
DNA Methylation Patterns According to Fatty Liver Index and Longitudinal Changes from the Korean Genome and Epidemiology Study (KoGES)


Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects and Data Source
2.2. Study Design
2.3. DNA Methylation and Bioinformatics Analysis
2.4. Bioinformatics Analysis
3. Results
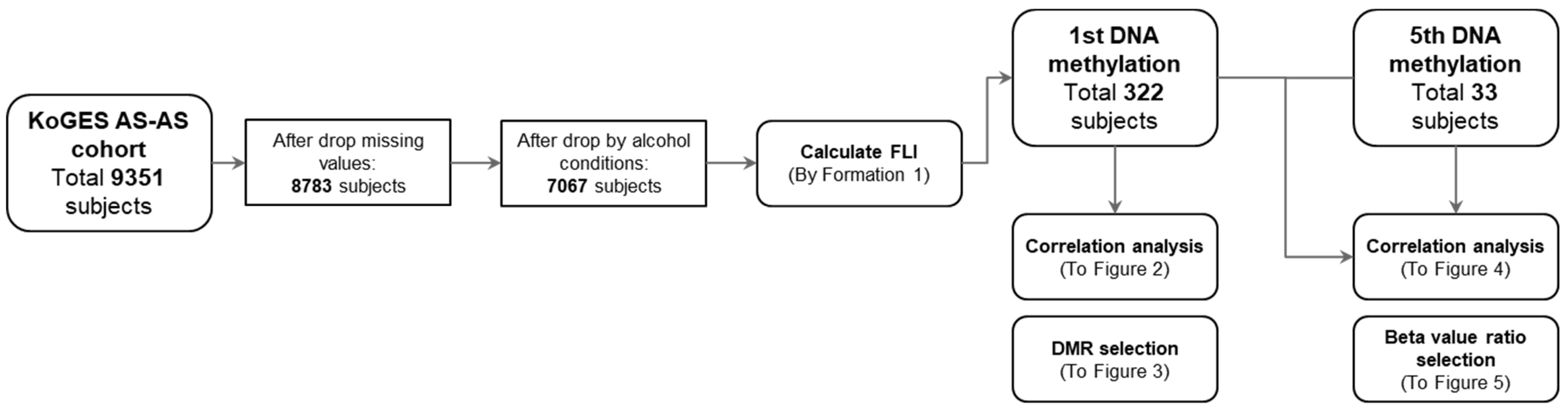
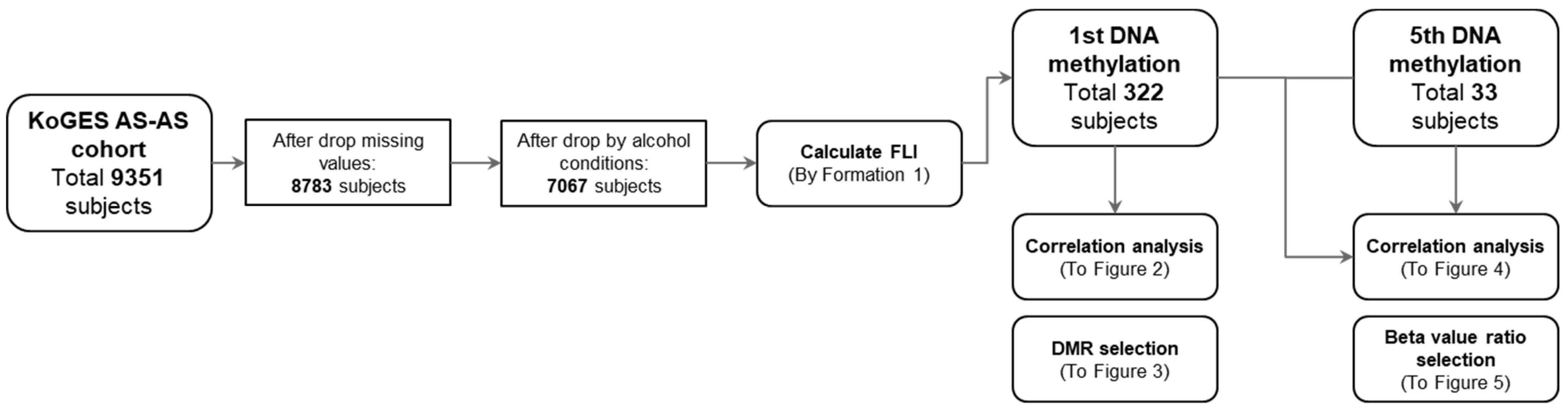
3.1. Study Processes
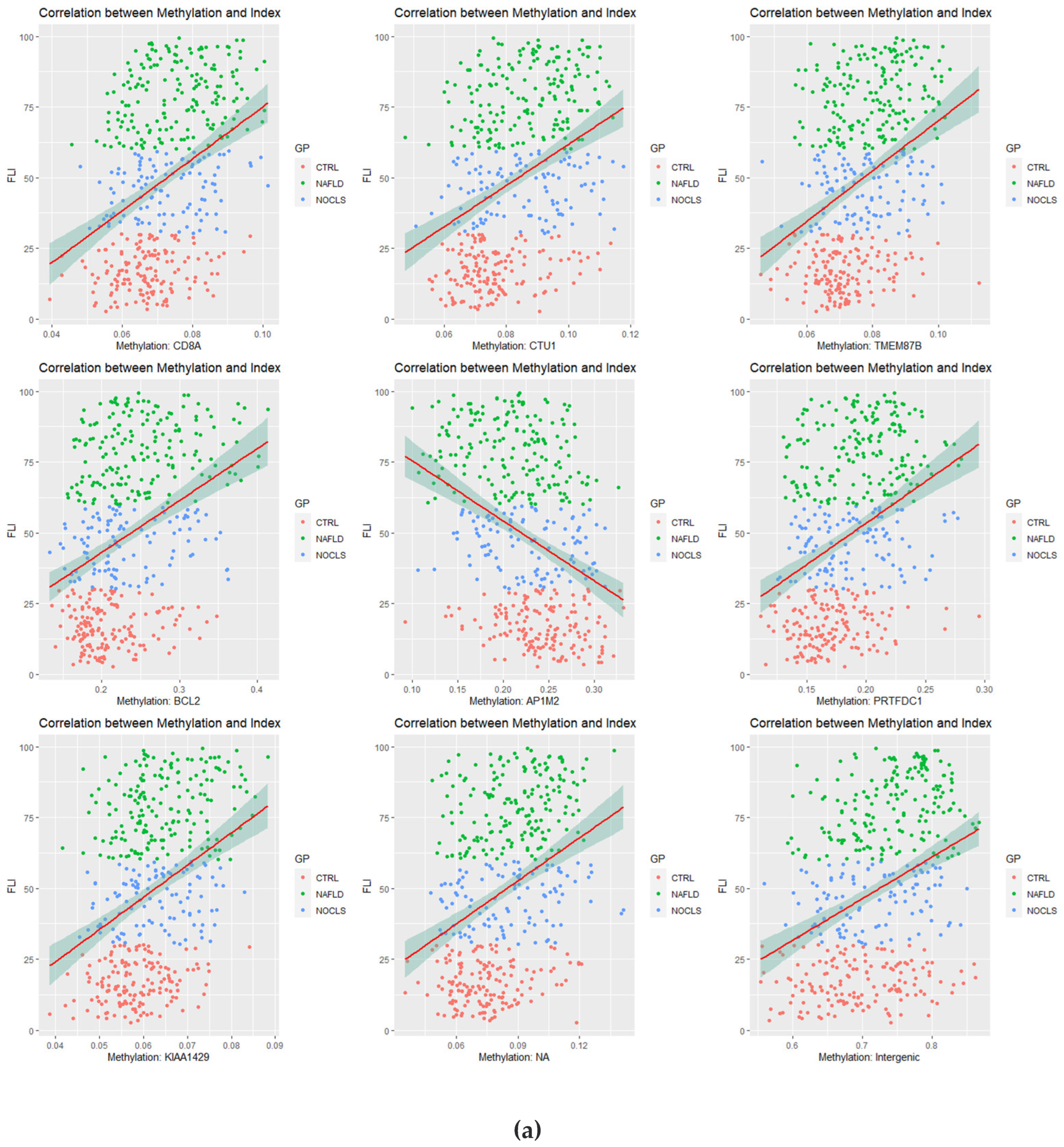
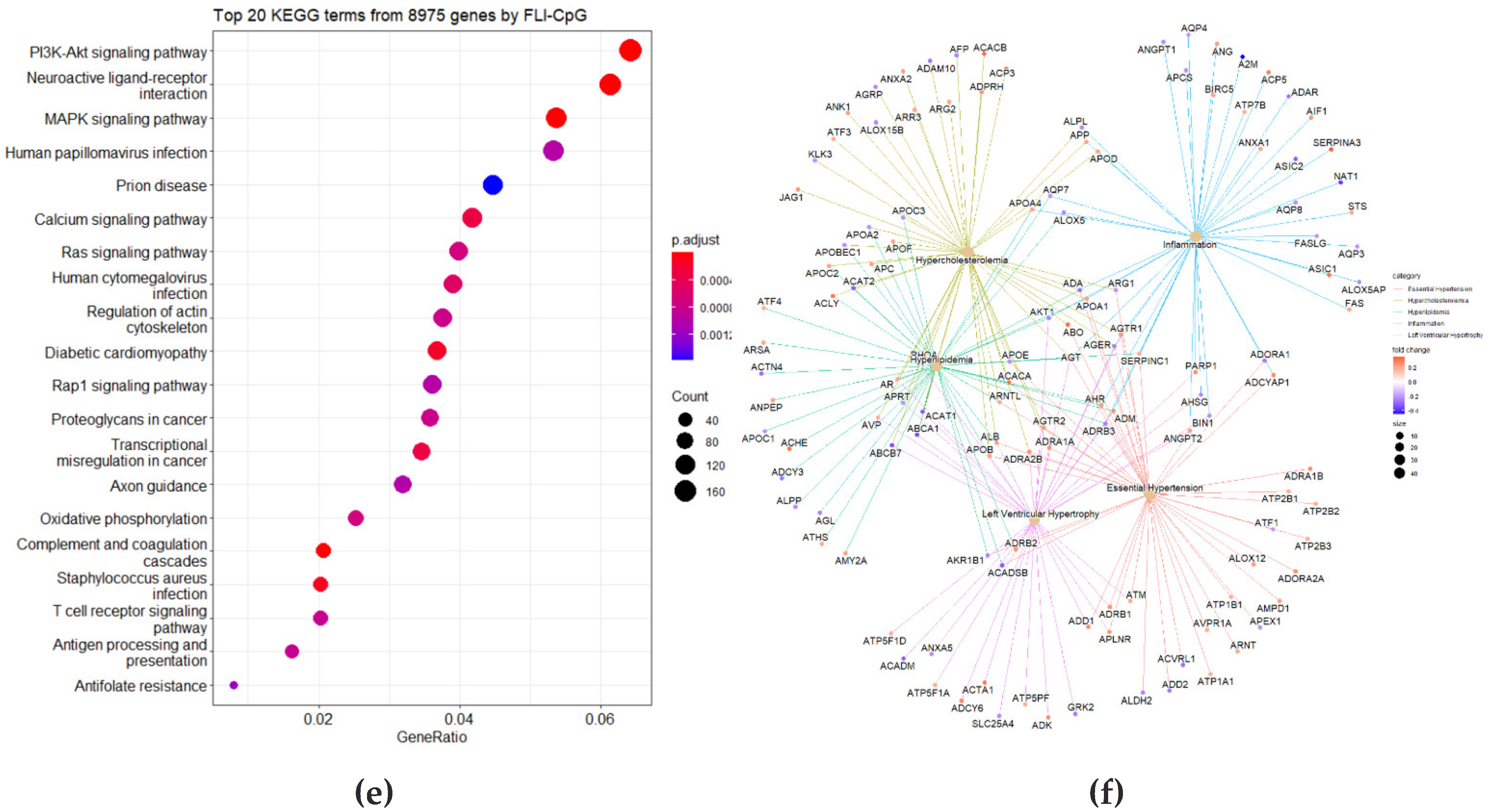
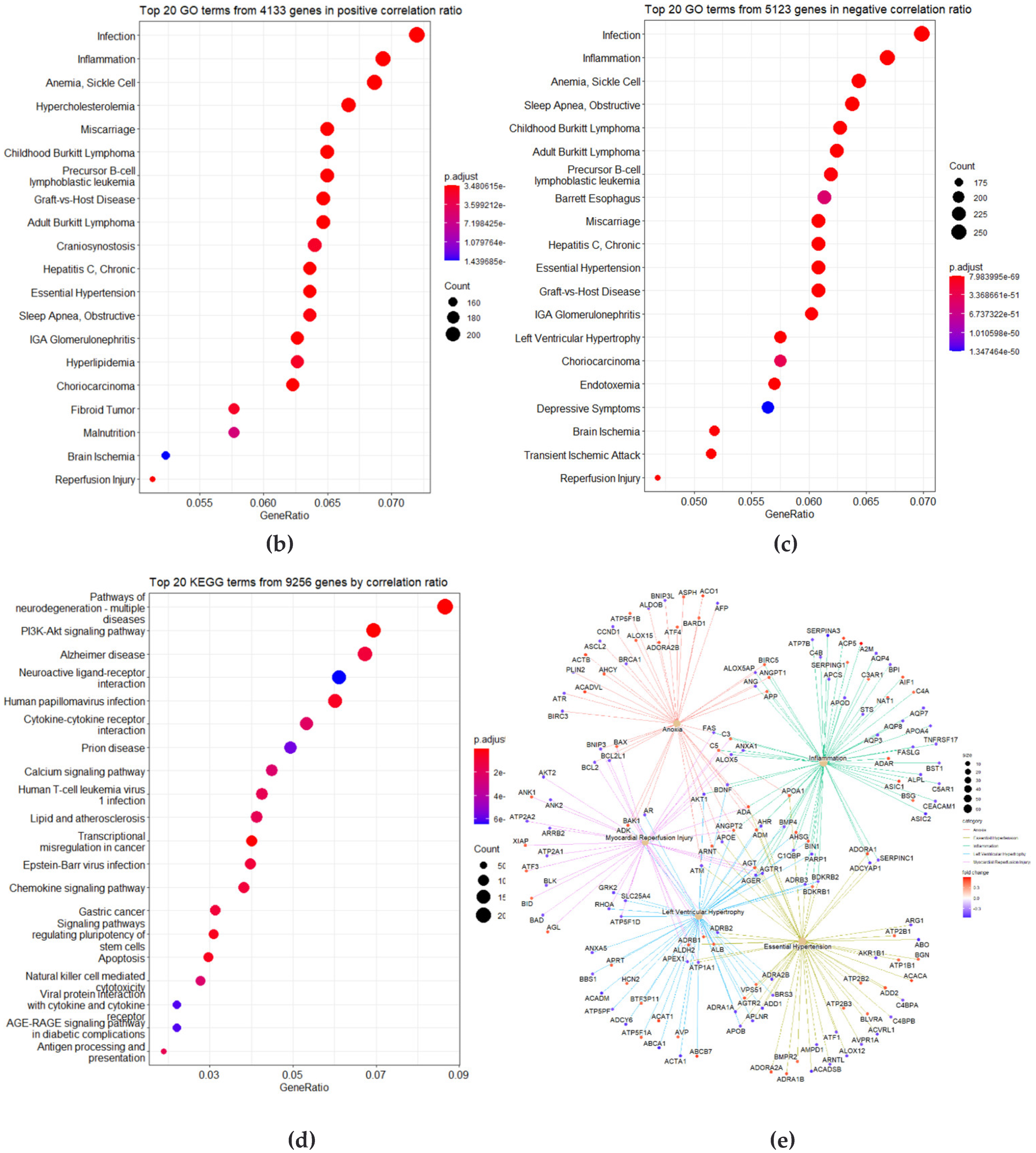
3.2. Correlation Analysis between CpG Site Methylation and FLI
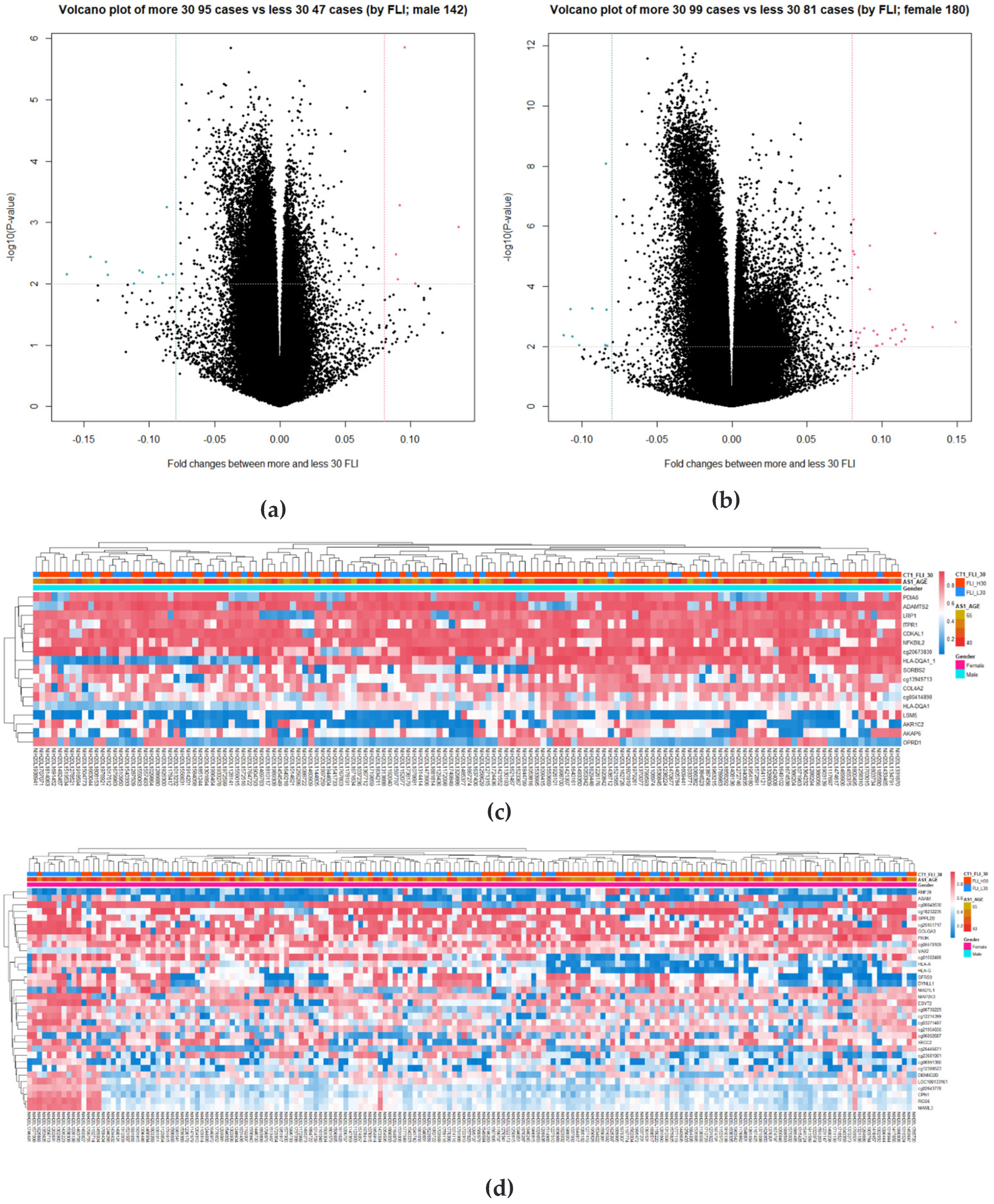
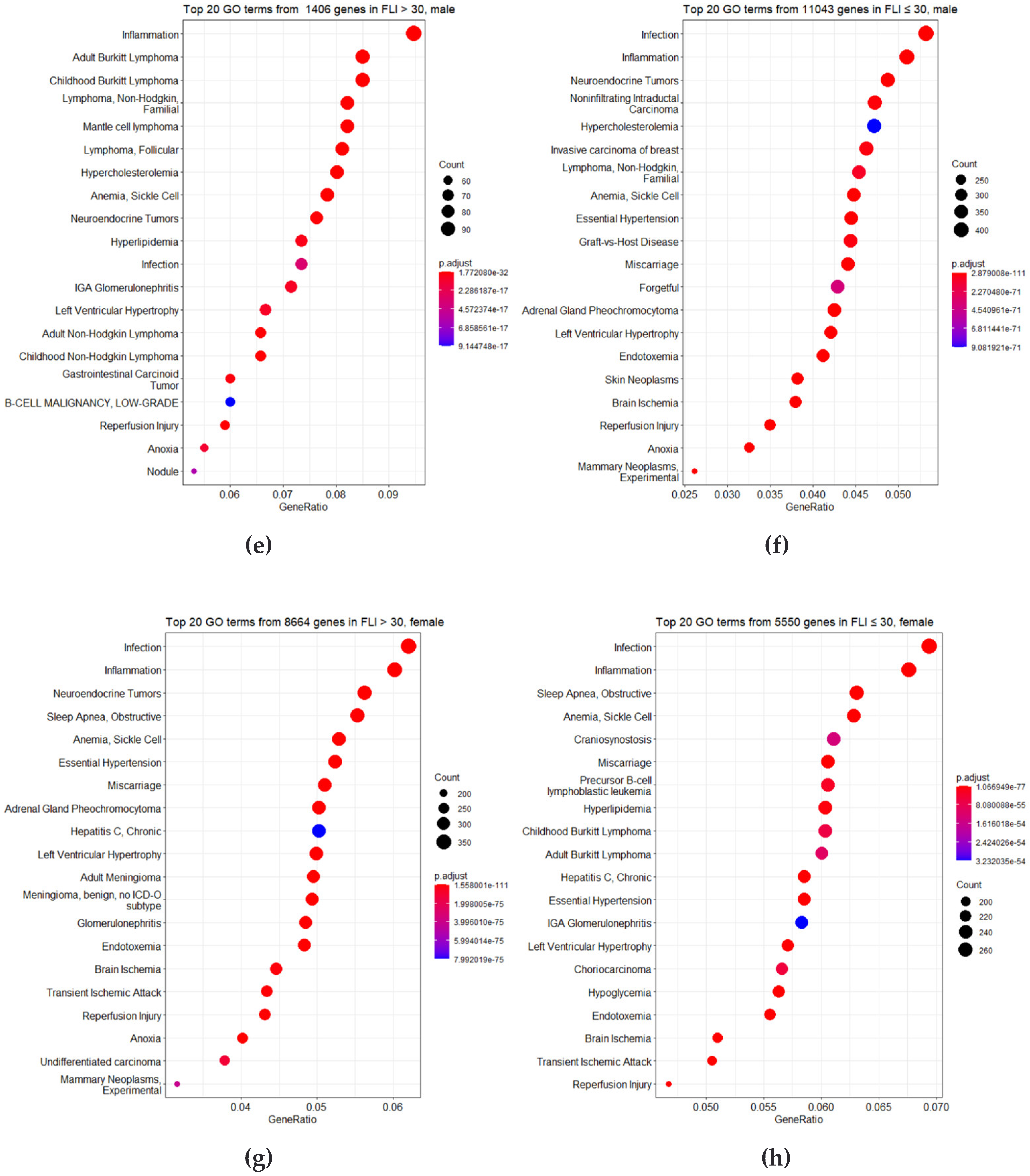
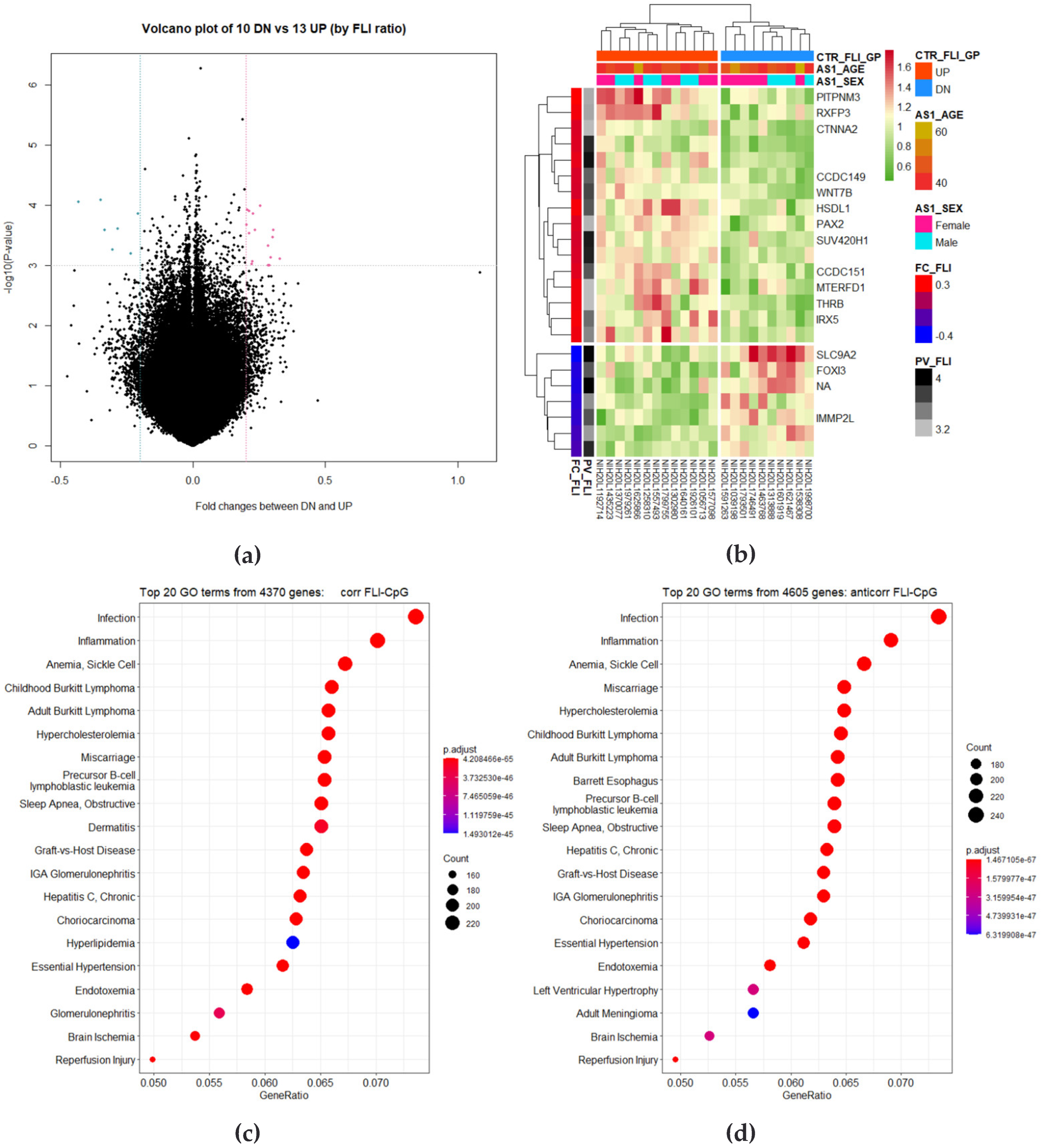
3.3. Identification of DMRs
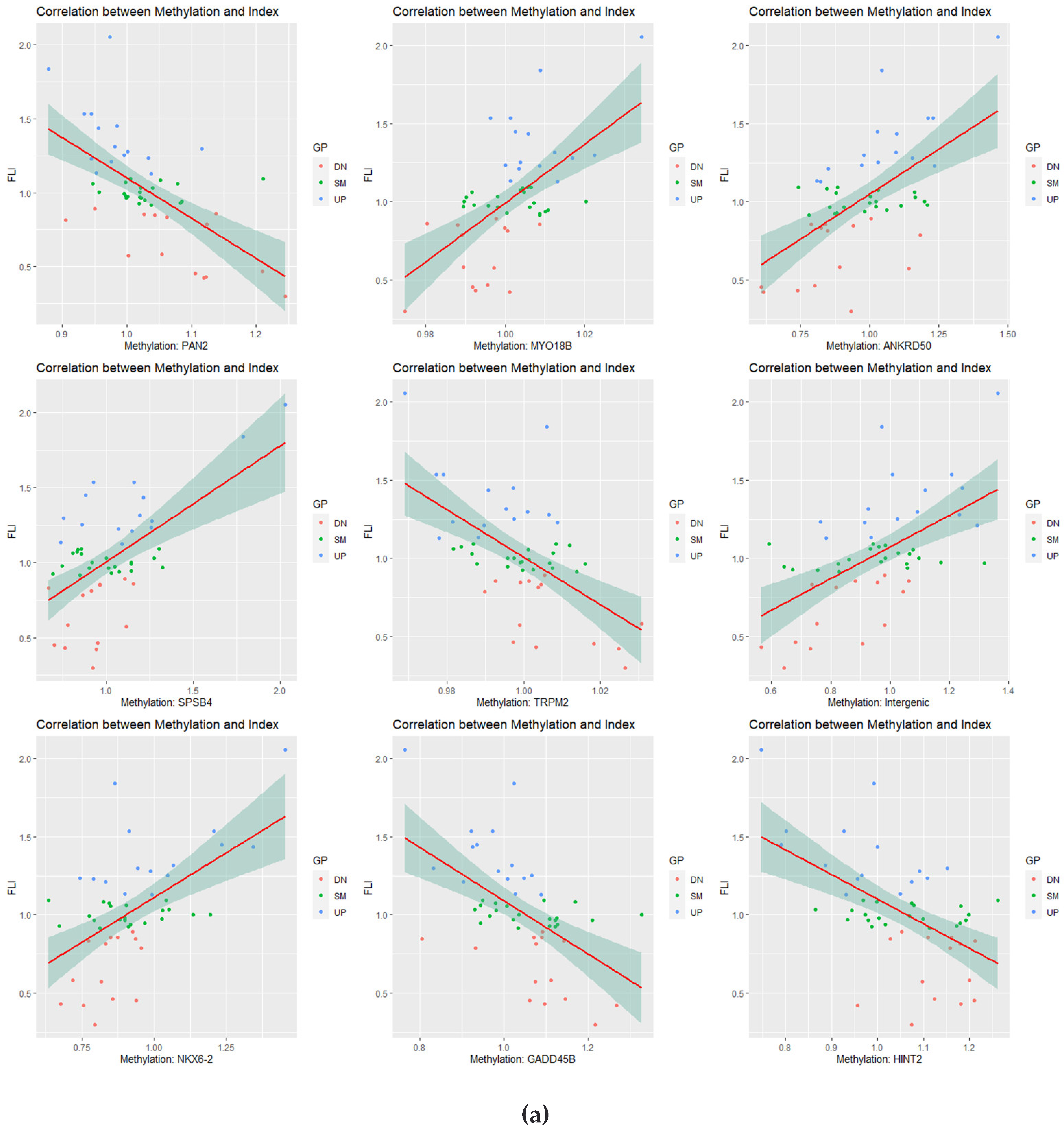
3.4. Correlation Analysis of Longitudinal Data
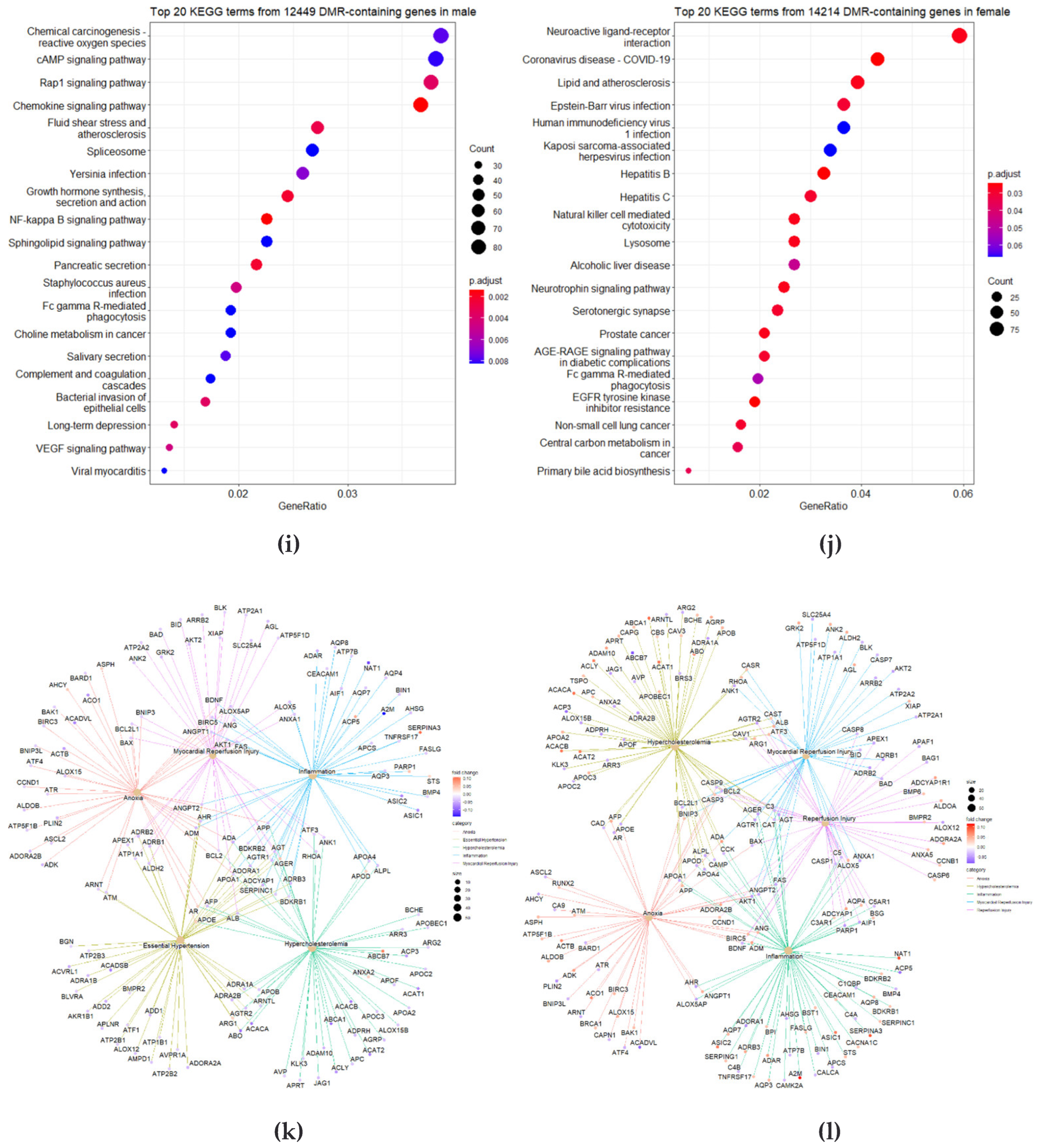
3.5. Identification of Differently Changed Regions
3.6. Integration of Four Analyses as Circos Plot
4. Discussion
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.-J.; Kim, W.; Kim, M.Y.; Jun, D.W.; Kim, S.G.; Yeon, J.-E.; Lee, J.W.; Cho, Y.K.; Park, S.H.; Sohn, J.H. Recent research trends and updates on nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2019, 25, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E. Nonalcoholic fatty liver disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Dulai, P.S.; Singh, S.; Patel, J.; Soni, M.; Prokop, L.J.; Younossi, Z.; Sebastiani, G.; Ekstedt, M.; Hagstrom, H.; Nasr, P. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 2017, 65, 1557–1565. [Google Scholar] [CrossRef] [PubMed]
- Vernon, G.; Baranova, A.; Younossi, Z. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef]
- Lee, Y.-S.; Lee, H.S.; Chang, S.W.; Lee, C.U.; Kim, J.S.; Jung, Y.K.; Kim, J.H.; Seo, Y.S.; Yim, H.J.; Lee, C.H. Underlying nonalcoholic fatty liver disease is a significant factor for breast cancer recurrence after curative surgery. Medicine 2019, 98, e17277. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.-A.; Lee, H.C.; Choe, J.; Kim, M.-J.; Lee, M.J.; Chang, H.-S.; Bae, I.Y.; Kim, H.-K.; An, J.; Shim, J.H. Association between non-alcoholic fatty liver disease and cancer incidence rate. J. Hepatol. 2018, 68, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Hagstrom, H.; Thiele, M.; Roelstraete, B.; Soderling, J.; Ludvigsson, J.F. Mortality in biopsy-proven alcohol-related liver disease: A population-based nationwide cohort study of 3453 patients. Gut 2021, 70, 170–179. [Google Scholar] [CrossRef]
- Duseja, A.; Chalasani, N. Epidemiology and risk factors of nonalcoholic fatty liver disease (NAFLD). Hepatol. Int. 2013, 7, 755–764. [Google Scholar] [CrossRef]
- Gim, J.-A.; Bang, S.M.; Lee, Y.-S.; Lee, Y.; Yim, S.Y.; Jung, Y.K.; Kim, H.; Kim, B.-H.; Kim, J.H.; Seo, Y.S. Evaluation of the severity of nonalcoholic fatty liver disease through analysis of serum exosomal miRNA expression. PLoS ONE 2021, 16, e0255822. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Lee, Y.; Lee, Y.-S.; Gim, J.-A.; Ko, E.; Yim, S.Y.; Jung, Y.K.; Kang, S.; Kim, M.Y.; Kim, H. Circulating miRNA is a useful diagnostic biomarker for nonalcoholic steatohepatitis in nonalcoholic fatty liver disease. Sci. Rep. 2021, 11, 14639. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American association for the study of liver diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Thampanitchawong, P.; Piratvisuth, T. Liver biopsy: Complications and risk factors. World J. Gastroenterol. 1999, 5, 301. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.-S.; Adams, L.A.; de Ledinghen, V.; Wong, G.L.-H.; Sookoian, S. Noninvasive biomarkers in NAFLD and NASH—current progress and future promise. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 461–478. [Google Scholar] [CrossRef] [PubMed]
- Bedogni, G.; Bellentani, S.; Miglioli, L.; Masutti, F.; Passalacqua, M.; Castiglione, A.; Tiribelli, C. The fatty liver index: A simple and accurate predictor of hepatic steatosis in the general population. BMC Gastroenterol. 2006, 6, 33. [Google Scholar] [CrossRef] [Green Version]
- Calori, G.; Lattuada, G.; Ragogna, F.; Garancini, M.P.; Crosignani, P.; Villa, M.; Bosi, E.; Ruotolo, G.; Piemonti, L.; Perseghin, G. Fatty liver index and mortality: The cremona study in the 15th year of follow-up. Hepatology 2011, 54, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Haflidadottir, S.; Jonasson, J.G.; Norland, H.; Einarsdottir, S.O.; Kleiner, D.E.; Lund, S.H.; Bjornsson, E.S. Long term follow-up and liver-related death rate in patients with non-alcoholic and alcoholic related fatty liver disease. BMC Gastroenterol. 2014, 14, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nano, J.; Ghanbari, M.; Wang, W.; de Vries, P.S.; Dhana, K.; Muka, T.; Uitterlinden, A.G.; van Meurs, J.B.; Hofman, A.; Franco, O.H. Epigenome-wide association study identifies methylation sites associated with liver enzymes and hepatic steatosis. Gastroenterology 2017, 153, 1096–1106.e1092. [Google Scholar] [CrossRef] [PubMed]
- Orozco, L.D.; Morselli, M.; Rubbi, L.; Guo, W.; Go, J.; Shi, H.; Lopez, D.; Furlotte, N.A.; Bennett, B.J.; Farber, C.R. Epigenome-wide association of liver methylation patterns and complex metabolic traits in mice. Cell Metab. 2015, 21, 905–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Han, B.-G.; Group, K. Cohort profile: The Korean genome and epidemiology study (KoGES) consortium. Int. J. Epidemiol. 2017, 46, e20. [Google Scholar] [CrossRef] [PubMed]
- Dugue, P.-A.; Jung, C.-H.; Joo, J.E.; Wang, X.; Wong, E.M.; Makalic, E.; Schmidt, D.F.; Baglietto, L.; Severi, G.; Southey, M.C. Smoking and blood DNA methylation: An epigenome-wide association study and assessment of reversibility. Epigenetics 2020, 15, 358–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zhao, W.; Ammous, F.; Turner, S.T.; Mosley, T.H.; Zhou, X.; Smith, J.A. Longitudinal analysis of epigenome-wide DNA methylation reveals novel smoking-related loci in African Americans. Epigenetics 2019, 14, 171–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicoletti, C.; Cortes-Oliveira, C.; Noronha, N.; Pinhel, M.; Dantas, W.; Jacome, A.; Marchini, J.; Gualano, B.; Crujeiras, A.; Nonino, C. DNA methylation pattern changes following a short-term hypocaloric diet in women with obesity. Eur. J. Clin. Nutr. 2020, 74, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Zhang, T.; Su, S.; Hao, G.; Chen, T.; Li, Q.-Z.; Bazzano, L.; He, J.; Wang, X.; Li, S. Body mass index drives changes in DNA methylation: A longitudinal study. Circ. Res. 2019, 125, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Witasp, A.; Luttropp, K.; Qureshi, A.R.; Barany, P.; Heimburger, O.; Wennberg, L.; Ekstrom, T.J.; Shiels, P.G.; Stenvinkel, P.; Nordfors, L. Longitudinal genome-wide DNA methylation changes in response to kidney failure replacement therapy. Sci. Rep. 2022, 12, 470. [Google Scholar] [CrossRef]
- Grant, C.D.; Jafari, N.; Hou, L.; Li, Y.; Stewart, J.D.; Zhang, G.; Lamichhane, A.; Manson, J.E.; Baccarelli, A.A.; Whitsel, E.A. A longitudinal study of DNA methylation as a potential mediator of age-related diabetes risk. Geroscience 2017, 39, 475–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.K.; Vanderlinden, L.A.; Dong, F.; Carry, P.M.; Seifert, J.; Waugh, K.; Shorrosh, H.; Fingerlin, T.; Frohnert, B.I.; Yang, I.V. Longitudinal DNA methylation differences precede type 1 diabetes. Sci. Rep. 2020, 10, 3721. [Google Scholar] [CrossRef] [Green Version]
- Ekstedt, M.; Franzen, L.E.; Mathiesen, U.L.; Thorelius, L.; Holmqvist, M.; Bodemar, G.; Kechagias, S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006, 44, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M.; Pagano, G. Meta-analysis: Natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann. Med. 2011, 43, 617–649. [Google Scholar] [CrossRef]
- Sanna, C.; Rosso, C.; Marietti, M.; Bugianesi, E. Non-alcoholic fatty liver disease and extra-hepatic cancers. Int. J. Mol. Sci. 2016, 17, 717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyun, J.; Jung, Y. DNA methylation in nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2020, 21, 8138. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Ahn, Y.-S.; Chang, S.-J.; Kim, C.-B.; Jeong, K.S.; Koh, S.-B.; Gim, J.-A. Variation in genotype and DNA methylation patterns based on alcohol use and cvd in the Korean genome and epidemiology study (KoGES). Genes 2022, 13, 172. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Liu, Y. DNA methylation in human diseases. Genes Dis. 2018, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- French, S.W. Epigenetic events in liver cancer resulting from alcoholic liver disease. Alcohol Res. 2013, 35, 57–67. [Google Scholar] [PubMed]
- Lyall, M.J.; Thomson, J.P.; Cartier, J.; Ottaviano, R.; Kendall, T.J.; Meehan, R.R.; Drake, A.J. Non-alcoholic fatty liver disease (NAFLD) is associated with dynamic changes in DNA hydroxymethylation. Epigenetics 2020, 15, 61–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borowa-Mazgaj, B.; de Conti, A.; Tryndyak, V.; Steward, C.R.; Jimenez, L.; Melnyk, S.; Seneshaw, M.; Mirshahi, F.; Rusyn, I.; Beland, F.A.; et al. Gene expression and DNA methylation alterations in the glycine N-methyltransferase gene in diet-induced nonalcoholic fatty liver disease-associated carcinogenesis. Toxicol. Sci. 2019, 170, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Falzone, L.; Salemi, R.; Travali, S.; Scalisi, A.; McCubrey, J.A.; Candido, S.; Libra, M. MMP-9 overexpression is associated with intragenic hypermethylation of MMP9 gene in melanoma. Aging 2016, 8, 933–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krause, C.; Grohs, M.; El Gammal, A.T.; Wolter, S.; Lehnert, H.; Mann, O.; Mittag, J.; Kirchner, H. Reduced expression of thyroid hormone receptor β in human nonalcoholic steatohepatitis. Endocr. Connect. 2018, 7, 1448–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, S.A.; Bashir, M.R.; Guy, C.D.; Zhou, R.; Moylan, C.A.; Frias, J.P.; Alkhouri, N.; Bansal, M.B.; Baum, S.; Neuschwander-tetri, B.A. resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2019, 394, 2012–2024. [Google Scholar] [CrossRef]
- Kannt, A.; Wohlfart, P.; Madsen, A.N.; Veidal, S.S.; Feigh, M.; Schmoll, D. Activation of thyroid hormone receptor-β improved disease activity and metabolism independent of body weight in a mouse model of non-alcoholic steatohepatitis and fibrosis. Br. J. Pharmacol. 2021, 178, 2412–2423. [Google Scholar] [CrossRef] [PubMed]
- Arguello, G.; Balboa, E.; Arrese, M.; Zanlungo, S. Recent insights on the role of cholesterol in non-alcoholic fatty liver disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 1765–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, R.A.; Singh, B.K.; Yen, P.M. Thyroid hormone regulation of hepatic lipid and carbohydrate metabolism. Trends Endocrinol. Metab. 2014, 25, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Pirola, C.J. Obstructive sleep apnea is associated with fatty liver and abnormal liver enzymes: A meta-analysis. Obes. Surg. 2013, 23, 1815–1825. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Total Analysis | Baseline | Follow-Up | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total (n = 322) | Male (n = 142) | Female (n = 180) | FLI High (n = 194) | FLI Low (n = 128) | Total (n = 33) | Upregulated (n = 13) | Similar (n = 10) | Downregulated (n = 10) | Total (n = 33) | Upregulated (n = 13) | Similar (n = 10) | Downregulated (n = 10) | |
| Age | 52.56 ± 8.45 | 51.02 ± 8.17 | 53.77 ± 8.5 | 53.39 ± 8.13 | 51.29 ± 8.8 | 45.21 ± 5.63 | 44.69 ± 5.66 | 43.5 ± 2.32 | 47.6 ± 7.4 | 53.15 ± 5.63 | 52.69 ± 5.57 | 51.5 ± 2.32 | 55.4 ± 7.55 |
| Sex | 142/180 (44.1/55.9) | 142/0 (100/0) | 0/180 (0/100) | 95/99 (49/51) | 47/81 (37/63) | 16/17 (48.48/51.52) | 6/7 (46/54) | 6/4 (60/40) | 4/6 (40/60) | 16/17 (48.48/51.52) | 6/7 (46/54) | 6/4 (60/40) | 4/6 (40/60) |
| Diabetes | 304/18 (94.41/5.59) | 135/7 (95/5) | 169/11 (94/6) | 180/14 (93/7) | 124/4 (97/3) | 33/0 (100/0) | 13/0 (100/0) | 10/0 (100/0) | 10/0 (100/0) | 29/4 (87.88/12.12) | 11/2 (85/15) | 8/2 (80/20) | 10/0 (100/0) |
| Hypertension | 267/55 (82.92/17.08) | 122/20 (86/14) | 145/35 (81/19) | 144/50 (74/26) | 123/5 (96/4) | 32/1 (96.97/3.03) | 12/1 (92/8) | 10/0 (100/0) | 10/0 (100/0) | 33/0 (100/0) | 13/0 (100/0) | 10/0 (100/0) | 10/0 (100/0) |
| LipidBlood | 318/4 (98.76/1.24) | 141/1 (99/1) | 177/3 (98/2) | 192/2 (99/1) | 126/2 (98/2) | 33/0 (100/0) | 13/0 (100/0) | 10/0 (100/0) | 10/0 (100/0) | 32/1 (96.97/3.03) | 12/1 (92/8) | 10/0 (100/0) | 10/0 (100/0) |
| BMI | 24.52 ± 3.35 | 24.05 ± 3.28 | 24.89 ± 3.37 | 26.16 ± 2.99 | 22.03 ± 2.12 | 23.65 ± 2.63 | 23.26 ± 1.84 | 24.96 ± 2.87 | 22.86 ± 2.99 | 23.56 ± 2.62 | 23.87 ± 2.18 | 24.56 ± 2.37 | 22.17 ± 3.02 |
| Hb | 13.56 ± 1.55 | 14.73 ± 1.14 | 12.63 ± 1.15 | 13.92 ± 1.42 | 13 ± 1.58 | 13.66 ± 1.42 | 13.48 ± 1.02 | 13.76 ± 1.96 | 13.81 ± 1.37 | 14.07 ± 1.53 | 13.94 ± 1.51 | 14.36 ± 1.81 | 13.95 ± 1.35 |
| Hematocrit | 41.02 ± 4.44 | 44.48 ± 3.3 | 38.28 ± 3.12 | 41.94 ± 4.19 | 39.62 ± 4.46 | 41.27 ± 4.24 | 40.25 ± 3.19 | 42.1 ± 5 | 41.75 ± 4.79 | 41.76 ± 3.83 | 41.43 ± 3.58 | 42.49 ± 4.69 | 41.45 ± 3.49 |
| Platelet | 271.96 ± 64.65 | 260.91 ± 65.51 | 280.67 ± 62.78 | 278.19 ± 64.34 | 262.51 ± 64.22 | 260.67 ± 54.27 | 265.08 ± 64.48 | 259 ± 41.48 | 256.6 ± 56.21 | 246.58 ± 67.66 | 273.62 ± 87.19 | 239.1 ± 42.47 | 218.9 ± 48.65 |
| AST | 27.93 ± 9.7 | 30.18 ± 11.16 | 26.15 ± 7.97 | 29.45 ± 10.78 | 25.62 ± 7.24 | 26.61 ± 8.33 | 30.31 ± 9.64 | 24.7 ± 6.63 | 23.7 ± 6.73 | 26.88 ± 8.27 | 28.92 ± 10.9 | 24.9 ± 8.31 | 26.2 ± 2.2 |
| ALT | 27.07 ± 16.71 | 32.62 ± 20.38 | 22.69 ± 11.39 | 31.47 ± 18.49 | 20.41 ± 10.58 | 26.67 ± 12.54 | 29.38 ± 12.98 | 26 ± 11.6 | 23.8 ± 13.4 | 26.58 ± 14.63 | 29.54 ± 14.81 | 28.5 ± 19.58 | 20.8 ± 5.61 |
| r-GTP | 28.41 ± 32.58 | 41.98 ± 43.1 | 17.71 ± 13.37 | 37.66 ± 38.54 | 14.39 ± 9.86 | 30.48 ± 27.51 | 37.31 ± 32.85 | 33.2 ± 29.68 | 18.9 ± 12.08 | NA | NA | NA | NA |
| Bilirubin | 0.61 ± 0.35 | 0.72 ± 0.41 | 0.53 ± 0.26 | 0.61 ± 0.34 | 0.62 ± 0.37 | 0.88 ± 0.41 | 0.95 ± 0.54 | 0.84 ± 0.27 | 0.82 ± 0.37 | NA | NA | NA | NA |
| Creatinine | 0.84 ± 0.17 | 0.96 ± 0.17 | 0.75 ± 0.11 | 0.86 ± 0.18 | 0.81 ± 0.16 | 0.93 ± 0.2 | 0.92 ± 0.2 | 0.94 ± 0.18 | 0.94 ± 0.24 | 0.96 ± 0.16 | 0.97 ± 0.17 | 0.95 ± 0.12 | 0.95 ± 0.19 |
| CRP | 0.2 ± 0.33 | 0.19 ± 0.22 | 0.2 ± 0.39 | 0.22 ± 0.28 | 0.17 ± 0.39 | 0.18 ± 0.22 | 0.11 ± 0.1 | 0.31 ± 0.34 | 0.14 ± 0.07 | 2.98 ± 7.98 | 3.91 ± 10.85 | 4.28 ± 7.69 | 0.47 ± 0.46 |
| HDL | 44.15 ± 9.1 | 43.35 ± 9.15 | 44.77 ± 9.04 | 41.59 ± 7.91 | 48.02 ± 9.45 | 46.06 ± 7.66 | 45 ± 5.74 | 45.6 ± 9.24 | 47.9 ± 8.57 | 46.64 ± 10.39 | 41.15 ± 6.04 | 44.6 ± 8.33 | 55.8 ± 11.15 |
| TG | 157.86 ± 91.54 | 165.3 ± 104.61 | 151.99 ± 79.54 | 191.65 ± 100.03 | 106.64 ± 39.77 | 129.52 ± 77.49 | 123.85 ± 50.05 | 143.2 ± 125.7 | 123.2 ± 43.74 | 147.27 ± 131.55 | 205.15 ± 186.48 | 130.6 ± 71.83 | 88.7 ± 32.22 |
| Glu0 | 91.55 ± 27.85 | 94.96 ± 26.51 | 88.86 ± 28.65 | 97.11 ± 30.72 | 83.13 ± 20.19 | 87.58 ± 12.64 | 86.69 ± 9.65 | 89.4 ± 19.34 | 86.9 ± 8.05 | 99.61 ± 33.58 | 93.38 ± 7.15 | 116.6 ± 58.5 | 90.7 ± 7.33 |
| Drinking | 5 ± 15.43 | 10.84 ± 21.82 | 0.39 ± 1.95 | 6.85 ± 19.06 | 2.2 ± 6.07 | 6.36 ± 9.98 | 6.19 ± 10.78 | 8.35 ± 10.56 | 4.58 ± 8.94 | 7.54 ± 14.18 | 6.53 ± 13.43 | 11.3 ± 15.84 | 5.09 ± 14.12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ko, Y.K.; Kim, H.; Lee, Y.; Lee, Y.-S.; Gim, J.-A. DNA Methylation Patterns According to Fatty Liver Index and Longitudinal Changes from the Korean Genome and Epidemiology Study (KoGES). Curr. Issues Mol. Biol. 2022, 44, 1149-1168. https://doi.org/10.3390/cimb44030075
Ko YK, Kim H, Lee Y, Lee Y-S, Gim J-A. DNA Methylation Patterns According to Fatty Liver Index and Longitudinal Changes from the Korean Genome and Epidemiology Study (KoGES). Current Issues in Molecular Biology. 2022; 44(3):1149-1168. https://doi.org/10.3390/cimb44030075
Chicago/Turabian StyleKo, Young Kyung, Hayeon Kim, Yoonseok Lee, Young-Sun Lee, and Jeong-An Gim. 2022. "DNA Methylation Patterns According to Fatty Liver Index and Longitudinal Changes from the Korean Genome and Epidemiology Study (KoGES)" Current Issues in Molecular Biology 44, no. 3: 1149-1168. https://doi.org/10.3390/cimb44030075
APA StyleKo, Y. K., Kim, H., Lee, Y., Lee, Y.-S., & Gim, J.-A. (2022). DNA Methylation Patterns According to Fatty Liver Index and Longitudinal Changes from the Korean Genome and Epidemiology Study (KoGES). Current Issues in Molecular Biology, 44(3), 1149-1168. https://doi.org/10.3390/cimb44030075