
Personalized 3-Gene Panel for Prostate Cancer Target Therapy



Abstract
1. Introduction
2. Materials and Methods
2.1. Prostate Tissues and Cell Lines
2.2. Transcriptomic Characteristics of Individual Genes
2.3. Transcriptomic Alterations in Cancer
3. Results
3.1. Overview
3.2. Independent Variables
3.3. The Power of the Weighted Individual (Gene) Regulation (WIR) Score to Identify the Main Contributors to the Cancer Phenotype
3.4. Each Cancer Nodule Has Its Own “Transcriptomic Signature”
3.5. Tumor Heterogeneity of Gene Networks
3.6. Gene Master Regulators
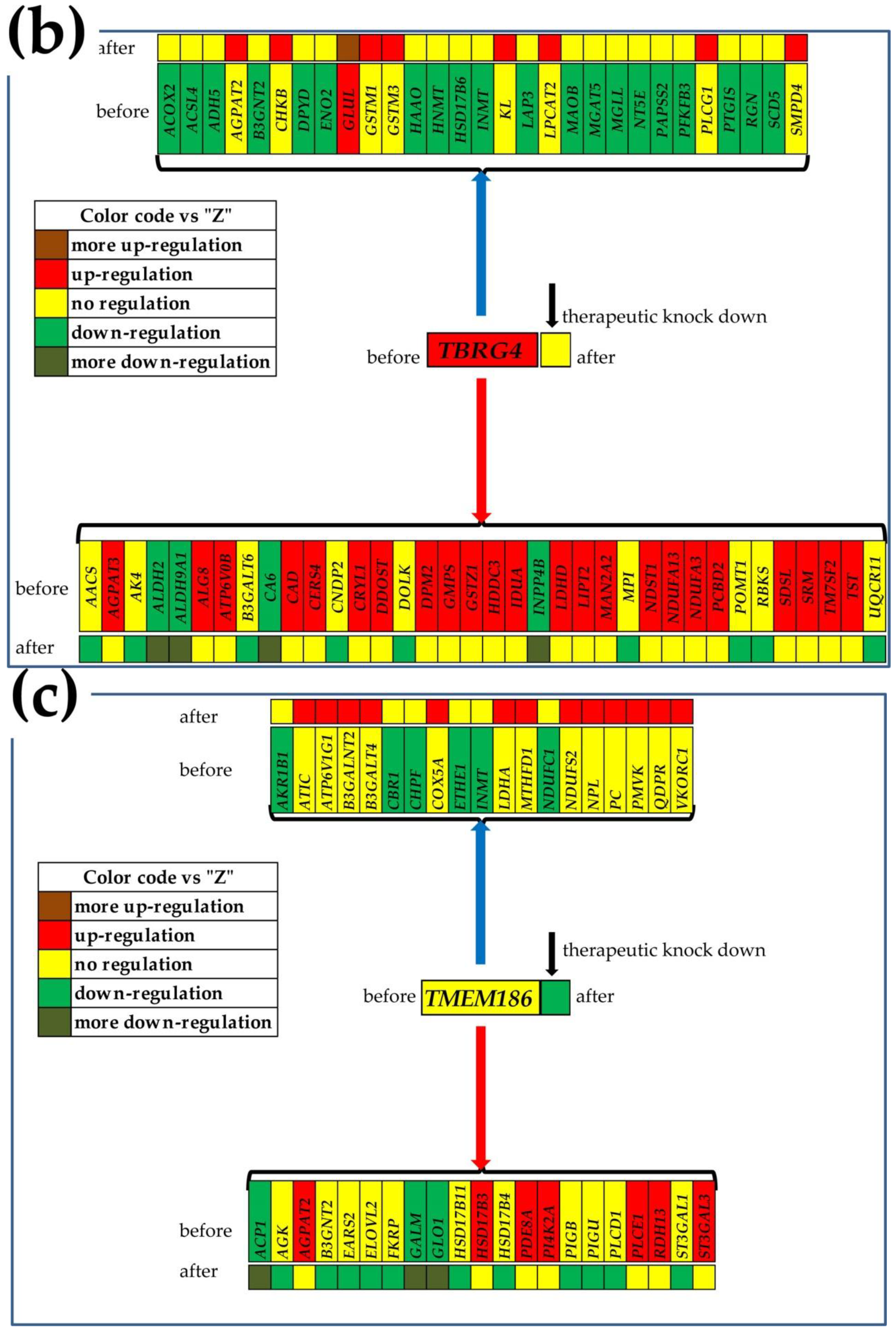
3.7. What Experimental Manipulation of the GMR Would Do to the Cancer Nodule’s Metabolism?
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hatano, K.; Nonomura, N. Genomic Profiling of Prostate Cancer: An Updated Review. World J. Men’s Health 2021, 39. [Google Scholar] [CrossRef]
- Spratt, D.E. Prostate Cancer Transcriptomic Subtypes. Adv. Exp. Med. Biol. 2019, 1210, 111–120. [Google Scholar] [CrossRef]
- Manceau, C.; Fromont, G.; Beauval, J.-B.; Barret, E.; Brureau, L.; Créhange, G.; Dariane, C.; Fiard, G.; Gauthé, M.; Mathieu, R.; et al. Biomarker in Active Surveillance for Prostate Cancer: A Systematic Review. Cancers 2021, 13, 4251. [Google Scholar] [CrossRef]
- Evans, R.; Hawkins, N.; Dequen-O’Byrne, P.; McCrea, C.; Muston, D.; Gresty, C.; Ghate, S.R.; Fan, L.; Hettle, R.; Abrams, K.R.; et al. Exploring the Impact of Treatment Switching on Overall Survival from the PROfound Study in Homologous Recombination Repair (HRR)-Mutated Metastatic Castration-Resistant Prostate Cancer (mCRPC). Target. Oncol. 2021, 16, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Rogers, O.C.; Rosen, D.M.; Antony, L.; Harper, H.M.; Das, D.; Yang, X.; Minn, I.; Mease, R.C.; Pomper, M.G.; Denmeade, S.R. Targeted delivery of cytotoxic proteins to prostate cancer via conjugation to small molecule urea-based PSMA inhibitors. Sci. Rep. 2021, 11, 14925. [Google Scholar] [CrossRef]
- Qureshi, Z.; Ahmad, M.; Yang, W.-X.; Tan, F.-Q. Kinesin 12 (KIF15) contributes to the development and tumorigenicity of prostate cancer. Biochem. Biophys. Res. Commun. 2021, 576, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Stachurska, A.; Elbanowski, J.; Kowalczyńska, H.M. Role of α5β1 and αvβ3 integrins in relation to adhesion and spreading dynamics of prostate cancer cells interacting with fibronectin underin vitroconditions. Cell Biol. Int. 2012, 36, 883–892. [Google Scholar] [CrossRef]
- Moghaddam, S.; Jalali, A.; O’Neill, A.; Murphy, L.; Gorman, L.; Reilly, A.-M.; Heffernan, Á.; Lynch, T.; Power, R.; O’Malley, K.J.; et al. Integrating Serum Biomarkers into Prediction Models for Biochemical Recurrence Following Radical Prostatectomy. Cancers 2021, 13, 4162. [Google Scholar] [CrossRef]
- Jillson, L.K.; Rider, L.C.; Rodrigues, L.U.; Romero, L.; Karimpour-Fard, A.; Nieto, C.; Gillette, C.; Torkko, K.; Danis, E.; Smith, E.E.; et al. MAP3K7 Loss Drives Enhanced Androgen Signaling and Independently Confers Risk of Recurrence in Prostate Cancer with Joint Loss of CHD1. Mol. Cancer Res. 2021, 19, 1123–1136. [Google Scholar] [CrossRef] [PubMed]
- Kulac, I.; Roudier, M.P.; Haffner, M.C. Molecular Pathology of Prostate Cancer. Surg. Pathol. Clin. 2021, 14, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Tolkach, Y.; Kristiansen, G. The Heterogeneity of Prostate Cancer: A Practical Approach. Pathobiology 2018, 85, 108–116. [Google Scholar] [CrossRef]
- Tu, S.-M.; Zhang, M.; Wood, C.G.; Pisters, L.L. Stem Cell Theory of Cancer: Origin of Tumor Heterogeneity and Plasticity. Cancers 2021, 13, 4006. [Google Scholar] [CrossRef]
- Berglund, E.; Maaskola, J.; Schultz, N.; Friedrich, S.; Marklund, M.; Bergenstråhle, J.; Tarish, F.; Tanoglidi, A.; Vickovic, S.; Larsson, L.; et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat. Commun. 2018, 9, 2419. [Google Scholar] [CrossRef]
- Brady, L.; Kriner, M.; Coleman, I.; Morrissey, C.; Roudier, M.; True, L.D.; Gulati, R.; Plymate, S.R.; Zhou, Z.; Birditt, B.; et al. Inter- and intra-tumor heterogeneity of metastatic prostate cancer determined by digital spatial gene expression profiling. Nat. Commun. 2021, 12, 1426. [Google Scholar] [CrossRef]
- Melo, C.M.; Vidotto, T.; Chaves, L.P.; Lautert-Dutra, W.; dos Reis, R.B.; Squire, J.A. The Role of Somatic Mutations on the Immune Response of the Tumor Microenvironment in Prostate Cancer. Int. J. Mol. Sci. 2021, 22, 9550. [Google Scholar] [CrossRef] [PubMed]
- Makino, T.; Izumi, K.; Iwamoto, H.; Mizokami, A. Treatment Strategies for High-Risk Localized and Locally Advanced and Oligometastatic Prostate Cancer. Cancers 2021, 13, 4470. [Google Scholar] [CrossRef] [PubMed]
- Burgess, L.; Roy, S.; Morgan, S.; Malone, S. A Review on the Current Treatment Paradigm in High-Risk Prostate Cancer. Cancers 2021, 13, 4257. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Mgbemena, V.E.; Iacobas, S.; Menezes, K.M.; Wang, H.; Saganti, P.B. Genomic Fabric Remodeling in Metastatic Clear Cell Renal Cell Carcinoma (ccRCC): A New Paradigm and Proposal for a Personalized Gene Therapy Approach. Cancers 2020, 12, 3678. [Google Scholar] [CrossRef]
- Mateo, J.; McKay, R.; Abida, W.; Aggarwal, R.; Alumkal, J.; Alva, A.; Feng, F.; Gao, X.; Graff, J.; Hussain, M.; et al. Accelerating precision medicine in metastatic prostate cancer. Nat. Rev. Cancer 2020, 1, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Iacobas, D.A.; Tuli, N.; Iacobas, S.; Rasamny, J.K.; Moscatello, A.; Geliebter, J.; Tiwari, R.M. Gene master regulators of papillary and anaplastic thyroid cancer phenotypes. Oncotarget 2018, 9, 2410–2424. [Google Scholar] [CrossRef]
- Iacobas, D.A. Biomarkers, Master Regulators and Genomic Fabric Remodeling in a Case of Papillary Thyroid Carcinoma. Genes 2020, 11, 1030. [Google Scholar] [CrossRef] [PubMed]
- Iacobas, S.; Ede, N.; Iacobas, D.A. The Gene Master Regulators (GMR) Approach Provides Legitimate Targets for Personalized, Time-Sensitive Cancer Gene Therapy. Genes 2019, 10, 560. [Google Scholar] [CrossRef]
- Iacobas, D.A. The Genomic Fabric Perspective on the Transcriptome Between Universal Quantifiers and Personalized Genomic Medicine. Biol. Theory 2016, 11, 123–137. [Google Scholar] [CrossRef]
- Iacobas, D.A. Powerful quantifiers for cancer transcriptomics. World J. Clin. Oncol. 2020, 11, 679–704. [Google Scholar] [CrossRef] [PubMed]
- Iacobas, D.A.; Iacobas, S.; Lee, P.R.; Cohen, J.E.; Fields, R.D. Coordinated Activity of Transcriptional Networks Responding to the Pattern of Action Potential Firing in Neurons. Genes 2019, 10, 754. [Google Scholar] [CrossRef] [PubMed]
- Petrucci, R.H.; Harwood, W.S.; Herring, F.G. General Chemistry: Principles and Modern Applications, 8th ed.; Prentice Hall: Hoboken, NJ, USA, 2002; p. 37. ISBN 978-0-13-014329-7. [Google Scholar]
- Horoszewicz, J.S.; Leong, S.S.; Kawinski, E.; Karr, J.P.; Rosenthal, H.; Chu, T.M.; Mirand, E.A.; Murphy, G.P. LNCaP model of human prostatic carcinoma. Cancer Res. 1983, 43, 1809–1818. [Google Scholar] [PubMed]
- Alimirah, F.; Chen, J.; Basrawala, Z.; Xin, H.; Choubey, D. DU-145 and PC-3 human prostate cancer cell lines express androgen receptor: Implications for the androgen receptor functions and regulation. FEBS Lett. 2006, 580, 2294–2300. [Google Scholar] [CrossRef]
- Remodeling of DNA Transcription Genomic Fabric in Capridine-Treated LNCaP Human Prostate Cancer Cell Line. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE72414 (accessed on 1 September 2021).
- Remodeling of Major Genomic Fabrics and Their Interplay in Capridine-Treated DU145 Classic Human Prostate Cancer. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE72333 (accessed on 1 September 2021).
- Gene Commanding Height (GCH) Hierarchy in the Cancer Nucleus and Cancer-Free Resection Margins from a Surgically Removed Prostatic Adenocarcinoma of a 65y Old Black Man. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?&acc=GSE133906 (accessed on 1 September 2021).
- Genomic Fabric Remodeling in Prostate Cancer. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE168718 (accessed on 1 September 2021).
- Iacobas, S.; Iacobas, D. A Personalized Genomics Approach of the Prostate Cancer. Cells 2021, 10, 1644. [Google Scholar] [CrossRef]
- Gene Commanding Height (GCH) Hierarchy in the Cancer Nucleus and Cancer-Free Resection Margins from a Surgically Removed Prostatic Adenocarcinoma of a 47y Old White Man. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE13389 (accessed on 1 September 2021).
- Transcriptomic Heterogeneity of the Prostate Cancer. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE183889 (accessed on 9 December 2021).
- Agilent-026652 Whole Human Genome Microarray 4 × 44K v2. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL13497 (accessed on 1 September 2021).
- Stranger, B.E.; Forrest, M.S.; Clark, A.G.; Minichiello, M.J.; Deutsch, S.; Lyle, R.; Hunt, S.; Kahl, B.; Antonarakis, S.E.; Tavaré, S.; et al. Genome-Wide Associations of Gene Expression Variation in Humans. PLoS Genet. 2005, 1, e78. [Google Scholar] [CrossRef]
- Guan, J.; Han, S.; Wu, J.; Zhang, Y.; Bai, M.; Abdullah, S.W.; Sun, S.; Guo, H. Ribosomal Protein L13 Participates in Innate Immune Response Induced by Foot-and-Mouth Disease Virus. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Xu, H.; Liu, P.; Yan, Y.; Fang, K.; Liang, D.; Hou, X.; Zhang, X.; Wu, S.; Ma, J.; Wang, R.; et al. FKBP9 promotes the malignant behavior of glioblastoma cells and confers resistance to endoplasmic reticulum stress inducers. J. Exp. Clin. Cancer Res. 2020, 39, 44. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.-N.; Dai, L.-J.; Yang, S.-B.; Wu, Y.-D.; Liang, Y.-X.; Yin, X.-L.; Zou, C.-Y.; Zhong, W.-D. Increasing of FKBP9 can predict poor prognosis in patients with prostate cancer. Pathol.—Res. Pract. 2020, 216, 152732. [Google Scholar] [CrossRef]
- Macauda, A.; Piredda, C.; Clay-Gilmour, A.I.; Sainz, J.; Buda, G.; Markiewicz, M.; Barington, T.; Ziv, E.; Hildebrandt, M.A.T.; Belachew, A.A.; et al. Expression quantitative trait loci of genes predicting outcome are associated with survival of multiple myeloma patients. Int. J. Cancer 2021, 149, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Luo, Q.; Liu, M.; Zhang, C.; Jia, Y.; Tong, R.; Yang, L.; Fu, X. TBRG4 silencing promotes progression of squamous cell carcinoma via regulation of CAV-1 expression and ROS formation. Cell. Mol. Biol. 2020, 66, 157–164. [Google Scholar] [CrossRef]
- Huang, F.; Zhou, P.; Wang, Z.; Zhang, X.-L.; Liao, F.-X.; Hu, Y.; Chang, J. Knockdown of TBRG4 suppresses proliferation, invasion and promotes apoptosis of osteosarcoma cells by downregulating TGF-β1 expression and PI3K/AKT signaling pathway. Arch. Biochem. Biophys. 2020, 686, 108351. [Google Scholar] [CrossRef]
- Varghese, R.T.; Liang, Y.; Guan, T.; Franck, C.T.; Kelly, D.F.; Sheng, Z. Survival kinase genes present prognostic significance in glioblastoma. Oncotarget 2016, 7, 20140–20151. [Google Scholar] [CrossRef] [PubMed]
- Bashanfer, S.A.A.; Saleem, M.; Heidenreich, O.; Moses, E.J.; Yusoff, N.M. Disruption of MAPK1 expression in the ERK signalling pathway and the RUNX1-RUNX1T1 fusion gene attenuate the differentiation and proliferation and induces the growth arrest in t(8;21) leukaemia cells. Oncol. Rep. 2019, 41, 2027–2040. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Zhao, C.; Liu, X.; Su, W.; Duan, G.; Xie, Z.; Chu, S.; Gao, Y. Knockdown of TBRG4 affects tumorigenesis in human H1299 lung cancer cells by regulating DDIT3, CAV1 and RRM2. Oncol. Lett. 2018, 15, 121–128. [Google Scholar] [CrossRef]
- Rao, Y.; Liu, H.; Yan, X.; Wang, J. In Silico Analysis Identifies Differently Expressed lncRNAs as Novel Biomarkers for the Prognosis of Thyroid Cancer. Comput. Math. Methods Med. 2020, 2020, 3651051. [Google Scholar] [CrossRef]
- Danielpour, D.; Purighalla, S.; Wang, E.; Zmina, P.M.; Sarkar, A.; Zhou, G. JAB1/COPS5 is a putative oncogene that controls critical oncoproteins deregulated in prostate cancer. Biochem. Biophys. Res. Commun. 2019, 518, 374–380. [Google Scholar] [CrossRef] [PubMed]
- mTOR Signaling Pathway. Available online: https://www.genome.jp/pathway/hsa04150 (accessed on 1 September 2021).
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef]
- Gong, C.; Qu, S.; Lv, X.-B.; Liu, B.; Tan, W.; Nie, Y.; Su, F.; Liu, Q.; Yao, H.; Song, E. BRMS1L suppresses breast cancer metastasis by inducing epigenetic silence of FZD10. Nat. Commun. 2014, 5, 5406. [Google Scholar] [CrossRef] [PubMed]
- Wendel, H.-G.; Silva, R.L.; Malina, A.; Mills, J.R.; Zhu, H.; Ueda, T.; Watanabe-Fukunaga, R.; Fukunaga, R.; Teruya-Feldstein, J.; Pelletier, J.; et al. Dissecting eIF4E action in tumorigenesis. Genes Dev. 2007, 21, 3232–3237. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas. Available online: https://portal.gdc.cancer.gov/ (accessed on 20 October 2021).
- Oncotype IQ. Available online: https://www.oncotypeiq.com/en-US/prostate-cancer/patients-and-caregivers/early-stage-gps/why-oncotype-dx-gps (accessed on 11 February 2021).
- Foundation Medicine. Available online: https://www.foundationmedicine.com/genomic-testing (accessed on 12 July 2020).
- Prostate Cancer is Manageable. Available online: https://decipherbio.com/ (accessed on 8 February 2021).
- Prostate Cancer Pathway. Available online: https://www.genome.jp/pathway/hsa05215 (accessed on 1 September 2021).
- KEGG Pathway Database. Available online: https://www.genome.jp/kegg/pathway.html (accessed on 1 September 2021).
- QIAGEN Ingenuity Pathway Analysis. Available online: https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/ (accessed on 20 October 2021).
- The Database for Annotation, Visualization and Integrated Discovery (DAVID). Available online: https://david.ncifcrf.gov/ (accessed on 20 October 2021).
- Prickett, D.; Watson, M. Use of GenMAPP and MAPPFinder to analyse pathways involved in chickens infected with the protozoan parasite Eimeria. BMC Proc. 2009, 3, S7. [Google Scholar] [CrossRef] [PubMed]
- What Is Gene Therapy? Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/what-gene-therapy (accessed on 1 September 2021).
- Long Term Follow-Up After Administration of Human Gene Therapy Products. Guidance for Industry. Available online: https://www.fda.gov/media/113768/download (accessed on 20 October 2021).
- U.S.A. National Cancer Institute: Targeted Cancer Therapies. Available online: https://www.cancer.gov/about-cancer/treatment/types/targeted-therapies/targeted-therapies-fact-sheet (accessed on 20 October 2021).
- Harrington, K.J.; Spitzweg, C.; Bateman, A.R.; Morris, J.C.; Vile, R.G. Gene therapy for prostate cancer: Current status and future prospects. J. Urol. 2001, 166, 1220–1233. [Google Scholar] [CrossRef]
- Ikegami, S.; Tadakuma, T.; Ono, T.; Suzuki, S.; Yoshimura, I.; Asano, T.; Hayakawa, M. Treatment efficiency of a suicide gene therapy using prostate-specific membrane antigen promoter/enhancer in a castrated mouse model of prostate cancer. Cancer Sci. 2004, 95, 367–370. [Google Scholar] [CrossRef]
- Satoh, T.; Irie, A.; Egawa, S.; Baba, S. In Situ Gene Therapy for Prostate Cancer. Curr. Gene Ther. 2005, 5, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Lv, H.; Cao, W.; Zhou, C.; Liu, Q.; Li, H.; Zhou, F. Targeting strategies of adenovirus-mediated gene therapy and virotherapy for prostate cancer. Mol. Med. Rep. 2017, 16, 6443–6458. [Google Scholar] [CrossRef][Green Version]
- Talkar, S.S. Gene Therapy for Prostate Cancer: A Review. Endocrine, Metab. Immune Disord.—Drug Targets 2021, 21, 385–396. [Google Scholar] [CrossRef]
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration, FDA. FDA Approves Olaparib for Hrr Gene-Mutated Metastatic Castration-Resistant Prostate Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-hrr-gene-mutated-metastatic-castration-resistant-prostate-cancer (accessed on 1 September 2021).
- U.S. Food and Drug Administration, FDA. Approved Treatments for Prostate Cancer. Available online: https://search.usa.gov/search?utf8=%E2%9C%93&affiliate=fda&sort_by=&query=prostate+cancer&commit=Search (accessed on 20 September 2021).
- Zhang, J.; Sun, J.; Bakht, S.; Hassan, W. Recent Development and Future Prospects of Molecular Targeted Therapy in Prostate Cancer. Curr. Mol. Pharmacol. 2021, 14. [Google Scholar] [CrossRef]
- Morgan, R.; da Silveira, W.A.; Kelly, R.C.; Overton, I.; Allott, E.H.; Hardiman, G. Long non-coding RNAs and their potential impact on diagnosis, prognosis, and therapy in prostate cancer: Racial, ethnic, and geographical considerations. Expert Rev. Mol. Diagn. 2021, 21, 1257–1271. [Google Scholar] [CrossRef] [PubMed]
- Oh-Hohenhorst, S.J.; Lange, T. Role of Metastasis-Related microRNAs in Prostate Cancer Progression and Treatment. Cancers 2021, 13, 4492. [Google Scholar] [CrossRef]
- FDA Approves Enzalutamide for Metastatic Castration-Sensitive Prostate Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-enzalutamide-metastatic-castration-sensitive-prostate-cancer (accessed on 20 October 2021).
- Leblanc, E.; Ban, F.; Cavga, A.D.; Lawn, S.; Huang, C.-C.F.; Mohan, S.; Chang, M.E.K.; Flory, M.R.; Ghaidi, F.; Lingadahalli, S.; et al. Development of 2-(5,6,7-Trifluoro-1H-Indol-3-yl)-quinoline-5-carboxamide as a Potent, Selective, and Orally Available Inhibitor of Human Androgen Receptor Targeting Its Binding Function-3 for the Treatment of Castration-Resistant Prostate Cancer. J. Med. Chem. 2021, 64, 14968–14982. [Google Scholar] [CrossRef]
- FDA Approves Darolutamide for Non-Metastatic Castration-Resistant Prostate Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-darolutamide-non-metastatic-castration-resistant-prostate-cancer (accessed on 20 October 2021).
- Devos, G.; Devlies, W.; De Meerleer, G.; Baldewijns, M.; Gevaert, T.; Moris, L.; Milonas, D.; Van Poppel, H.; Berghen, C.; Everaerts, W.; et al. Neoadjuvant hormonal therapy before radical prostatectomy in high-risk prostate cancer. Nat. Rev. Urol. 2021, 18, 739–762. [Google Scholar] [CrossRef] [PubMed]
- Iacobas, D.; Wen, J.; Iacobas, S.; Schwartz, N.; Putterman, C. Remodeling of Neurotransmission, Chemokine, and PI3K-AKT Signaling Genomic Fabrics in Neuropsychiatric Systemic Lupus Erythematosus. Genes 2021, 12, 251. [Google Scholar] [CrossRef]
- Iacobas, S.; Amuzescu, B.; Iacobas, D.A. Transcriptomic uniqueness and commonality of the ion channels and transporters in the four heart chambers. Sci. Rep. 2021, 11, 2743. [Google Scholar] [CrossRef] [PubMed]
- Iacobas, D.A.; Wen, J.; Iacobas, S.; Putterman, C.; Schwartz, N. TWEAKing the Hippocampus: The Effects of TWEAK on the Genomic Fabric of the Hippocampus in a Neuropsychiatric Lupus Mouse Model. Genes 2021, 12, 1172. [Google Scholar] [CrossRef]
- Pratap, J.; Lian, J.B.; Javed, A.; Barnes, G.L.; Van Wijnen, A.J.; Stein, J.L.; Stein, G.S. Regulatory roles of Runx2 in metastatic tumor and cancer cell interactions with bone. Cancer Metastasis Rev. 2006, 25, 589–600. [Google Scholar] [CrossRef]
- Orr-Urtreger, A.; Bar-Shira, A.; Matzkin, H.; Mabjeesh, N.J. The homozygous P582S mutation in the oxygen-dependent degradation domain of HIF-1α is associated with increased risk for prostate cancer. Prostate 2006, 67, 8–13. [Google Scholar] [CrossRef]
- Kinyamu, H.K.; Collins, J.B.; Grissom, S.F.; Hebbar, P.B.; Archer, T.K. Genome wide transcriptional profiling in breast cancer cells reveals distinct changes in hormone receptor target genes and chromatin modifying enzymes after proteasome inhibition. Mol. Carcinog. 2008, 47, 845–885. [Google Scholar] [CrossRef] [PubMed]
- Latchman, D.S. Transcription factors: An overview. Int. J. Exp. Pathol. 1993, 74, 417–422. [Google Scholar] [CrossRef]
- Kawaji, H. Computational predictions of transcription factor binding sites. Tanpakushitsu Kakusan Koso. 2004, 49, 2877–2881. [Google Scholar]
- Ji, H.; Wong, W.H. Computational Biology: Toward Deciphering Gene Regulatory Information in Mammalian Genomes. Biometrics 2006, 62, 645–663. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Approved Cellular and Gene Therapy Products. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products (accessed on 20 September 2021).
- American Cancer Society: Key Statistics for Prostate Cancer. Available online: https://www.cancer.org/cancer/prostate-cancer/about/key-statistics.html (accessed on 20 October 2021).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Description | AVE-P | AVE-Q | AVE-M | AVE-Z |
| CYTB | mitochondrially encoded cytochrome b | 231 | 237 | 186 | 266 |
| RPL13 | ribosomal protein L13 | 215 | 2365 | 269 | 366 |
| ACTG2 | actin, gamma 2, smooth muscle, enteric | 204 | 126 | 119 | 325 |
| NPY | neuropeptide Y | 76 | 471 | 10 | 123 |
| RPL13 | ribosomal protein L13 | 215 | 265 | 269 | 366 |
| RPL7A | ribosomal protein L7a | 106 | 310 | 141 | 241 |
| RPL13 | ribosomal protein L13 | 215 | 265 | 269 | 366 |
| ZNF865 | zinc finger protein 865 | 182 | 267 | 203 | 292 |
| PQLC2 | PQ loop repeat containing 2 | 172 | 179 | 199 | 203 |
| RPL13 | ribosomal protein L13 | 215 | 265 | 269 | 366 |
| ACTG2 | actin, gamma 2, smooth muscle, enteric | 204 | 126 | 119 | 325 |
| MYH11 | myosin, heavy chain 11, smooth muscle | 137 | 101 | 139 | 307 |
| Gene | Description | AVE-A | AVE-B | AVE-C | AVE-N |
| RPL13 | ribosomal protein L13 | 288 | 477 | 415 | 621 |
| PQLC2 | PQ loop repeat containing 2 | 227 | 266 | 338 | 490 |
| CYTB | mitochondrially encoded cytochrome b | 215 | 173 | 146 | 382 |
| RPL13 | ribosomal protein L13 | 288 | 477 | 415 | 621 |
| RPS27 | ribosomal protein S27 | 158 | 365 | 294 | 212 |
| RPL32 | ribosomal protein L32 | 164 | 315 | 299 | 359 |
| RPL13 | ribosomal protein L13 | 288 | 477 | 415 | 621 |
| ZNF865 | zinc finger protein 865 | 132 | 215 | 395 | 600 |
| RPS8 | ribosomal protein S8 | 153 | 233 | 348 | 504 |
| RPL13 | ribosomal protein L13 | 288 | 477 | 415 | 621 |
| ZNF865 | zinc finger protein 865 | 132 | 215 | 395 | 600 |
| RPS2 | ribosomal protein S2 | 164 | 260 | 337 | 517 |
| Gene | Description | REV-P | REV-Q | REV-M | REV-Z |
| FKBP9 | FK506 binding protein 9 | 0.32 | 8.80 | 10.87 | 9.27 |
| ZBTB2 | zinc finger and BTB domain containing 2 | 0.50 | 12.39 | 5.03 | 16.51 |
| NUBPL | nucleotide binding protein-like | 0.69 | 12.19 | 5.48 | 10.73 |
| TBRG4 | transforming growth factor beta regulator 4 | 11.07 | 0.59 | 1.56 | 8.06 |
| DNAJC24 | DnaJ (Hsp40) homolog, subfamily C, member 24 | 11.62 | 0.85 | 7.19 | 4.16 |
| UBE3B | ubiquitin protein ligase E3B | 5.32 | 1.00 | 6.51 | 9.46 |
| TMEM186 | transmembrane protein 186 | 9.32 | 20.45 | 0.28 | 12.49 |
| NDUFA6-AS1 | NDUFA6 antisense RNA 1 (head to head) | 28.15 | 8.46 | 0.50 | 15.82 |
| LMAN2L | lectin, mannose-binding 2-like | 8.05 | 8.20 | 0.52 | 8.00 |
| COPS5 | COP9 signalosome subunit 5 | 51.46 | 27.83 | 7.37 | 0.12 |
| ARPC5L | actin related protein 2/3 complex, subunit 5-like | 19.90 | 22.18 | 7.13 | 0.14 |
| DAZAP1 | DAZ associated protein 1 | 7.14 | 18.87 | 3.84 | 0.22 |
| Gene | Description | REV-A | REV-B | REV-C | REV-N |
| ENTPD2 | ectonucleoside triphosphate diphosphohydrolase 2 | 0.29 | 33.36 | 15.97 | 8.66 |
| COMMD9 | COMM domain containing 9 | 1.04 | 23.01 | 4.49 | 3.82 |
| MIEN1 | migration and invasion enhancer 1 | 1.14 | 13.78 | 9.01 | 8.67 |
| SSX3 | synovial sarcoma, X breakpoint 3 | 39.80 | 0.94 | 17.16 | 12.18 |
| FCRL5 | Fc receptor-like 5 | 121.40 | 1.41 | 36.36 | 38.96 |
| RANBP2 | RAN binding protein 2 | 48.44 | 1.63 | 11.21 | 10.48 |
| BAIAP2L1 | BAI1-associated protein 2-like 1 | 64.94 | 11.35 | 0.40 | 8.03 |
| FAM71E1 | family with sequence similarity 71, member E1 | 41.61 | 48.01 | 0.81 | 11.93 |
| LILRB3 | leukocyte immunoglobulin-like receptor, subfamily B, member 3 | 22.77 | 32.51 | 0.91 | 27.55 |
| MRPS12 | mitochondrial ribosomal protein S12 | 34.96 | 42.93 | 11.21 | 0.32 |
| TOR1A | torsin family 1, member A | 11.09 | 17.64 | 6.28 | 0.42 |
| DENND1B | DENN/MADD domain containing 1B | 8.18 | 45.86 | 15.79 | 0.47 |
| Gene | Description | WIR-P | WIR-Q | WIR-M |
| PSCA | prostate stem cell antigen | 54 | 138 | 0 |
| KLK12 | kallikrein-related peptidase 12 | 40 | 121 | 0 |
| BASP1 | brain abundant, membrane attached signal protein 1 | 32 | 39 | 0 |
| RPS8 | ribosomal protein S8 | −383 | 0 | −123 |
| CNN1 | calponin 1, basic, smooth muscle | −403 | −680 | −568 |
| RNA28S5 | RNA, 28S ribosomal 5 | −604 | −314 | −26 |
| MARC1 | mitochondrial amidoxime reducing component 1 | 0 | 9180 | 0 |
| NPY | neuropeptide Y | −50 | 348 | −1345 |
| PSCA | prostate stem cell antigen | 54 | 138 | 0 |
| CNN1 | calponin 1, basic, smooth muscle | −403 | −680 | −568 |
| LTF | lactotransferrin | −301 | −1682 | 22 |
| RLN1 | relaxin 1 | −86 | −2658 | −46 |
| IGKC | immunoglobulin kappa constant | −51 | −144 | 60 |
| IGHG1 | immunoglobulin heavy constant gamma 1 | −29 | −83 | 52 |
| SNORD3B-1 | small nucleolar RNA, C/D box 3B-1 | −3 | −35 | 49 |
| ACTG2 | actin, gamma 2, smooth muscle, enteric | −170 | −514 | −561 |
| CNN1 | calponin 1, basic, smooth muscle | −403 | −680 | −568 |
| NPY | neuropeptide Y | −50 | 348 | −1345 |
| Gene | Description | WIR-A | WIR-B | WIR-C |
| IGLL5 | immunoglobulin lambda-like polypeptide 5 | 59 | 141 | 56 |
| TPM2 | tropomyosin 2 (beta) | 50 | 9 | −9 |
| ACTG2 | actin, gamma 2, smooth muscle, enteric | 46 | 2 | −29 |
| RPS8 | ribosomal protein S8 | −1139 | −575 | −190 |
| RPL14 | ribosomal protein L14 | −1182 | −734 | −85 |
| ZNF865 | zinc finger protein 865 | −2073 | −1027 | −239 |
| IGLL5 | immunoglobulin lambda-like polypeptide 5 | 59 | 141 | 56 |
| MDK | midkine (neurite growth-promoting factor 2) | 28 | 133 | 148 |
| RPS27 | ribosomal protein S27 | −51 | 127 | 56 |
| NPIPB5 | nuclear pore complex interacting protein family, member B5 | −937 | −1077 | −98 |
| KLK3 | kallikrein-related peptidase 3 | −585 | −1248 | −439 |
| SPON2 | spondin 2, extracellular matrix protein | −332 | −1575 | −1288 |
| MDK | midkine (neurite growth-promoting factor 2) | 28 | 133 | 148 |
| IFI27 | interferon, alpha-inducible protein 27 | 13 | 55 | 123 |
| HMGN2 | high mobility group nucleosomal binding domain 2 | 6 | 107 | 116 |
| CYTB | mitochondrially encoded cytochrome b | −238 | −446 | −600 |
| LOC101929612 | mitochondrially encoded cytochrome c oxidase III | −97 | −279 | −800 |
| SPON2 | spondin 2, extracellular matrix protein | −332 | −1575 | −1288 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iacobas, S.; Iacobas, D.A. Personalized 3-Gene Panel for Prostate Cancer Target Therapy. Curr. Issues Mol. Biol. 2022, 44, 360-382. https://doi.org/10.3390/cimb44010027
Iacobas S, Iacobas DA. Personalized 3-Gene Panel for Prostate Cancer Target Therapy. Current Issues in Molecular Biology. 2022; 44(1):360-382. https://doi.org/10.3390/cimb44010027
Chicago/Turabian StyleIacobas, Sanda, and Dumitru Andrei Iacobas. 2022. "Personalized 3-Gene Panel for Prostate Cancer Target Therapy" Current Issues in Molecular Biology 44, no. 1: 360-382. https://doi.org/10.3390/cimb44010027
APA StyleIacobas, S., & Iacobas, D. A. (2022). Personalized 3-Gene Panel for Prostate Cancer Target Therapy. Current Issues in Molecular Biology, 44(1), 360-382. https://doi.org/10.3390/cimb44010027