
A Comparative Study between Spanish and British SARS-CoV-2 Variants


Abstract
:1. Introduction
2. Materials and Methods
2.1. Alignment of the Two Genomes
2.2. Translation of the Nucleotide Sequence into Aminoacidic Sequences
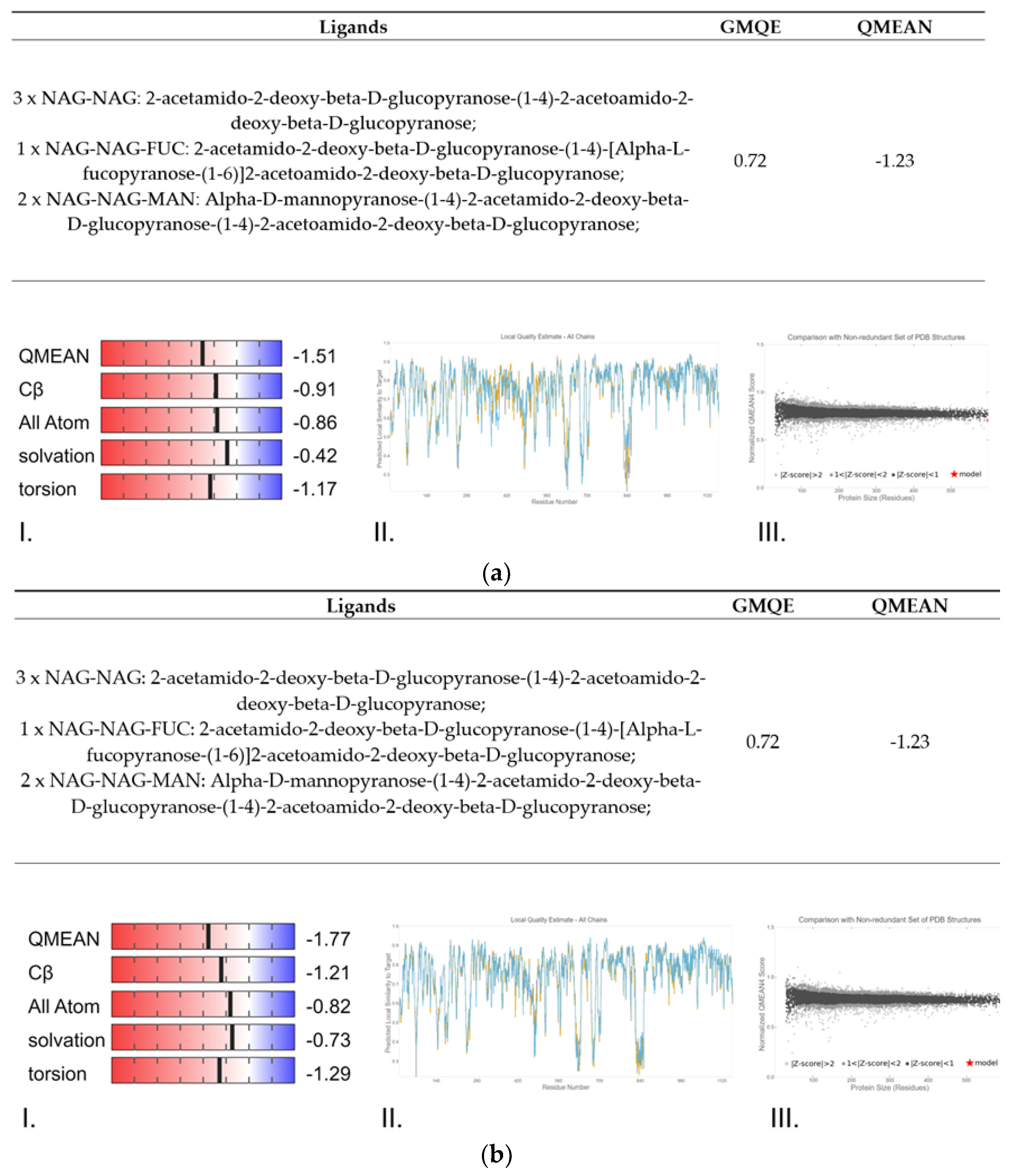
2.3. Protein Structure Homology-Modelling, Simulation and Comparations
2.4. Antigenicity and Binding Receptor Domain Interaction with ACE2
2.5. Computational Prediction of Protein-Protein Docking Results
3. Results
3.1. Alignment of the Two Genomes
3.2. Translation of the Nucleotide Sequence into Aminoacidic Sequences
3.3. Protein Structure Homology-Modelling, Simulation and Comparations
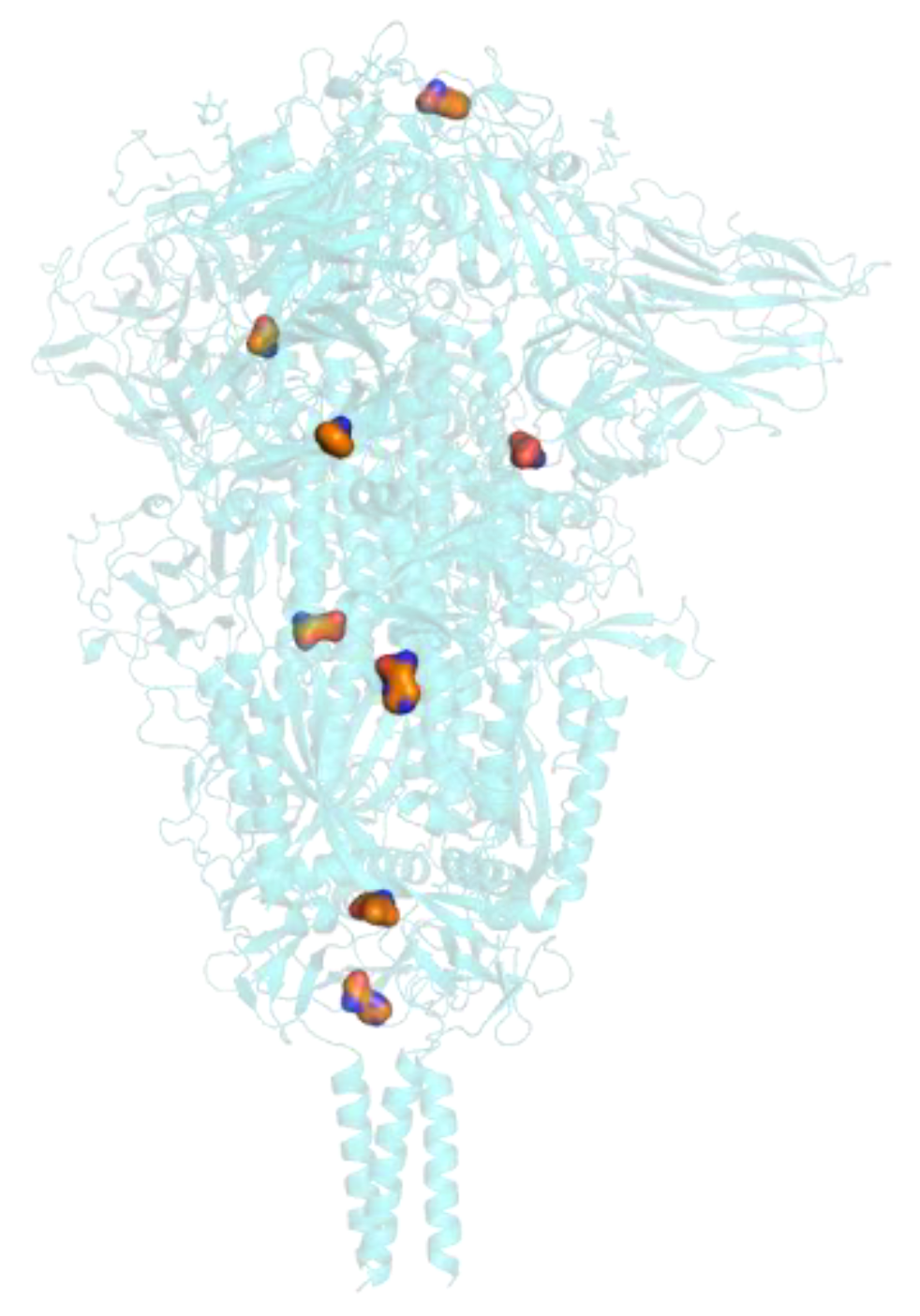
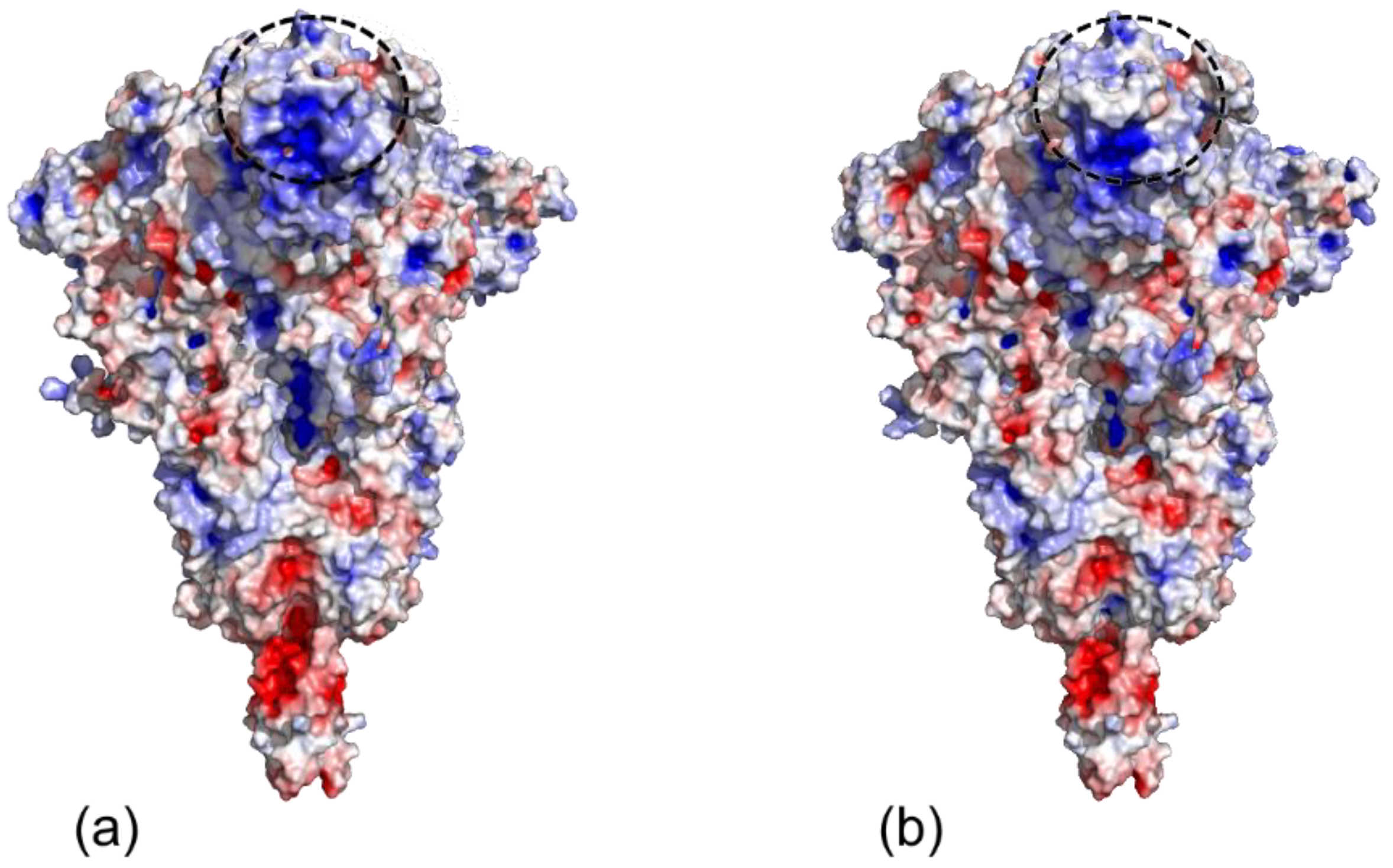
3.4. Antigenicity
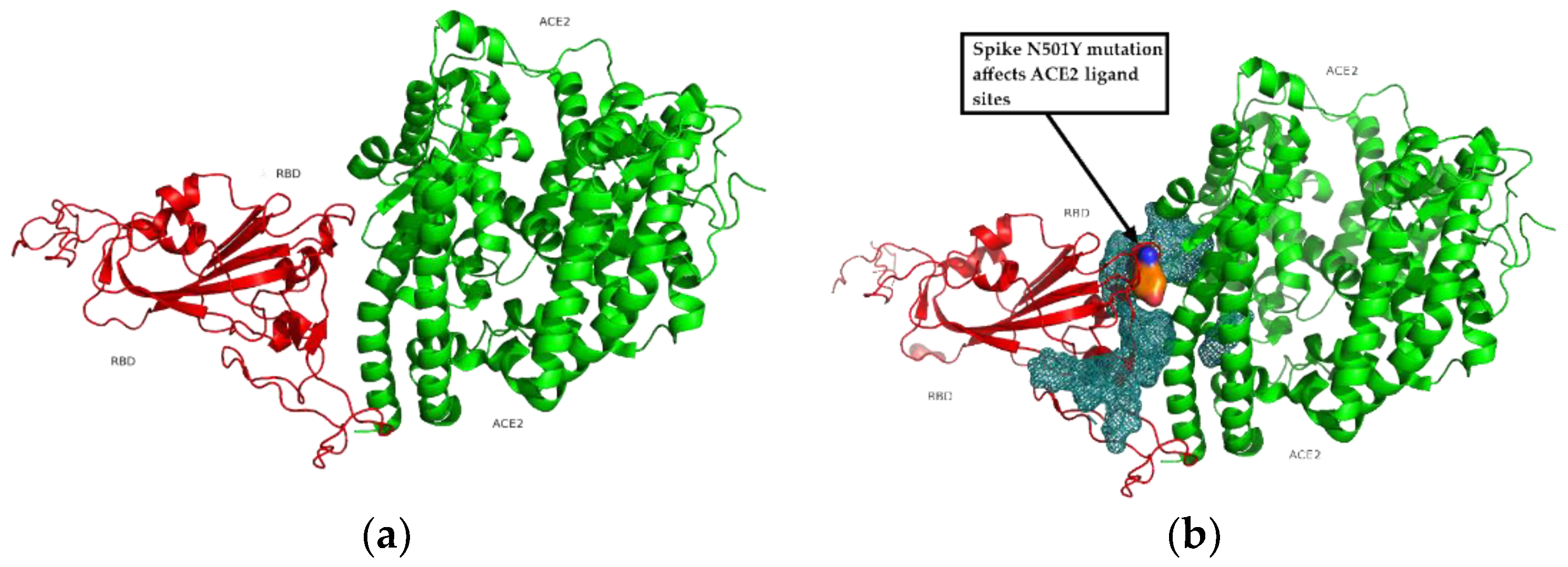
3.5. Binding Receptor Domain Interaction with ACE2
3.6. Computational Prediction of Protein-Protein Docking Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in china. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Forchette, L.; Sebastian, W.; Liu, T. A comprehensive review of covid-19 virology, vaccines, variants, and therapeutics. Curr. Med. Sci. 2021, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Khailany, R.A.; Safdar, M.; Ozaslan, M. Genomic characterization of a novel sars-cov-2. Gene Rep. 2020, 19, 100682. [Google Scholar] [CrossRef] [PubMed]
- Gorkhali, R.; Koirala, P.; Rijal, S.; Mainali, A.; Baral, A.; Bhattarai, H.K. Structure and function of major sars-cov-2 and sars-cov proteins. Bioinform. Biol. Insights 2021, 15, 11779322211025876. [Google Scholar] [CrossRef]
- Verma, J.; Subbarao, N. Insilico study on the effect of sars-cov-2 rbd hotspot mutants’ interaction with ace2 to understand the binding affinity and stability. Virology 2021, 561, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Ostrov, D.A. Structural consequences of variation in sars-cov-2 b.1.1.7. J. Cell Immunol. 2021, 3, 103–108. [Google Scholar] [CrossRef]
- Villoutreix, B.O.; Calvez, V.; Marcelin, A.G.; Khatib, A.M. In silico investigation of the new uk (b.1.1.7) and south african (501y.V2) sars-cov-2 variants with a focus at the ace2-spike rbd interface. Int. J. Mol. Sci. 2021, 22, 1695. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Plante, K.S.; Plante, J.A.; Xie, X.; Zhang, X.; Ku, Z.; An, Z.; Scharton, D.; Schindewolf, C.; et al. The n501y spike substitution enhances sars-cov-2 transmission. bioRxiv 2021. [Google Scholar] [CrossRef]
- Eddy, S.R. Where did the blosum62 alignment score matrix come from? Nat. Biotechnol. 2004, 22, 1035–1036. [Google Scholar] [CrossRef]
- Gonnet, G.H.; Cohen, M.A.; Benner, S.A. Exhaustive matching of the entire protein sequence database. Science 1992, 256, 1443–1445. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal w and clustal x version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McWilliam, H.; Li, W.; Uludag, M.; Squizzato, S.; Park, Y.M.; Buso, N.; Cowley, A.P.; Lopez, R. Analysis tool web services from the embl-ebi. Nucleic Acids Res. 2013, 41, W597–W600. [Google Scholar] [CrossRef] [Green Version]
- IUPAC-IUB Commission on Biochemical Nomenclature. A one-letter notation for amino acid sequences. Tentative rules. J. Biol. Chem. 1968, 243, 3557–3559. [Google Scholar] [CrossRef]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. Swiss-model: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biasini, M.; Schmidt, T.; Bienert, S.; Mariani, V.; Studer, G.; Haas, J.; Johner, N.; Schenk, A.D.; Philippsen, A.; Schwede, T. Openstructure: An integrated software framework for computational structural biology. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 701–709. [Google Scholar] [CrossRef] [Green Version]
- Studer, G.; Rempfer, C.; Waterhouse, A.M.; Gumienny, R.; Haas, J.; Schwede, T. Qmeandisco-distance constraints applied on model quality estimation. Bioinformatics 2020, 36, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Mura, C.; McCrimmon, C.M.; Vertrees, J.; Sawaya, M.R. An introduction to biomolecular graphics. PLoS Comput. Biol. 2010, 6. [Google Scholar] [CrossRef] [Green Version]
- Fiorucci, S.; Zacharias, M. Prediction of protein-protein interaction sites using electrostatic desolvation profiles. Biophys. J. 2010, 98, 1921–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretti, R.; Bender, B.J.; Allison, B.; Meiler, J. Rosetta and the design of ligand binding sites. Methods Mol. Biol. 2016, 1414, 47–62. [Google Scholar] [CrossRef] [Green Version]
- Barradas-Bautista, D.; Rosell, M.; Pallara, C.; Fernandez-Recio, J. Structural prediction of protein-protein interactions by docking: Application to biomedical problems. Adv. Protein Chem. Struct. Biol. 2018, 110, 203–249. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-em structure of the 2019-ncov spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the sars-cov-2 spike receptor-binding domain bound to the ace2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Deng, W.; Li, S.; Yang, X. Advances in research on ace2 as a receptor for 2019-ncov. Cell Mol. Life Sci. 2021, 78, 531–544. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the sars-cov-2 spike glycoprotein. Cell 2020, 181, 281–292.e286. [Google Scholar] [CrossRef]
- Conti, P.; Caraffa, A.; Gallenga, C.E.; Kritas, S.K.; Frydas, I.; Younes, A.; Di Emidio, P.; Tetè, G.; Pregliasco, F.; Ronconi, G. The british variant of the new coronavirus-19 (sars-cov-2) should not create a vaccine problem. J. Biol. Regul. Homeost. Agents 2021, 35, 1–4. [Google Scholar] [CrossRef]
- Arashkia, A.; Jalilvand, S.; Mohajel, N.; Afchangi, A.; Azadmanesh, K.; Salehi-Vaziri, M.; Fazlalipour, M.; Pouriayevali, M.H.; Jalali, T.; Mousavi Nasab, S.D.; et al. Severe acute respiratory syndrome-coronavirus-2 spike (s) protein based vaccine candidates: State of the art and future prospects. Rev. Med. Virol. 2021, 31, e2183. [Google Scholar] [CrossRef] [PubMed]
- Koyama, T.; Platt, D.; Parida, L. Variant analysis of sars-cov-2 genomes. Bull. World Health Organ. 2020, 98, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Bindayna, K.M.; Crinion, S. Variant analysis of sars-cov-2 genomes in the middle east. Microb. Pathog. 2021, 153, 104741. [Google Scholar] [CrossRef]
- Tchesnokova, V.; Kulasekara, H.; Larson, L.; Bowers, V.; Rechkina, E.; Kisiela, D.; Sledneva, Y.; Choudhury, D.; Maslova, I.; Deng, K.; et al. Acquisition of the l452r mutation in the ace2-binding interface of spike protein triggers recent massive expansion of sars-cov-2 variants. J. Clin. Microbiol. 2021, 59, e0092121. [Google Scholar] [CrossRef]
- Yi, C.; Sun, X.; Ye, J.; Ding, L.; Liu, M.; Yang, Z.; Lu, X.; Zhang, Y.; Ma, L.; Gu, W.; et al. Key residues of the receptor binding motif in the spike protein of sars-cov-2 that interact with ace2 and neutralizing antibodies. Cell Mol. Immunol. 2020, 17, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of sars-cov-2 lineage b.1.1.7 in england. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.K.; Wang, B.; Sam, A.; Hoop, C.L.; Case, D.A.; Baum, J. Molecular dynamics analysis of a flexible loop at the binding interface of the sars-cov-2 spike protein receptor-binding domain. Proteins 2021. [Google Scholar] [CrossRef]
- Rattanapisit, K.; Bulaon, C.J.I.; Khorattanakulchai, N.; Shanmugaraj, B.; Wangkanont, K.; Phoolcharoen, W. Plant-produced sars-cov-2 receptor binding domain (rbd) variants showed differential binding efficiency with anti-spike specific monoclonal antibodies. PLoS ONE 2021, 16, e0253574. [Google Scholar] [CrossRef]
- Lou, Y.; Zhao, W.; Wei, H.; Chu, M.; Chao, R.; Yao, H.; Su, J.; Li, Y.; Li, X.; Cao, Y.; et al. Cross-neutralization of rbd mutant strains of sars-cov-2 by convalescent patient derived antibodies. Biotechnol. J. 2021, 16, e2100207. [Google Scholar] [CrossRef] [PubMed]
- West Jr, A.P.; Wertheim, J.O.; Wang, J.C.; Vasylyeva, T.I.; Havens, J.L.; Chowdhury, M.A.; Gonzalez, E.; Fang, C.E.; Di Lonardo, S.S.; Hughes, S.; et al. Detection and characterization of the sars-cov-2 lineage b.1.526 in new york. Nat. Commun. 2021, 12, 4886. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, J.; Kim, H.J.; Ko, M.; Jee, Y.; Kim, S. Tmprss2 and rna-dependent rna polymerase are effective targets of therapeutic intervention for treatment of covid-19 caused by sars-cov-2 variants (b.1.1.7 and b.1.351). Microbiol. Spectr. 2021, 9, e0047221. [Google Scholar] [CrossRef]
- Koopman, J.S.; Simon, C.P.; Getz, W.M.; Salter, R. Modeling the population effects of escape mutations in sars-cov-2 to guide vaccination strategies. Epidemics 2021, 36, 100484. [Google Scholar] [CrossRef]
- Duerr, R.; Dimartino, D.; Marier, C.; Zappile, P.; Wang, G.; Lighter, J.; Elbel, B.; Troxel, A.B.; Heguy, A. Dominance of alpha and iota variants in sars-cov-2 vaccine breakthrough infections in new york city. J. Clin. Investig. 2021, 131, e152702. [Google Scholar] [CrossRef]
- Zelyas, N.; Pabbaraju, K.; Croxen, M.A.; Lynch, T.; Buss, E.; Murphy, S.A.; Shokoples, S.; Wong, A.; Kanji, J.N.; Tipples, G. Precision response to the rise of the sars-cov-2 b.1.1.7 variant of concern by combining novel pcr assays and genome sequencing for rapid variant detection and surveillance. Microbiol. Spectr. 2021, 9, e0031521. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spanish Variant | British Variant | |
|---|---|---|
| Virus name | hCoV-19/Spain/CN-ISCIII-201048/2020 | hCoV-19/England/MILK-9E05B3/2020 |
| Accession ID | EPI_ISL_539531 | EPI_ISL_601443 |
| Type | Betacoronavirus | Betacoronavirus |
| GISAID Clade | G | GR |
| Lineage | B.1 | B.1.1.7 |
| Mutations (AA) | Spike D614G, Spike E773V, Spike F32L, NSP3 A358V, NSP3 E427D, NSP4 H31N, NSP12 P323L, NSP16 G208R, NSP16 K277E, NSP16 V294A | Spike A570D, Spike D614G, Spike D1118H, Spike H69del, Spike N501Y, Spike P681H, Spike S982A, Spike T716I, Spike V70del, Spike Y145del, N D3L, N G204R, N R203K, N S235F, NS8 Q27stop, NS8 R52I, NS8 Y73C, NSP3 A890D, NSP3 I1412T, NSP3 T183I, NSP6 F108del, NSP6 G107del, NSP6 S106del, NSP12 P323L |
| Collection date | 20 February 2020 | 20 September 2020 |
| Location | Europe/Spain/Canary Islands | Europe/United Kingdom/England |
| Host | Human | Human |
| Originating laboratory | Hospital Universitario Nuestra Señora de Candelaria, Tenerife, Spain | Lighthouse Lab, Milton Keynes, United Kingdom |
| Submitting laboratory | Instituto de Salud Carlos III, Madrid, Spain | Wellcome Sanger Institute for the COVID-19 Genomics UK (COG-UK) consortium |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jimenez Ruiz, J.A.; Lopez Ramirez, C.; Lopez-Campos, J.L. A Comparative Study between Spanish and British SARS-CoV-2 Variants. Curr. Issues Mol. Biol. 2021, 43, 2036-2047. https://doi.org/10.3390/cimb43030140
Jimenez Ruiz JA, Lopez Ramirez C, Lopez-Campos JL. A Comparative Study between Spanish and British SARS-CoV-2 Variants. Current Issues in Molecular Biology. 2021; 43(3):2036-2047. https://doi.org/10.3390/cimb43030140
Chicago/Turabian StyleJimenez Ruiz, Jose A., Cecilia Lopez Ramirez, and Jose Luis Lopez-Campos. 2021. "A Comparative Study between Spanish and British SARS-CoV-2 Variants" Current Issues in Molecular Biology 43, no. 3: 2036-2047. https://doi.org/10.3390/cimb43030140
APA StyleJimenez Ruiz, J. A., Lopez Ramirez, C., & Lopez-Campos, J. L. (2021). A Comparative Study between Spanish and British SARS-CoV-2 Variants. Current Issues in Molecular Biology, 43(3), 2036-2047. https://doi.org/10.3390/cimb43030140