
Renoprotective Effect of Liraglutide Is Mediated via the Inhibition of TGF-Beta 1 in an LLC-PK1 Cell Model of Diabetic Nephropathy

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Assessment of Cell Viability
2.3. Assesment of the Oxidative Stress
2.4. Measurement of TGF-β Levels in an In Vitro Mimic Model of DN in Proximal Tubule Cells
2.5. Measurement of ECM Expression in an In Vitro Mimic Model of DN in Proximal Tubule Cells
2.6. Protein Extraction and Western Blot Method in the In Vitro Mimic Model of DN in Proximal Tubule Cells
2.7. Measurement of Treatment Effectiveness the and Influence on Cell Morphology by Visualizing the F-Actin Cytoskelet with Phalloidin Stain
3. Results
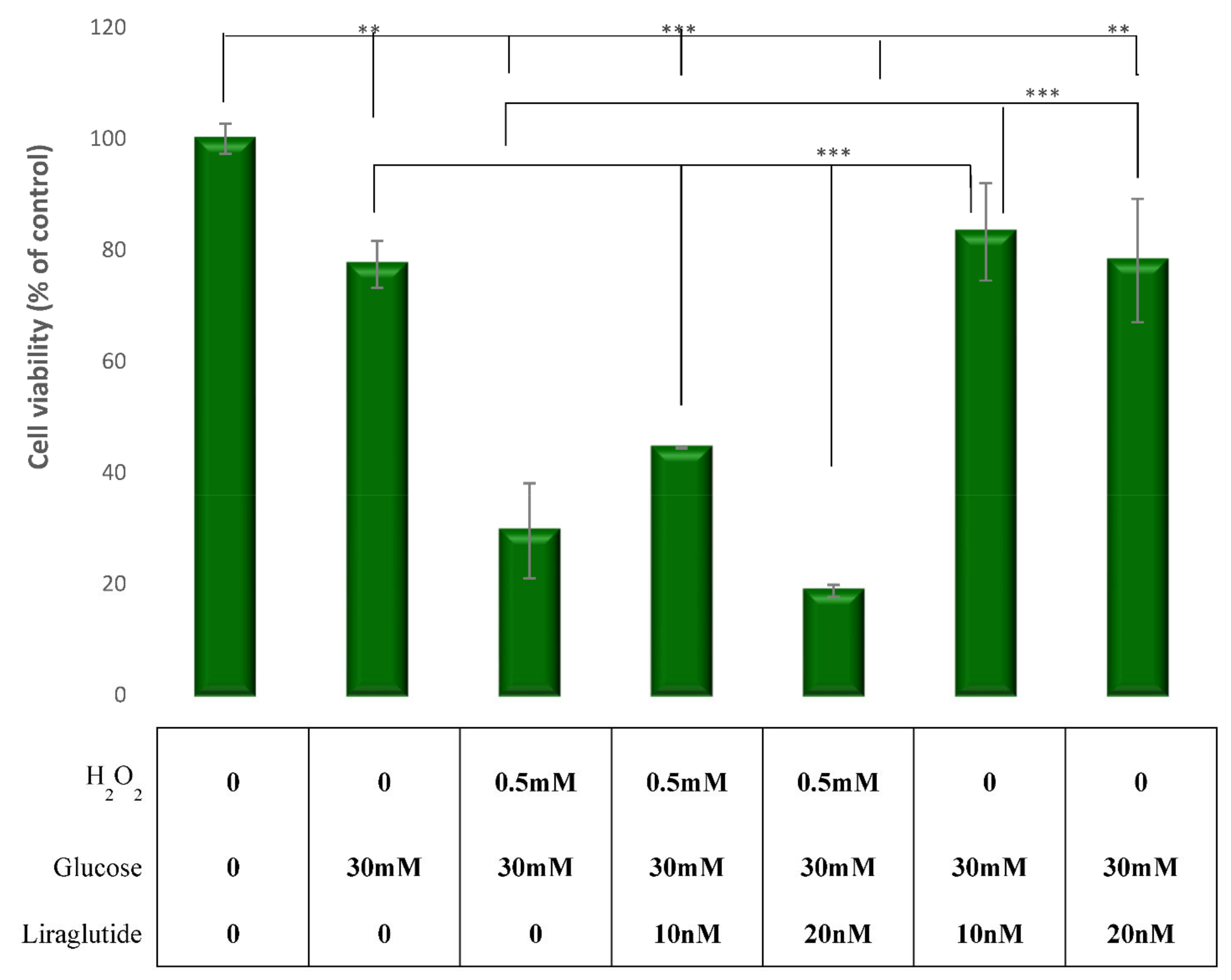
3.1. Cell Viability in the LLC-PK1 Cell Line
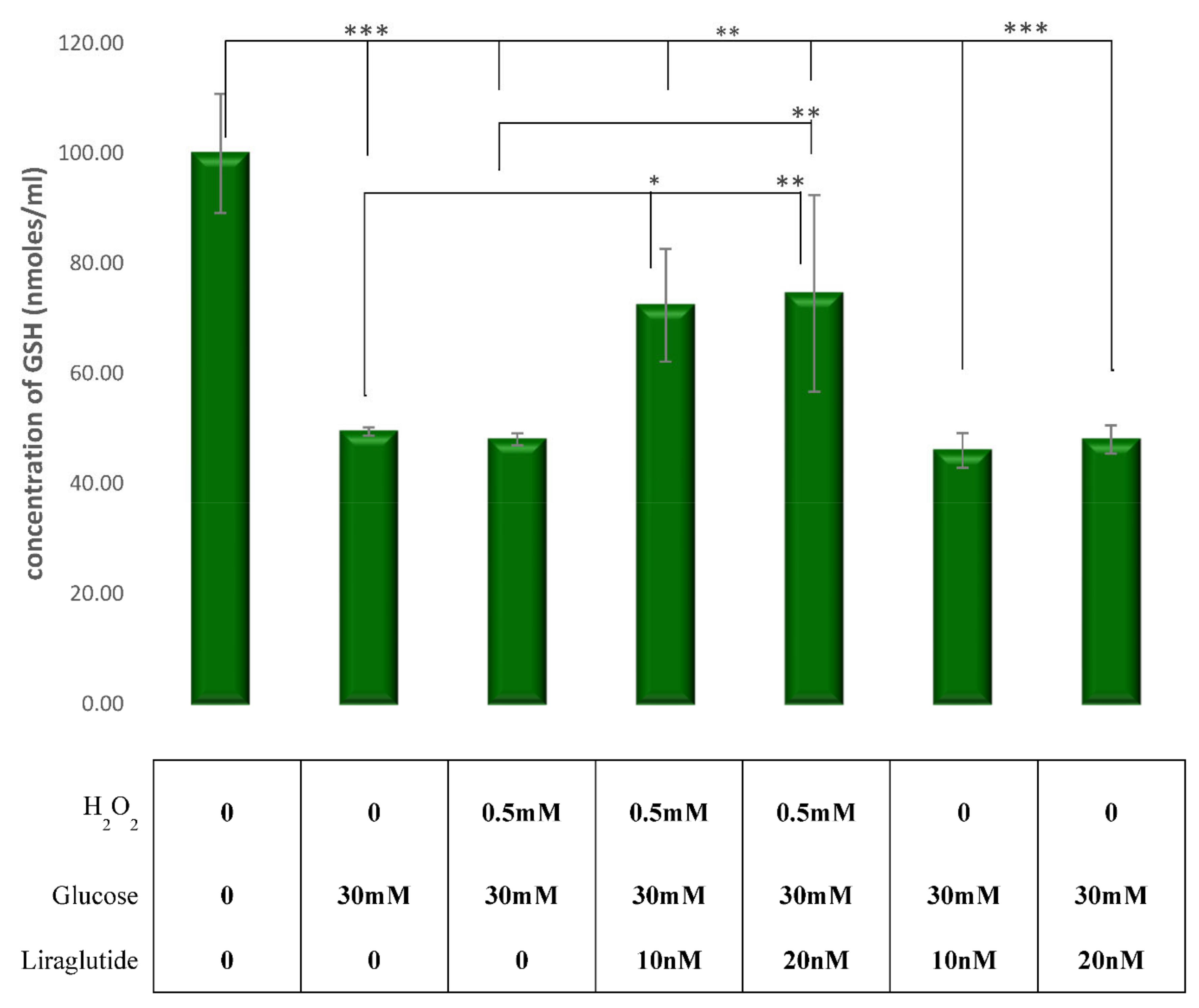
3.2. Measurement of Cellular Glutathione (GSH) Concentration in the LLC-PK1 Cell Line
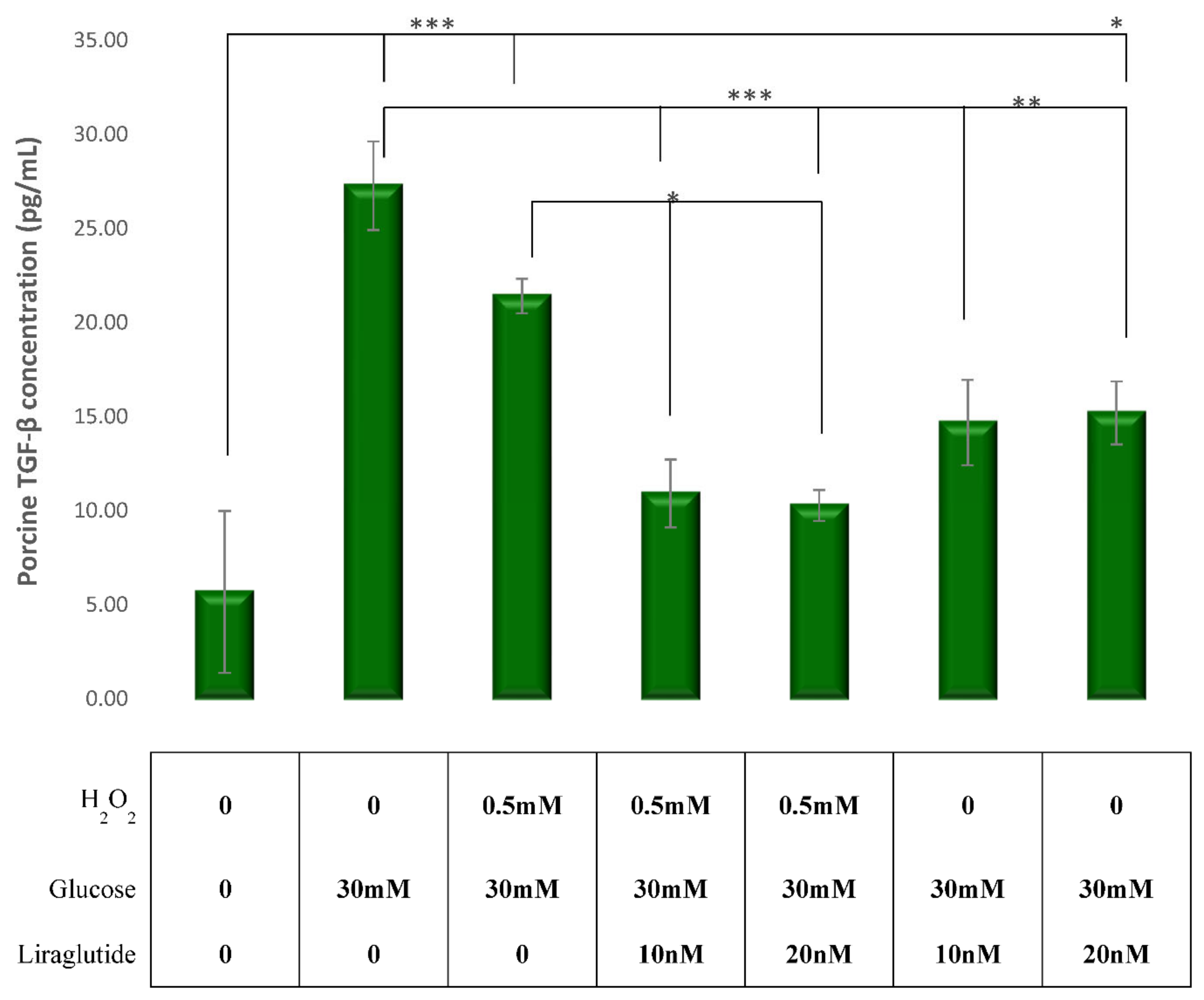
3.3. Effects of Glucose, H2O2 and Liraglutide on TGF-β1 Levels
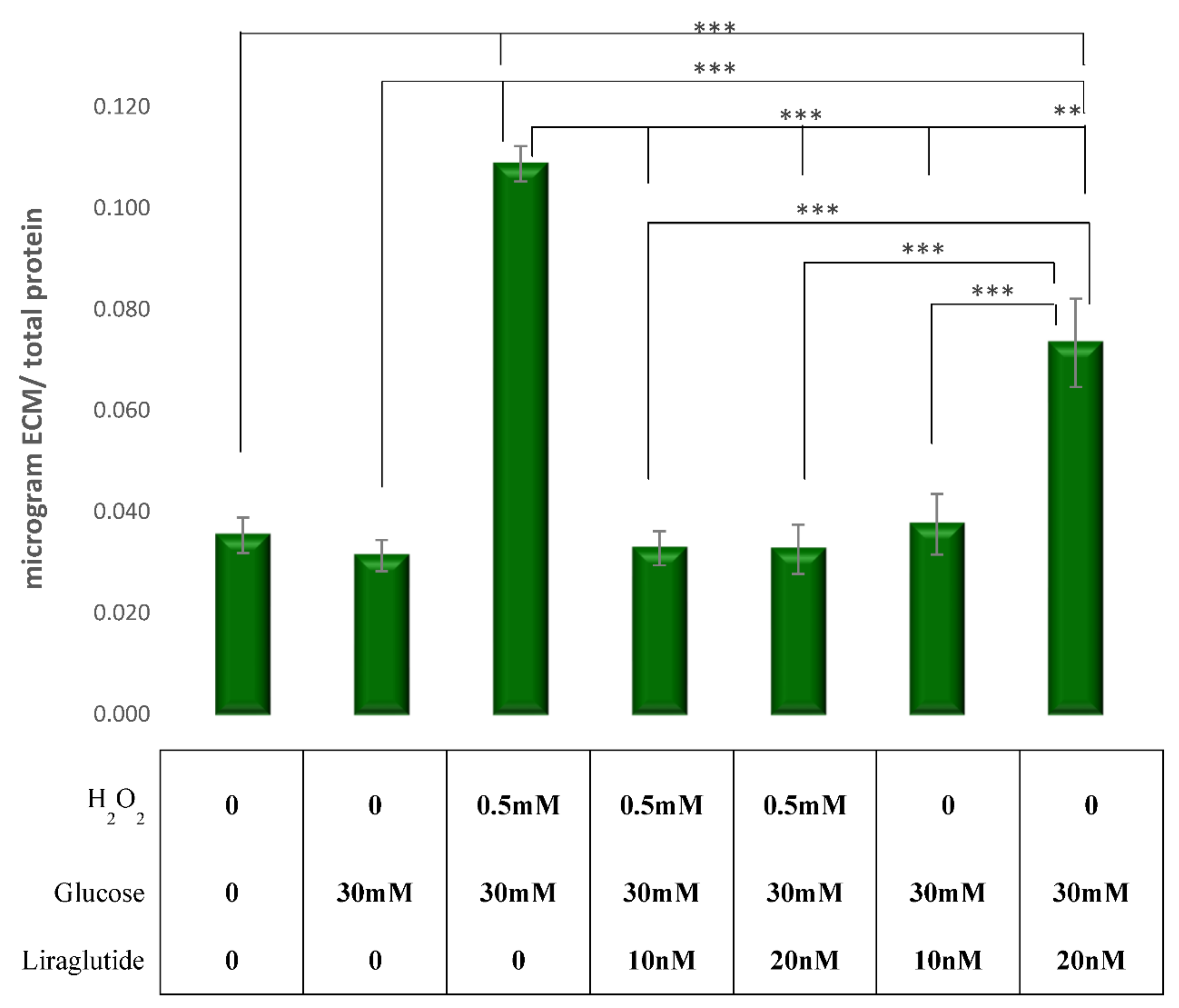
3.4. Measurement of ECM Expression in a Cell Culture Model of Diabetic Nephropathy
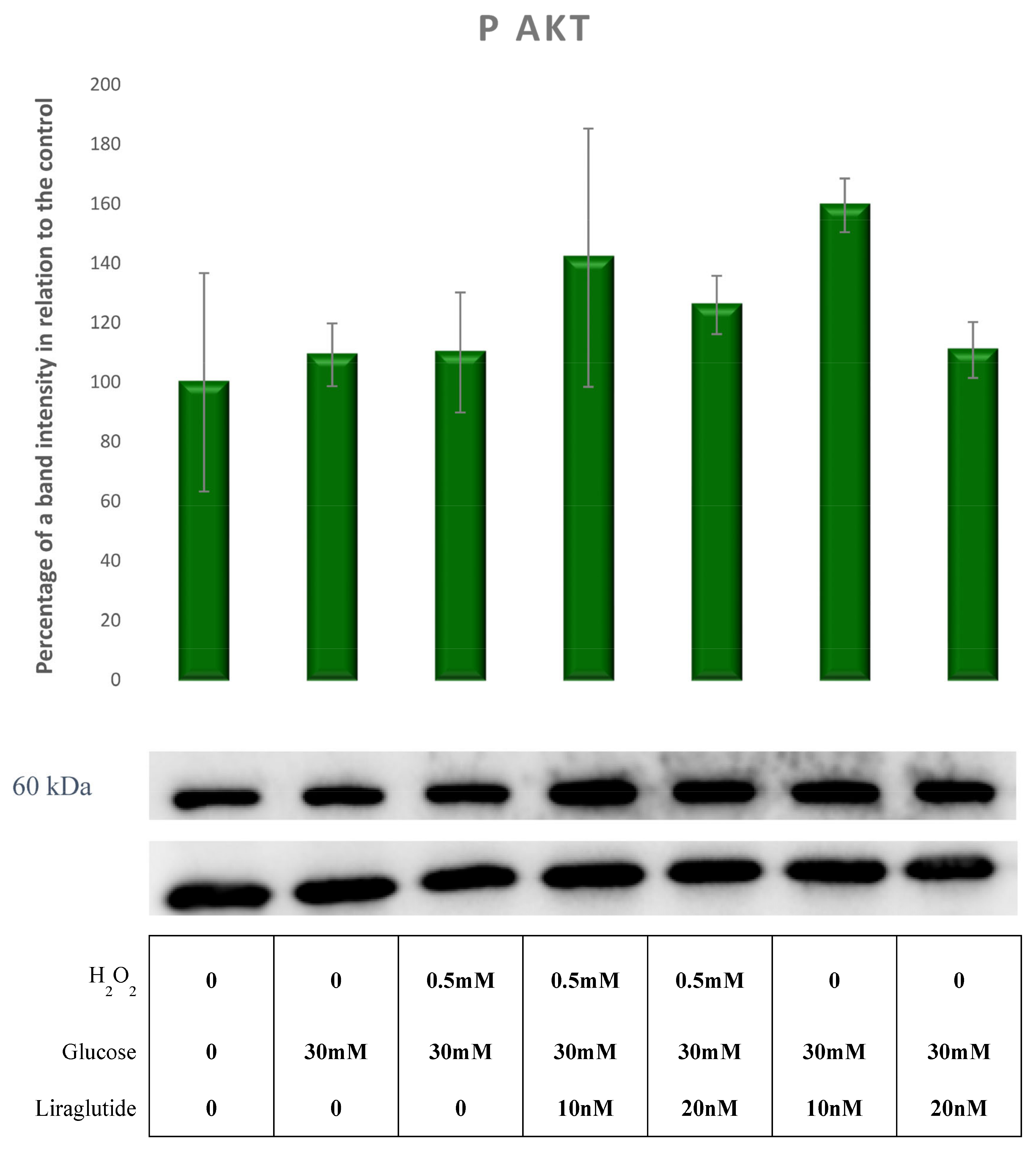
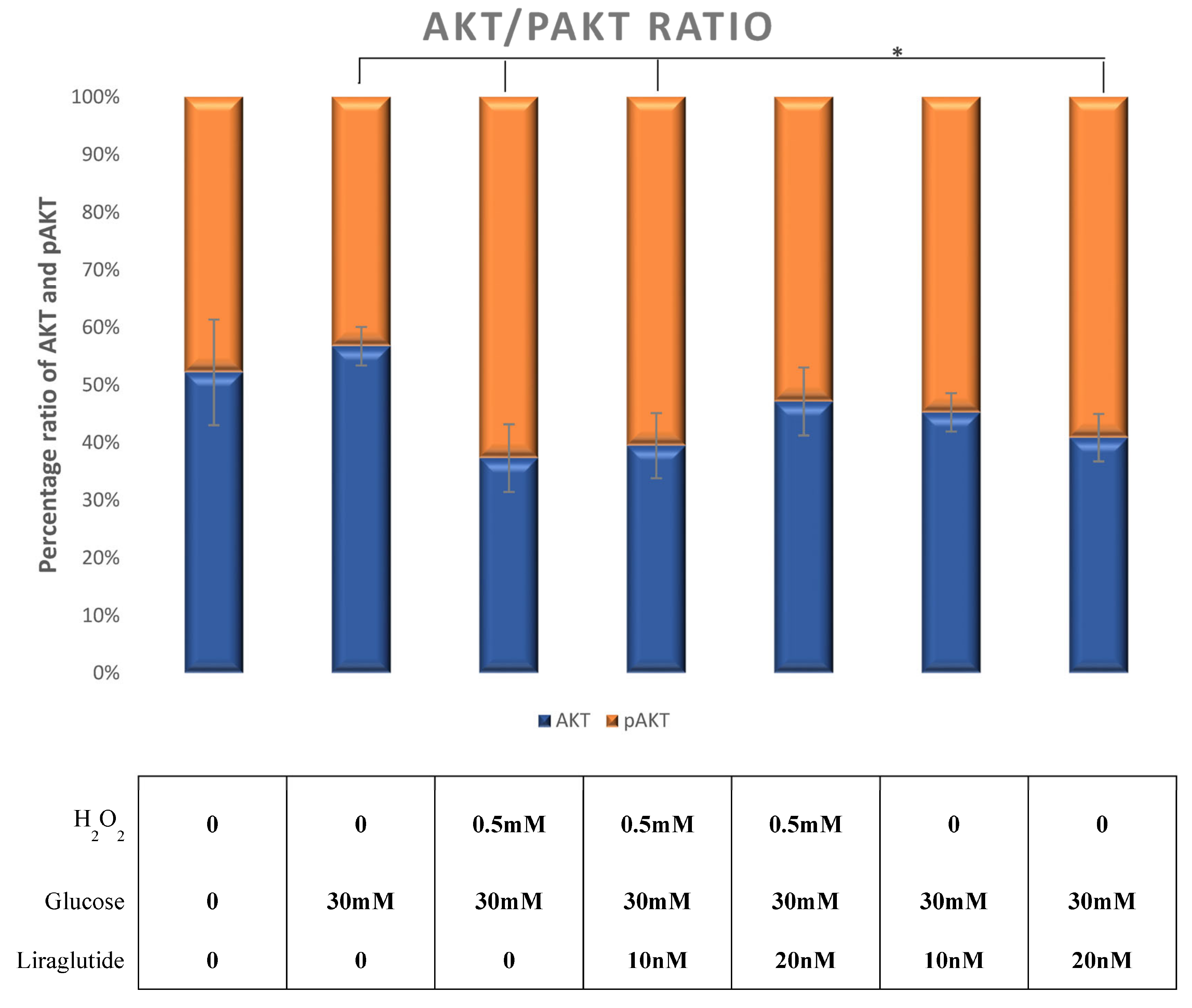
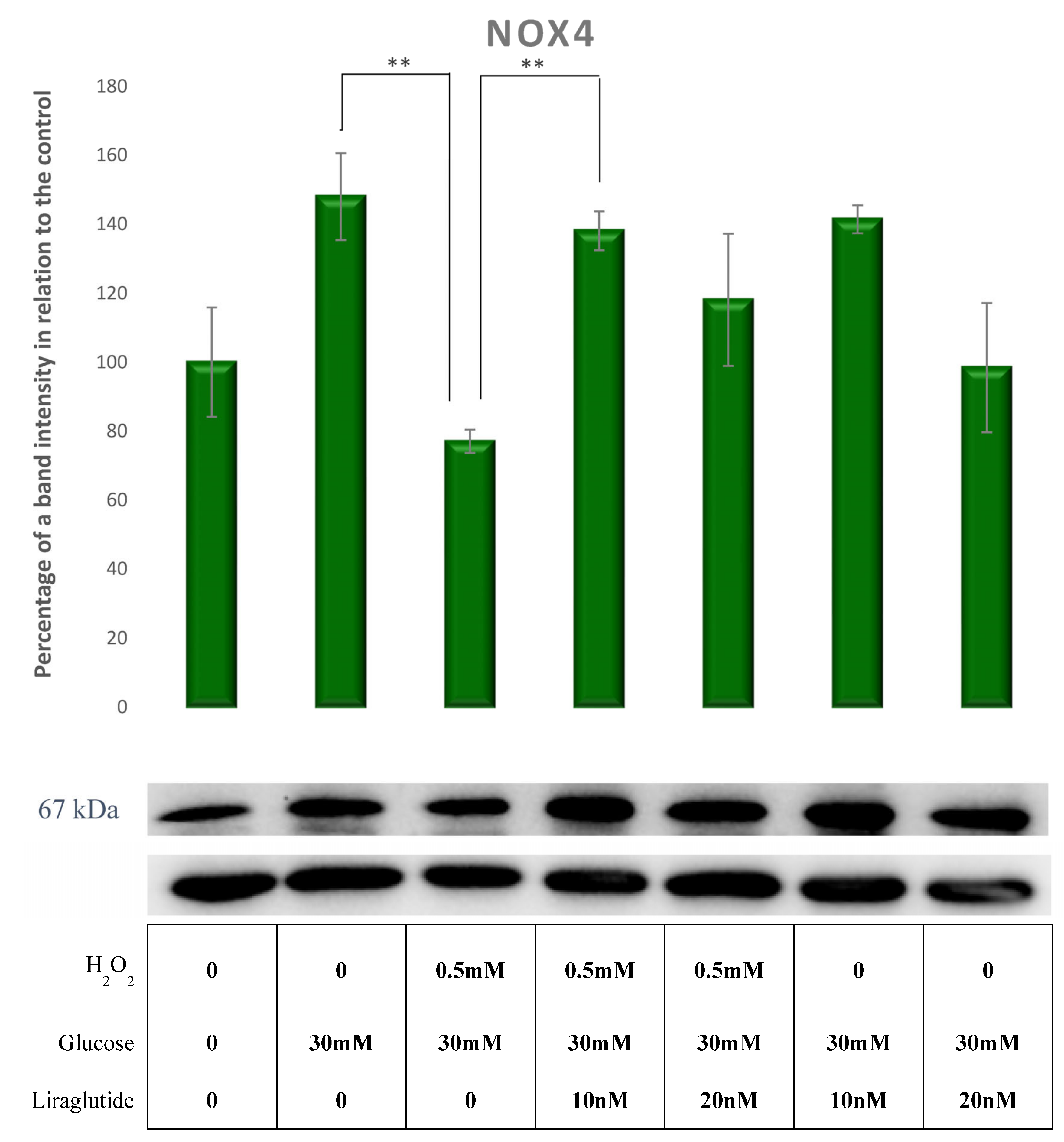
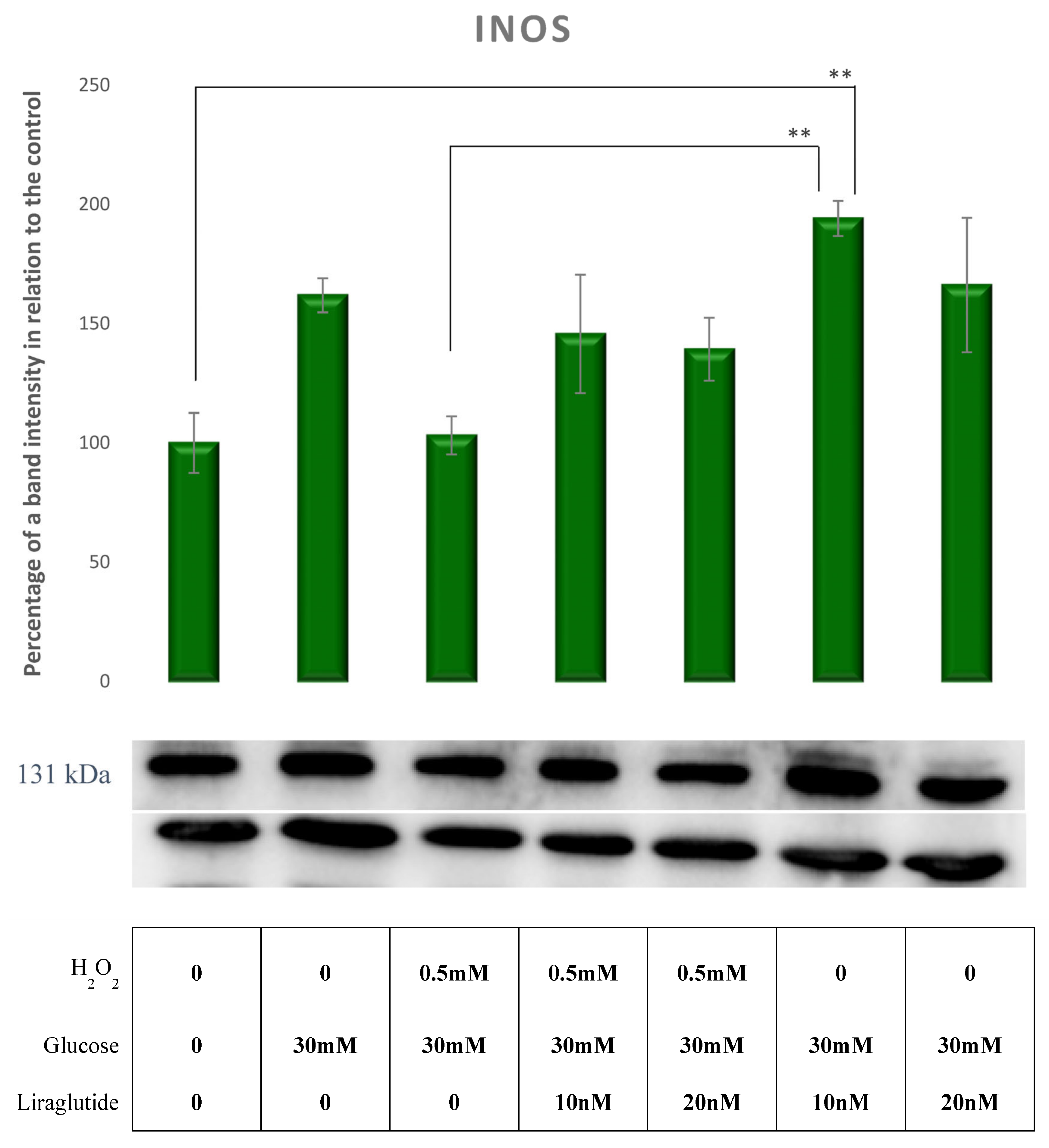
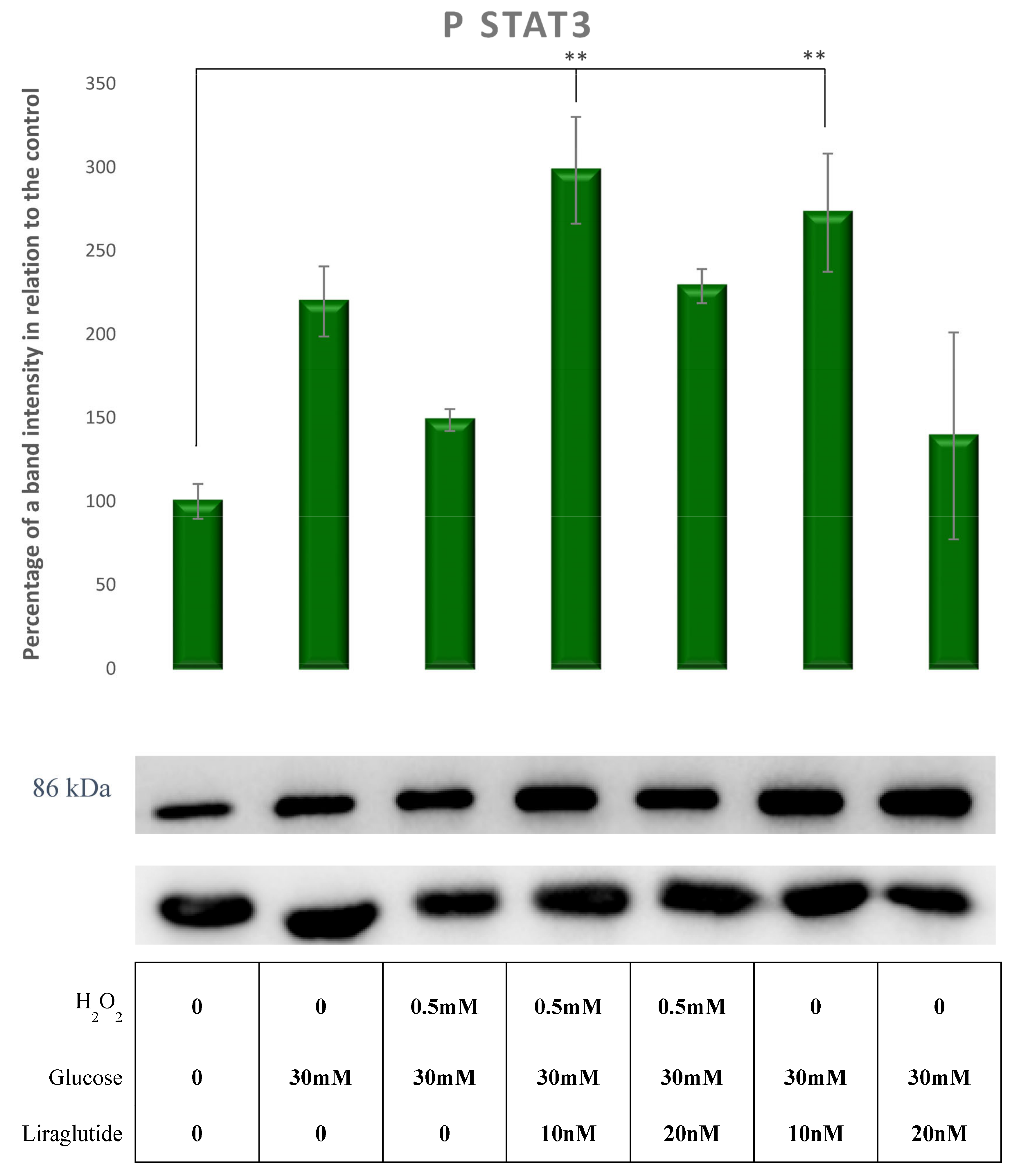
3.5. Protein Extraction and Western Blot Method in a Cell Culture Model of Diabetic Nephropathy
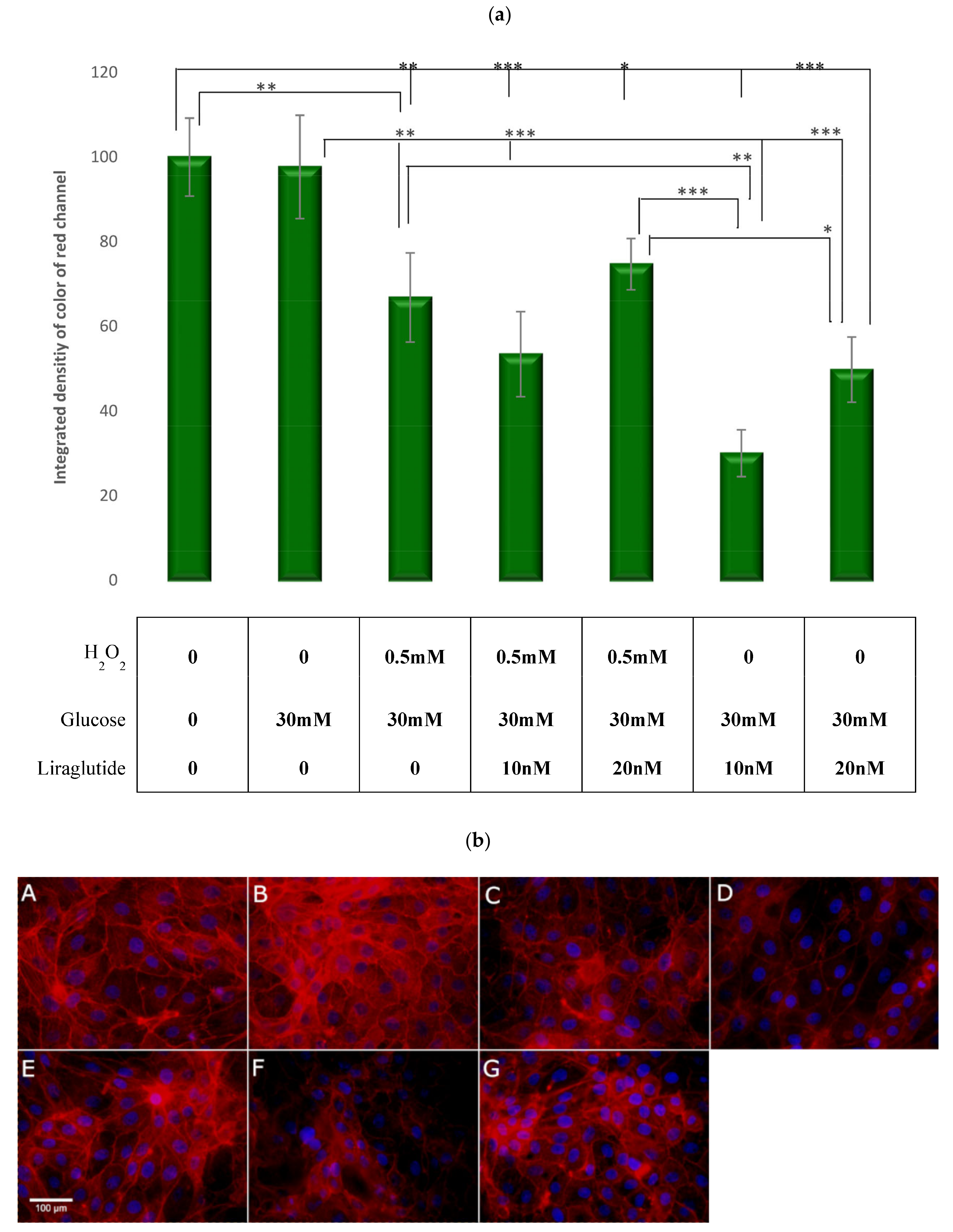
3.6. Visualization and Quantification of the F-Actin Cytoskelet with Phalloidin Stain
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thomas, B. The Global Burden of Diabetic Kidney Disease: Time Trends and Gender Gaps. Curr. Diabetes Rep. 2019, 19, 18. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.J.; Cardwell, C.R.; Patterson, C.C.; Maxwell, A.P.; Magee, G.M.; Young, R.J.; Matthews, B.; O’Donoghue, D.J.; Fogarty, D.G. Chronic kidney disease and diabetes in the national health service: A cross-sectional survey of the U.K. national diabetes audit. Diabetes Med. 2014, 31, 448–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosterd, C.M.; Bjornstad, P.; van Raalte, D.H. Nephroprotective effects of GLP-1 receptor agonists: Where do we stand? J. Nephrol. 2020, 33, 965–975. [Google Scholar] [CrossRef]
- Baggio, L.L.; Drucker, D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef]
- Kawanami, D.; Takashi, Y. GLP-1 Receptor Agonists in Diabetic Kidney Disease: From Clinical Outcomes to Mechanisms. Front. Pharmacol. 2020, 11, 967. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Xu, S.; Wang, Z.; Liu, H.; Peng, L.; Fang, Q.; Deng, T.; Zhang, W.; Lou, J. Recombinant human GLP-1(rhGLP-1) alleviating renal tubulointestitial injury in diabetic STZ-induced rats. Biochem. Biophys. Res. Commun. 2018, 495, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Hills, C.E.; Al-Rasheed, N.; Willars, G.B.; Brunskill, N.J. C-peptide reverses TGF-beta1-induced changes in renal proximal tubular cells: Implications for treatment of diabetic nephropathy. Am. J. Physiol. Renal. Physiol. 2009, 296, F614–F621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heldin, C.H.; Landström, M.; Moustakas, A. Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr. Opin. Cell. Biol. 2009, 21, 166–176. [Google Scholar] [CrossRef]
- Daniels, M.C.; McClain, D.A.; Crook, E.D. Transcriptional regulation of transforming growth factor beta1 by glucose: Investigation into the role of the hexosamine biosynthesis pathway. Am. J. Med. Sci. 2000, 319, 138–142. [Google Scholar] [CrossRef]
- Orphanides, C.; Fine, L.G.; Norman, J.T. Hypoxia stimulates proximal tubular cell matrix production via a TGF-beta1-independent mechanism. Kidney Int. 1997, 52, 637–647. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.C.; Saunders, H.J.; Pollock, C.A. High glucose increases growth and collagen synthesis in cultured human tubulointerstitial cells. Diabet. Med. 1999, 16, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Yang, Y.; Liu, Y.T.; Zhu, J. Liraglutide Regulates the Kidney and Liver in Diabetic Nephropathy Rats through the miR-34a/SIRT1 Pathway. J. Diabetes Res. 2021, 2021, 8873956. [Google Scholar] [CrossRef]
- Cho, H.; Thorvaldsen, J.L.; Chu, Q.; Feng, F.; Birnbaum, M.J. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J. Biol. Chem. 2001, 276, 38349–38352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, K.; Chen, Z.; Gao, J.; Shi, W.; Li, L.; Jiang, S.; Hu, H.; Liu, Z.; Xu, D.; Wu, L. The Key Roles of GSK-3β in Regulating Mitochondrial Activity. Cell. Physiol. Biochem. 2017, 44, 1445–1459. [Google Scholar] [CrossRef]
- Zhang, X.; Lu, Z.; Abdul, K.S.M.; Changping, M.A.; Tan, K.S.; Jovanovi, A.; Tan, W. Isosteviol sodium protects heart embryonic H9c2 cells against oxidative stress by activating Akt/GSK-3β signaling pathway. Pharmazie 2020, 75, 36–40. [Google Scholar] [CrossRef]
- Stark, G.R.; Darnell, J.E. The JAK-STAT pathway at twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darnell, J.E. STATs and gene regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef]
- Kawai, M.; Namba, N.; Mushiake, S.; Etani, Y.; Nishimura, R.; Makishima, M.; Ozono, K. Growth hormone stimulates adipogenesis of 3T3-L1 cells through activation of the Stat5A/5B-PPARgamma pathway. J. Mol. Endocrinol. 2007, 38, 19–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opazo-Ríos, L.; Sanchez Matus, Y.; Rodrigues-Díez, R.R.; Carpio, D.; Droguett, A.; Egido, J.; Gomez-Guerrero, C.; Mezzano, S. Anti-inflammatory, antioxidant and renoprotective effects of SOCS1 mimetic peptide in the BTBR ob/ob mouse model of type 2 diabetes. BMJ Open Diabetes Res. Care 2020, 8, e001242. [Google Scholar] [CrossRef]
- Laddha, A.P.; Kulkarni, Y.A. NADPH oxidase: A membrane-bound enzyme and its inhibitors in diabetic complications. Eur. J. Pharmacol. 2020, 881, 173206. [Google Scholar] [CrossRef] [PubMed]
- Gorin, Y.; Block, K. Nox4 and diabetic nephropathy: With a friend like this, who needs enemies? Free Radic. Biol. Med. 2013, 61, 130–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.Y.; Noh, H.J.; Gang, J.G.; Xu, Z.G.; Jeong, H.J.; Kang, S.W.; Choi, K.H.; Han, D.S. Inducible nitric oxide synthase (iNOS) expression is increased in lipopolysaccharide (LPS)-stimulated diabetic rat glomeruli: Effect of ACE inhibitor and angiotensin II receptor blocker. Yonsei Med. J. 2002, 43, 183–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, M.; Fujimoto, S.; Haruna, Y.; Arakawa, S.; Horike, H.; Komai, N.; Sasaki, T.; Tsujioka, K.; Makino, H.; Kashihara, N. NAD(P)H oxidase and uncoupled nitric oxide synthase are major sources of glomerular superoxide in rats with experimental diabetic nephropathy. Am. J. Physiol. Renal. Physiol. 2005, 288, F1144–F1152. [Google Scholar] [CrossRef]
- Wardle, E.N. Cellular oxidative processes in relation to renal disease. Am. J. Nephrol. 2005, 25, 13–22. [Google Scholar] [CrossRef]
- Gorin, Y.; Block, K.; Hernandez, J.; Bhandari, B.; Wagner, B.; Barnes, J.L.; Abboud, H.E. Nox4 NAD(P)H oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. J. Biol. Chem. 2005, 280, 39616–39626. [Google Scholar] [CrossRef] [Green Version]
- Han, D.C.; Isono, M.; Hoffman, B.B.; Ziyadeh, F.N. High glucose stimulates proliferation and collagen type I synthesis in renal cortical fibroblasts: Mediation by autocrine activation of TGF-beta. J. Am. Soc. Nephrol. 1999, 10, 1891–1899. [Google Scholar] [CrossRef] [PubMed]
- Mima, A.; Hiraoka-Yamomoto, J.; Li, Q.; Kitada, M.; Li, C.; Geraldes, P.; Matsumoto, M.; Mizutani, K.; Park, K.; Cahill, C.; et al. Protective effects of GLP-1 on glomerular endothelium and its inhibition by PKCβ activation in diabetes. Diabetes 2012, 61, 2967–2979. [Google Scholar] [CrossRef] [Green Version]
- Hendarto, H.; Inoguchi, T.; Maeda, Y.; Ikeda, N.; Zheng, J.; Takei, R.; Yokomizo, H.; Hirata, E.; Sonoda, N.; Takayanagi, R. GLP-1 analog liraglutide protects against oxidative stress and albuminuria in streptozotocin-induced diabetic rats via protein kinase A-mediated inhibition of renal NAD(P)H oxidases. Metabolism 2012, 61, 1422–1434. [Google Scholar] [CrossRef]
- Kodera, R.; Shikata, K.; Kataoka, H.U.; Takatsuka, T.; Miyamoto, S.; Sasaki, M.; Kajitani, N.; Nishishita, S.; Sarai, K.; Hirota, D.; et al. Glucagon-like peptide-1 receptor agonist ameliorates renal injury through its anti-inflammatory action without lowering blood glucose level in a rat model of type 1 diabetes. Diabetologia 2011, 54, 965–978. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.S.; Ma, M.C.; Chen, J.; Chen, C.F. Changes in the oxidant-antioxidant balance in the kidney of rats with nephrolithiasis induced by ethylene glycol. J. Urol. 2002, 167, 2584–2593. [Google Scholar] [CrossRef]
- Rizzo, M.; Abate, N.; Chandalia, M.; Rizvi, A.A.; Giglio, R.V.; Nikolic, D.; Marino Gammazza, A.; Barbagallo, I.; Isenovic, E.R.; Banach, M.; et al. Liraglutide reduces oxidative stress and restores heme oxygenase-1 and ghrelin levels in patients with type 2 diabetes: A prospective pilot study. J. Clin. Endocrinol. Metab. 2015, 100, 603–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krause, G.C.; Lima, K.G.; Dias, H.B.; da Silva, E.F.G.; Haute, G.V.; Basso, B.S.; Gassen, R.B.; Marczak, E.S.; Nunes, R.S.B.; de Oliveira, J.R. Liraglutide, a glucagon-like peptide-1 analog, induce autophagy and senescence in HepG2 cells. Eur. J. Pharmacol. 2017, 809, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Navarro-González, J.F.; Mora-Fernández, C. The role of inflammatory cytokines in diabetic nephropathy. J. Am. Soc. Nephrol. 2008, 19, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.C.; Chen, Y.L.; Pan, Y.H.; Ling, W.; Tian, F.; Zhang, X.X.; Zhao, H.L. Changes of transforming growth factor beta 1 in patients with type 2 diabetes and diabetic nephropathy: A PRISMA-compliant systematic review and meta-analysis. Medicine 2017, 96, e6583. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kimura, H.; Hirota, K.; Sugimoto, H.; Yoshida, H. Hypoxia reduces constitutive and TNF-alpha-induced expression of monocyte chemoattractant protein-1 in human proximal renal tubular cells. Biochem. Biophys. Res. Commun. 2005, 335, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, D.-X.; Wang, Z.-M.; Hu, X.-F.; Li, S.-L.; Tian, H.-Z.; Wang, M.-J.; Shu, Y.-W.; Yang, J. The glucagon-like peptide-1 (GLP-1) analog liraglutide attenuates renal fibrosis. Pharmacol. Res. 2018, 131, 102–111. [Google Scholar] [CrossRef]
- Huang, L.; Lin, T.; Shi, M.; Chen, X.; Wu, P. Liraglutide suppresses production of extracellular matrix proteins and ameliorates renal injury of diabetic nephropathy by enhancing Wnt/β-catenin signaling. Am. J. Physiol. Renal. Physiol. 2020, 319, F458–F468. [Google Scholar] [CrossRef]
- Morrisey, K.; Steadman, R.; Williams, J.D.; Phillips, A.O. Renal proximal tubular cell fibronectin accumulation in response to glucose is polyol pathway dependent. Kidney Int. 1999, 55, 2548–2572. [Google Scholar] [CrossRef]
- Iglesias-De La Cruz, M.C.; Ruiz-Torres, P.; Alcamí, J.; Díez-Marqués, L.; Ortega-Velázquez, R.; Chen, S.; Rodríguez-Puyol, M.; Ziyadeh, F.N.; Rodríguez-Puyol, D. Hydrogen peroxide increases extracellular matrix mRNA through TGF-beta in human mesangial cells. Kidney Int. 2001, 59, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palau, V.; Nugraha, B.; Benito, D.; Pascual, J.; Emmert, M.Y.; Hoerstrup, S.P.; Riera, M.; Soler, M.J. Both Specific Endothelial and Proximal Tubular Adam17 Deletion Protect against Diabetic Nephropathy. Int. J. Mol. Sci. 2021, 22, 5520. [Google Scholar] [CrossRef] [PubMed]
- Milani, L.; Galindo, C.M.; Turin de Oliveira, N.M.; Corso, C.R.; Adami, E.R.; Stipp, M.C.; Beltrame, O.C.; Acco, A. The GLP-1 analog liraglutide attenuates acute liver injury in mice. Ann. Hepatol. 2019, 18, 918–928. [Google Scholar] [CrossRef] [PubMed]
- Schunk, S.J.; Floege, J.; Fliser, D.; Speer, T. WNT-β-catenin signalling—A versatile player in kidney injury and repair. Nat. Rev. Nephrol. 2021, 17, 172–184. [Google Scholar] [CrossRef]
- Zhang, X.; Fu, Y.; Xu, X.; Li, M.; Du, L.; Han, Y.; Ge, Y. PERK pathway are involved in NO-induced apoptosis in endothelial cells cocultured with RPE under high glucose conditions. Nitric Oxide 2014, 40, 10–16. [Google Scholar] [CrossRef]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tessari, P.; Cecchet, D.; Cosma, A.; Vettore, M.; Coracina, A.; Millioni, R.; Iori, E.; Puricelli, L.; Avogaro, A.; Vedovato, M. Nitric oxide synthesis is reduced in subjects with type 2 diabetes and nephropathy. Diabetes 2010, 59, 2152–2159. [Google Scholar] [CrossRef] [Green Version]
- Goligorsky, M.S.; Chen, J.; Brodsky, S. Workshop: Endothelial cell dysfunction leading to diabetic nephropathy: Focus on nitric oxide. Hypertension 2001, 37, 744–748. [Google Scholar] [CrossRef] [Green Version]
- Heemskerk, S.; Masereeuw, R.; Russel, F.G.; Pickkers, P. Selective iNOS inhibition for the treatment of sepsis-induced acute kidney injury. Nat. Rev. Nephrol. 2009, 5, 629–640. [Google Scholar] [CrossRef]
- Tikoo, K.; Singh, K.; Kabra, D.; Sharma, V.; Gaikwad, A. Change in histone H3 phosphorylation, MAP kinase p38, SIR 2 and p53 expression by resveratrol in preventing streptozotocin induced type I diabetic nephropathy. Free Radic. Res. 2008, 42, 397–404. [Google Scholar] [CrossRef]
- Singh, D.K.; Winocour, P.; Farrington, K. Oxidative stress in early diabetic nephropathy: Fueling the fire. Nat. Rev. Endocrinol. 2011, 7, 176–184. [Google Scholar] [CrossRef]
- Sedeek, M.; Callera, G.; Montezano, A.; Gutsol, A.; Heitz, F.; Szyndralewiez, C.; Page, P.; Kennedy, C.R.; Burns, K.D.; Touyz, R.M.; et al. Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: Implications in type 2 diabetic nephropathy. Am. J. Physiol. Renal. Physiol. 2010, 299, F1348–F1358. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, J.F.; Bertels, I.M.; Gontijo, J.A. Actin cytoskeletal and functional studies of the proximal convoluted tubules after preservation. Transplant. Proc. 2008, 40, 3311–3315. [Google Scholar] [CrossRef]
- Chen, P.Y.; Shih, N.L.; Hao, W.R.; Chen, C.C.; Liu, J.C.; Sung, L.C. Inhibitory Effects of Momordicine I on High-Glucose-Induced Cell Proliferation and Collagen Synthesis in Rat Cardiac Fibroblasts. Oxidative Med. Cell Longev. 2018, 2018, 3939714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Meng, X.W.; Yang, X.S.; Liu, X.F.; Ou-Yang, C.H.; Liu, C. Curcumin administration suppresses collagen synthesis in the hearts of rats with experimental diabetes. Acta Pharmacol. Sin. 2018, 39, 195–204. [Google Scholar] [CrossRef]
- Xie, X.W. Liquiritigenin attenuates cardiac injury induced by high fructose-feeding through fibrosis and inflammation suppression. Biomed. Pharmacother. 2017, 86, 694–704. [Google Scholar] [CrossRef]
- DalleDonne, I.; Milzani, A.; Colombo, R. H2O2-treated actin: Assembly and polymer interactions with cross-linking proteins. Biophys. J. 1995, 69, 2710–2719. [Google Scholar] [CrossRef] [Green Version]
- Lv, Z.; Hu, M.; Ren, X.; Fan, M.; Zhen, J.; Chen, L.; Lin, J.; Ding, N.; Wang, Q.; Wang, R. Fyn Mediates High Glucose-Induced Actin Cytoskeleton Reorganization of Podocytes via Promoting ROCK Activation In Vitro. J. Diabetes Res. 2016, 2016, 5671803. [Google Scholar] [CrossRef] [Green Version]
- Nemoto, O.; Kawaguchi, M.; Yaoita, H.; Miyake, K.; Maehara, K.; Maruyama, Y. Left ventricular dysfunction and remodeling in streptozotocin-induced diabetic rats. Circ. J. 2006, 70, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Romanelli, G.; Varela, R.; Benech, J.C. Diabetes induces differences in the F-actin spatial organization of striated muscles. Cytoskeleton 2020, 77, 202–213. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody Label | Full Name of the Antibody | Antibody Classification | Host Species | Manufacturer and Catalog Number | Dilution Used for Western Blot |
|---|---|---|---|---|---|
| Akt | Anti-protein kinase B | IgG monoclonal | Mouse | Cell signaling, Danvers, MA, USA Cat. No. 2920S | 1:2000 |
| pAkt | Anti-protein kinase B—phosphorylated on serine 473 | IgG monoclonal | Rabbit | Cell signaling, Danvers, MA, USA Cat. No. 9271S | 1:2000 |
| GSK3 β | Anti-glycogen synthase kinase 3 alpha + beta | IgG monoclonal | Rabbit | Cell signaling, Danvers, MA, USA Cat. No. 5676S | 1:2000 |
| pGSK3 β | Anti-GSK3 beta—phosphorylated on tyrosine 216 + GSK3 alpha—phosphorylated on tyrosine 279 | IgG polyclonal | Rabbit | Abcam, Cambridge, UK Cat. No. ab75745 | 1:2000 |
| SOCS3 | Anti-suppressor of cytokine signaling-3 | IgG | Mouse | Abcam, Cambridge, UK Cat. No. ab14939 | 1:1000 |
| pSTAT3 | Anti-signal transducer and activator of transcription 3—phosphorylated on tyrosine 705 | IgG monoclonal | Rabbit | Cell signaling, Danvers, MA, USA Cat. No. 9145S | 1:2000 |
| NOX4 | Anti-NADPH oxidase 4 | IgG | Goat | Santa Cruz Biotechnology, Dallas, TX, USA Cat. No. 21860 | 1:1000 |
| iNOS | Anti-inducible nitric oxide synthase | IgG | Rabbit | Novus Biologicals, Littleton, CO, USA Cat. No. NB300-605 | 1:1000 |
| αGO-biotin | Anti-goat antibody labeled with biotin | IgG | Donkey | Abcam, Cambridge, UK Cat. No. ab6884 | 1:20,000 |
| αMO-biotin | Anti-mouse antibody labeled with biotin | IgG | Goat | Jackson Immunoresearch, West Grove, PA, USA Cat. No. 115-065-071 | 1:20,000 |
| αRB-HRP | Anti-rabbit antibody labeled with HRP | IgG | Goat | Jackson Immunoresearch, West Grove, PA, USA Cat. No. 111-035-144 | 1:20,000 |
| SA-HRP | Streptavidin peroxidase polymer, Ultrasensitive | - | - | Sigma-Aldrich, St. Louis, MO, USA Cat. No. S2438 | 1:1000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ninčević, V.; Zjalić, M.; Kolarić, T.O.; Smolić, M.; Kizivat, T.; Kuna, L.; Včev, A.; Tabll, A.; Ćurčić, I.B. Renoprotective Effect of Liraglutide Is Mediated via the Inhibition of TGF-Beta 1 in an LLC-PK1 Cell Model of Diabetic Nephropathy. Curr. Issues Mol. Biol. 2022, 44, 1087-1114. https://doi.org/10.3390/cimb44030072
Ninčević V, Zjalić M, Kolarić TO, Smolić M, Kizivat T, Kuna L, Včev A, Tabll A, Ćurčić IB. Renoprotective Effect of Liraglutide Is Mediated via the Inhibition of TGF-Beta 1 in an LLC-PK1 Cell Model of Diabetic Nephropathy. Current Issues in Molecular Biology. 2022; 44(3):1087-1114. https://doi.org/10.3390/cimb44030072
Chicago/Turabian StyleNinčević, Vjera, Milorad Zjalić, Tea Omanović Kolarić, Martina Smolić, Tomislav Kizivat, Lucija Kuna, Aleksandar Včev, Ashraf Tabll, and Ines Bilić Ćurčić. 2022. "Renoprotective Effect of Liraglutide Is Mediated via the Inhibition of TGF-Beta 1 in an LLC-PK1 Cell Model of Diabetic Nephropathy" Current Issues in Molecular Biology 44, no. 3: 1087-1114. https://doi.org/10.3390/cimb44030072
APA StyleNinčević, V., Zjalić, M., Kolarić, T. O., Smolić, M., Kizivat, T., Kuna, L., Včev, A., Tabll, A., & Ćurčić, I. B. (2022). Renoprotective Effect of Liraglutide Is Mediated via the Inhibition of TGF-Beta 1 in an LLC-PK1 Cell Model of Diabetic Nephropathy. Current Issues in Molecular Biology, 44(3), 1087-1114. https://doi.org/10.3390/cimb44030072