5749–1.5257) | 7.92 × 10−1 |
| Normal | 150 | 0.62 (0.376–1.0547) | 7.88 × 10−2 |
| Probe | Chromosome | Start | End | CGI Position |
|---|---|---|---|---|
| cg04793118 | chr16 | 58707974 | 58707975 | Open Sea |
| cg06070269 | chr16 | 58718235 | 58718236 | Open Sea |
| cg06624121 | chr16 | 58718247 | 58718248 | Open Sea |
| cg04471375 | chr16 | 58718261 | 58718262 | Open Sea |
| cg06302295 | chr16 | 58730982 | 58730983 | N Shelf |
| cg08348831 | chr16 | 58733959 | 58733960 | Island |
| cg13626907 | chr16 | 58733963 | 58733964 | Island |
| cg14863484 | chr16 | 58734106 | 58734107 | Island |
| cg09082840 | chr16 | 58734264 | 58734265 | Island |
| cg16406345 | chr16 | 58734350 | 58734351 | Island |
| cg08578141 | chr16 | 58734361 | 58734362 | Island |
| cg10055227 | chr16 | 58734416 | 58734417 | Island |
| cg08950929 | chr16 | 58734423 | 58734424 | Island |
| cg00028829 | chr16 | 58734507 | 58734508 | S Shore |
| cg10140957 | chr16 | 58734573 | 58734574 | S Shore |
| cg13883681 | chr16 | 58734661 | 58734662 | S Shore |
| cg08987251 | chr16 | 58735200 | 58735201 | S Shore |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, W.A.S.; de Oliveira, E.H.C. Expression of GOT2 Is Epigenetically Regulated by DNA Methylation and Correlates with Immune Infiltrates in Clear-Cell Renal Cell Carcinoma. Curr. Issues Mol. Biol. 2022, 44, 2472-2489. https://doi.org/10.3390/cimb44060169
Ferreira WAS, de Oliveira EHC. Expression of GOT2 Is Epigenetically Regulated by DNA Methylation and Correlates with Immune Infiltrates in Clear-Cell Renal Cell Carcinoma. Current Issues in Molecular Biology. 2022; 44(6):2472-2489. https://doi.org/10.3390/cimb44060169
Chicago/Turabian StyleFerreira, Wallax Augusto Silva, and Edivaldo Herculano Correa de Oliveira. 2022. "Expression of GOT2 Is Epigenetically Regulated by DNA Methylation and Correlates with Immune Infiltrates in Clear-Cell Renal Cell Carcinoma" Current Issues in Molecular Biology 44, no. 6: 2472-2489. https://doi.org/10.3390/cimb44060169
APA StyleFerreira, W. A. S., & de Oliveira, E. H. C. (2022). Expression of GOT2 Is Epigenetically Regulated by DNA Methylation and Correlates with Immune Infiltrates in Clear-Cell Renal Cell Carcinoma. Current Issues in Molecular Biology, 44(6), 2472-2489. https://doi.org/10.3390/cimb44060169