
Genome-Wide Identification of LRR-RLK Family in Saccharum and Expression Analysis in Response to Biotic and Abiotic Stress



Abstract
:1. Introduction
2. Materials and Methods
2.1. Arabidopsis LRR-RLKs and Saccharum spontaneum Genome Resources
2.2. Identification of LRR-RLKs in Saccharum spontaneum Genome
2.3. Multiple Sequence Alignments and Phylogenetic Tree Construction
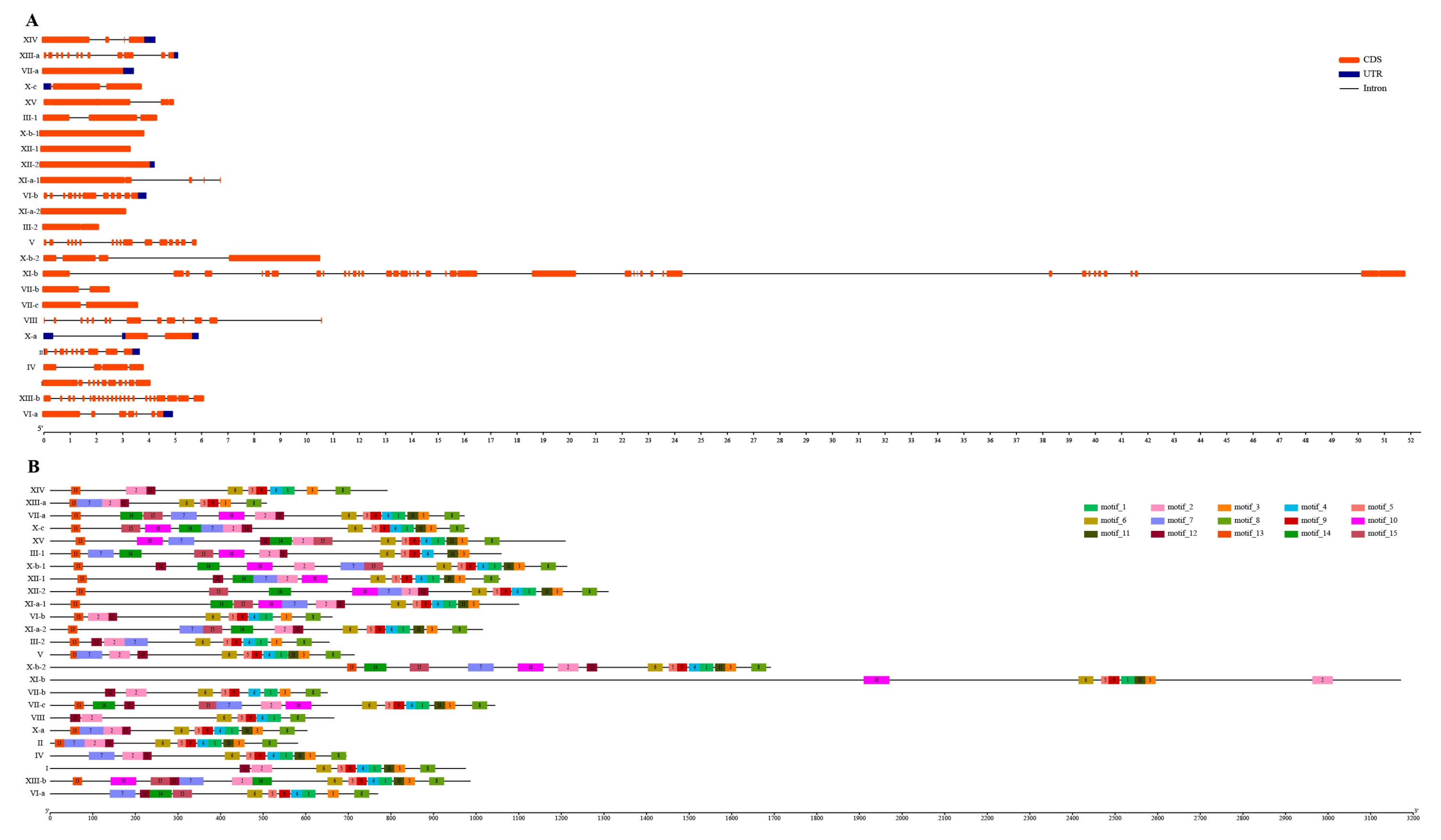
2.4. The Analysis of the Chromosome Distribution, Gene Structure, and Conserved Motif of the Sugarcane LRR-RLK Genes
2.5. Promoter and Regulatory Analysis of the Sugarcane LRR-RLK Genes
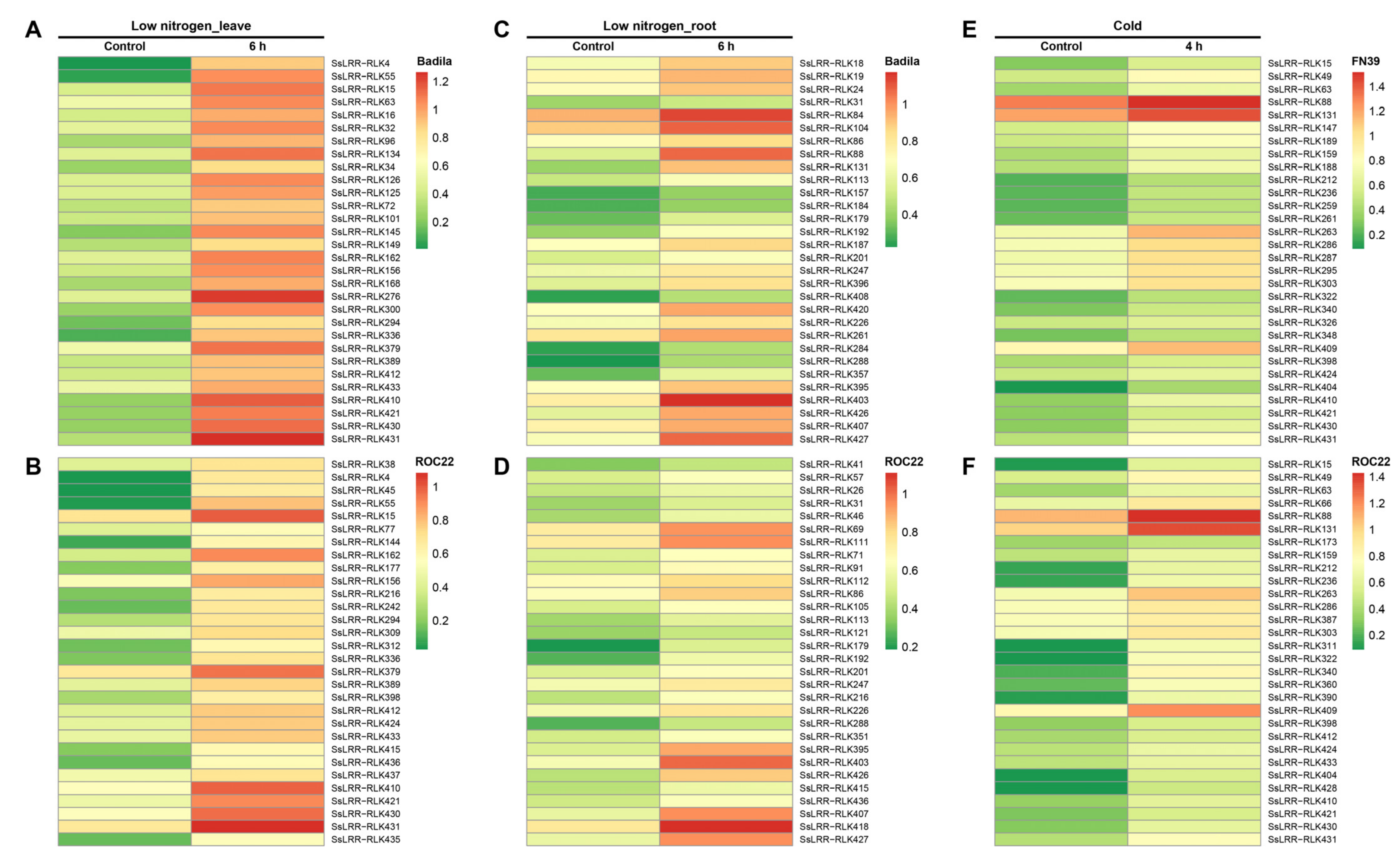
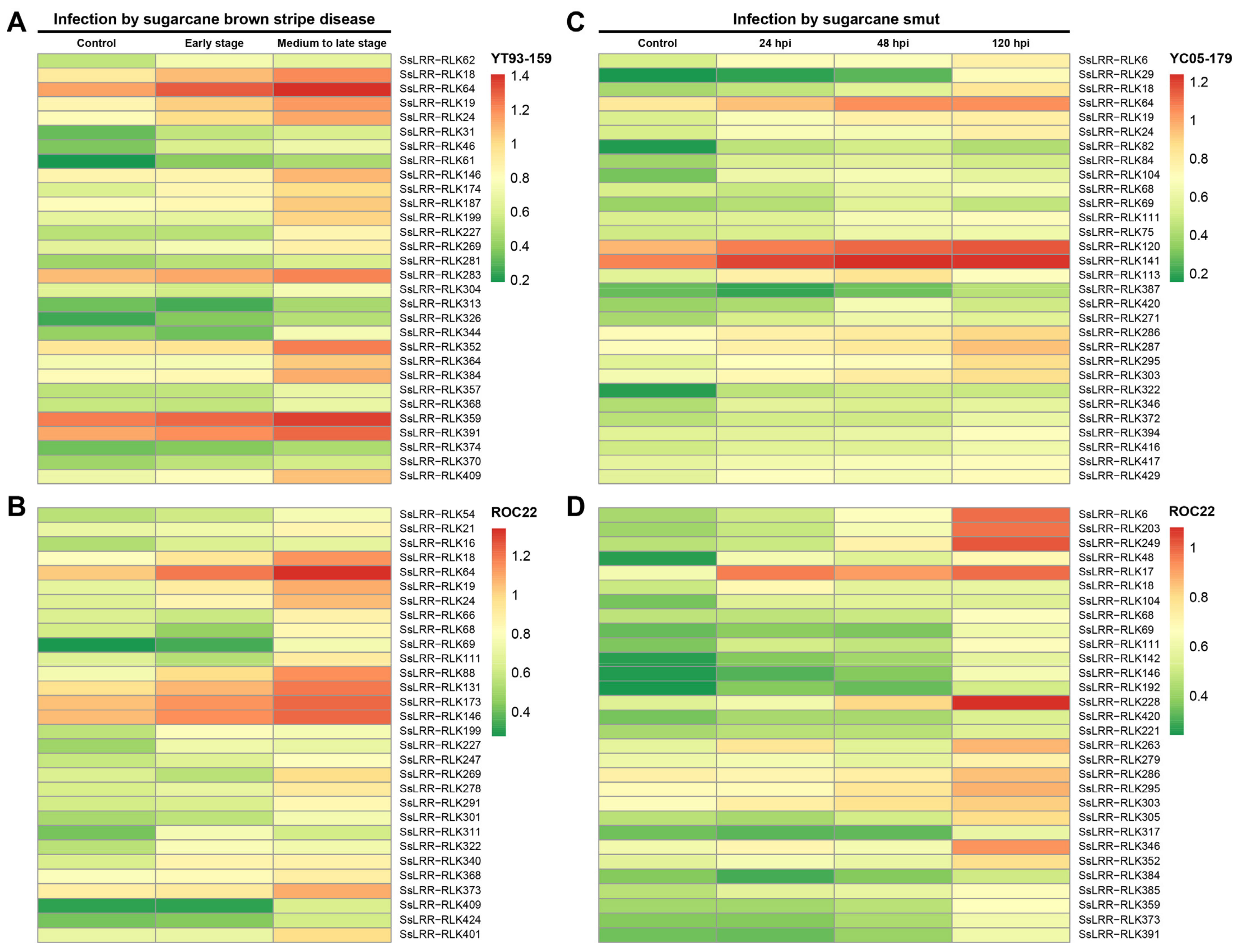
2.6. Expression Pattern Analysis of the Sugarcane LRR-RLK Genes
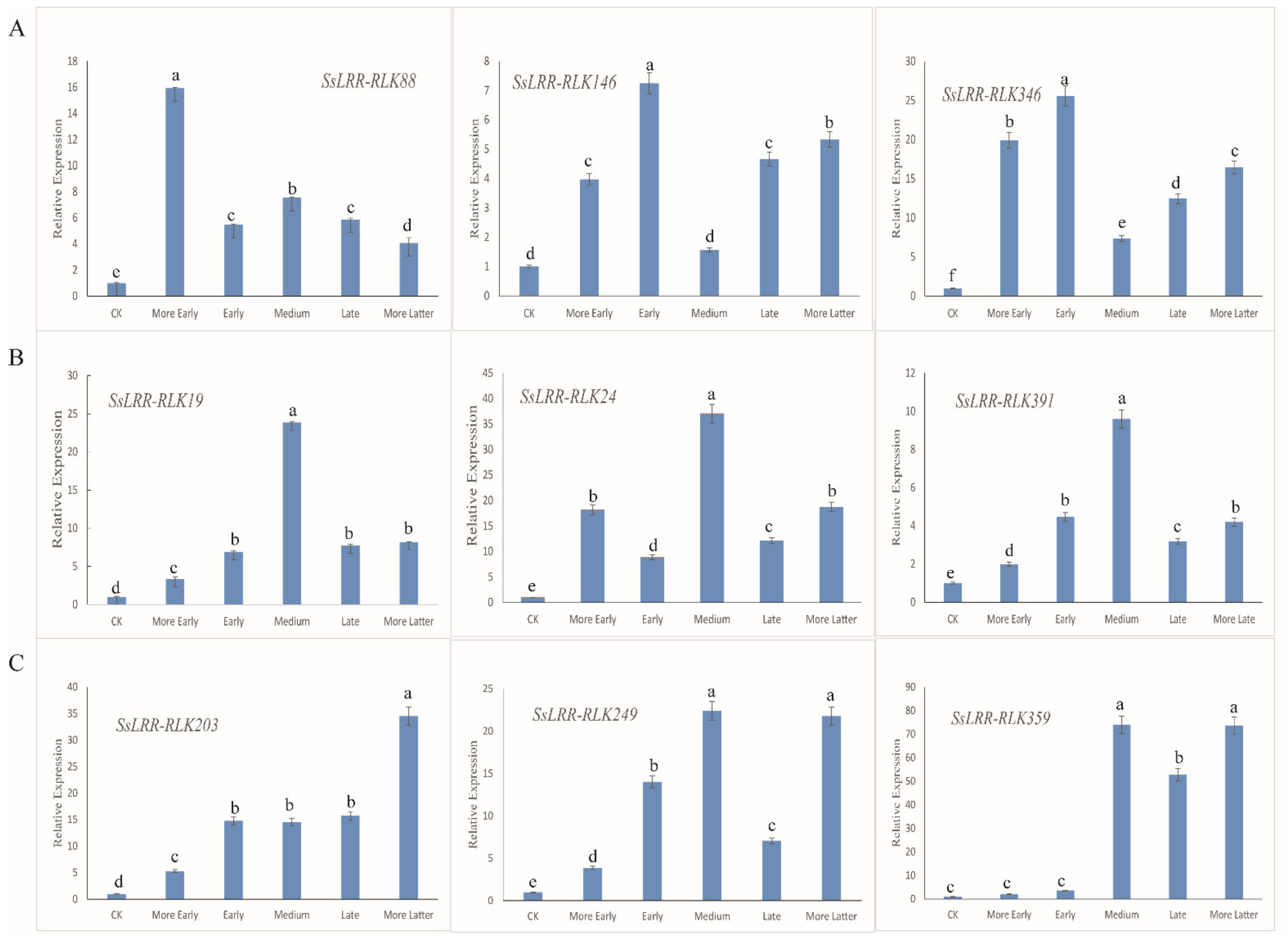
2.7. Plant Materials, RNA Extraction and qRT-PCR Analysis
3. Results
3.1. Identification and Distribution of LRR-RLK Genes in Saccharum spontaneum
3.2. Signal Peptide and Subcellular Localization Analysis of the LRR-RLK Family Proteins
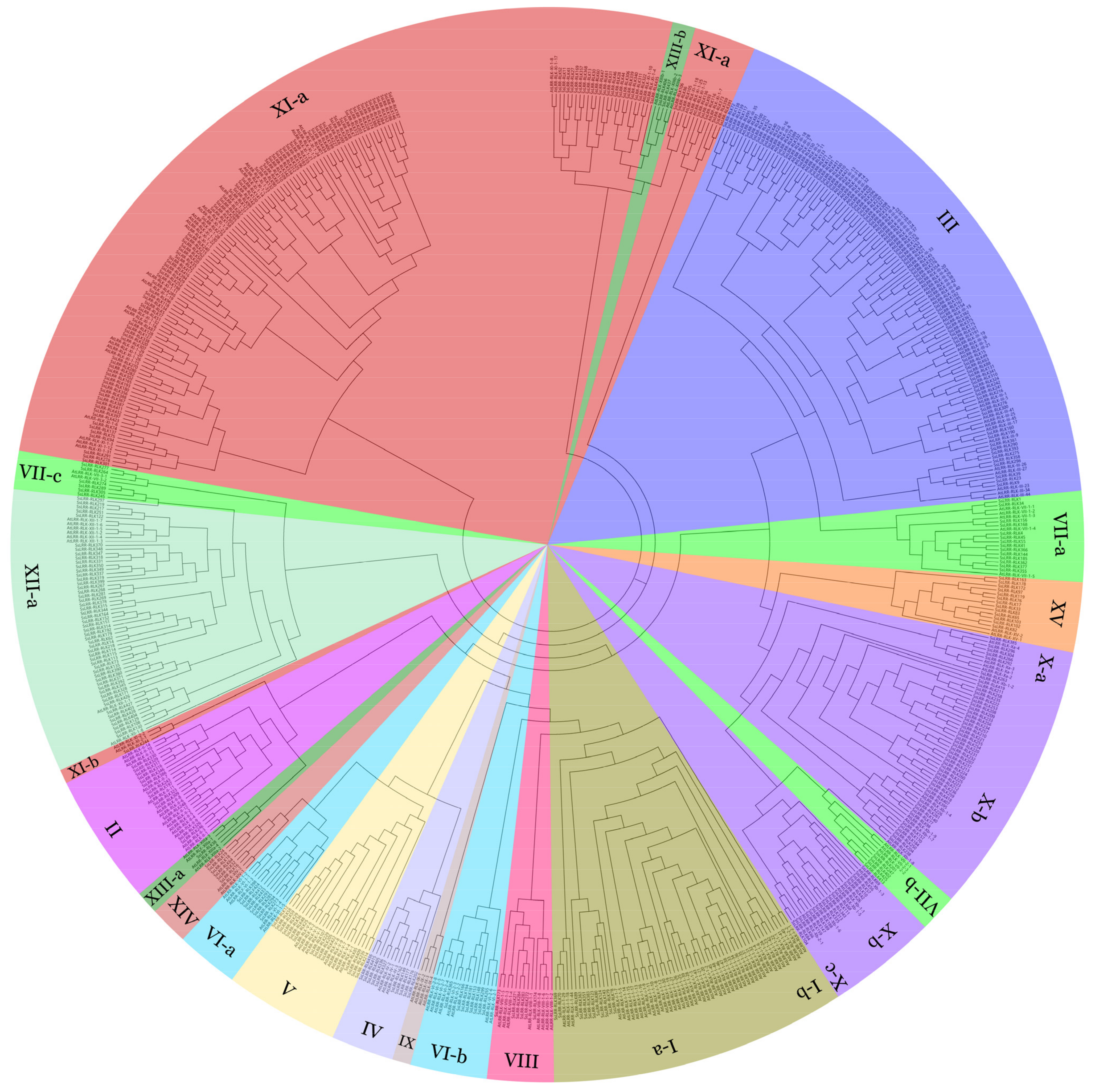
3.3. Phylogenetic Analysis of Sugarcane LRR-RLKs
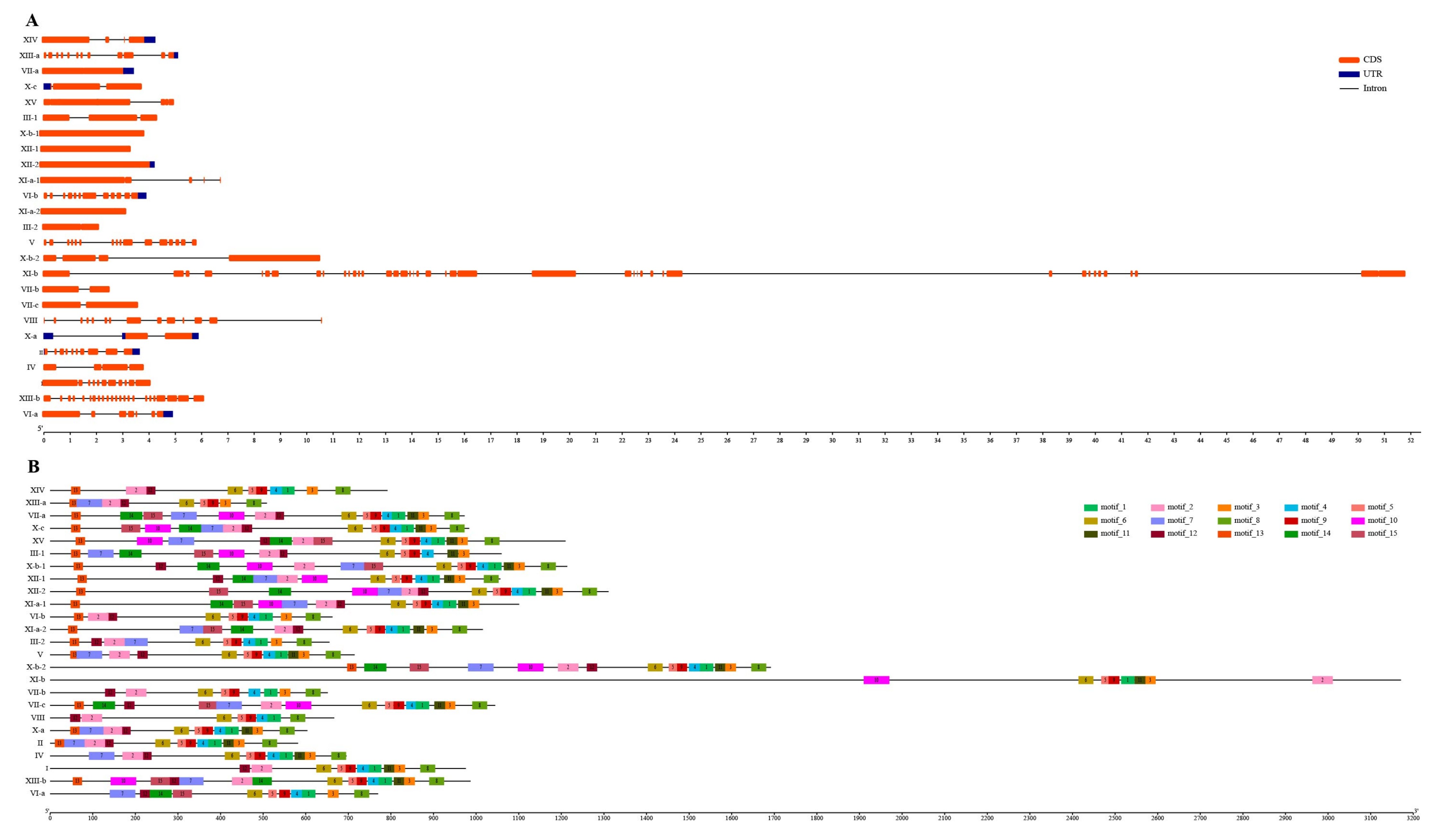
3.4. Exon–Intron Organization and Conserved Motifs of SsLRR-RLKs
3.5. A cis-Acting Regulatory Elements and TF Binding Sites Analysis of SsLRR-RLKs
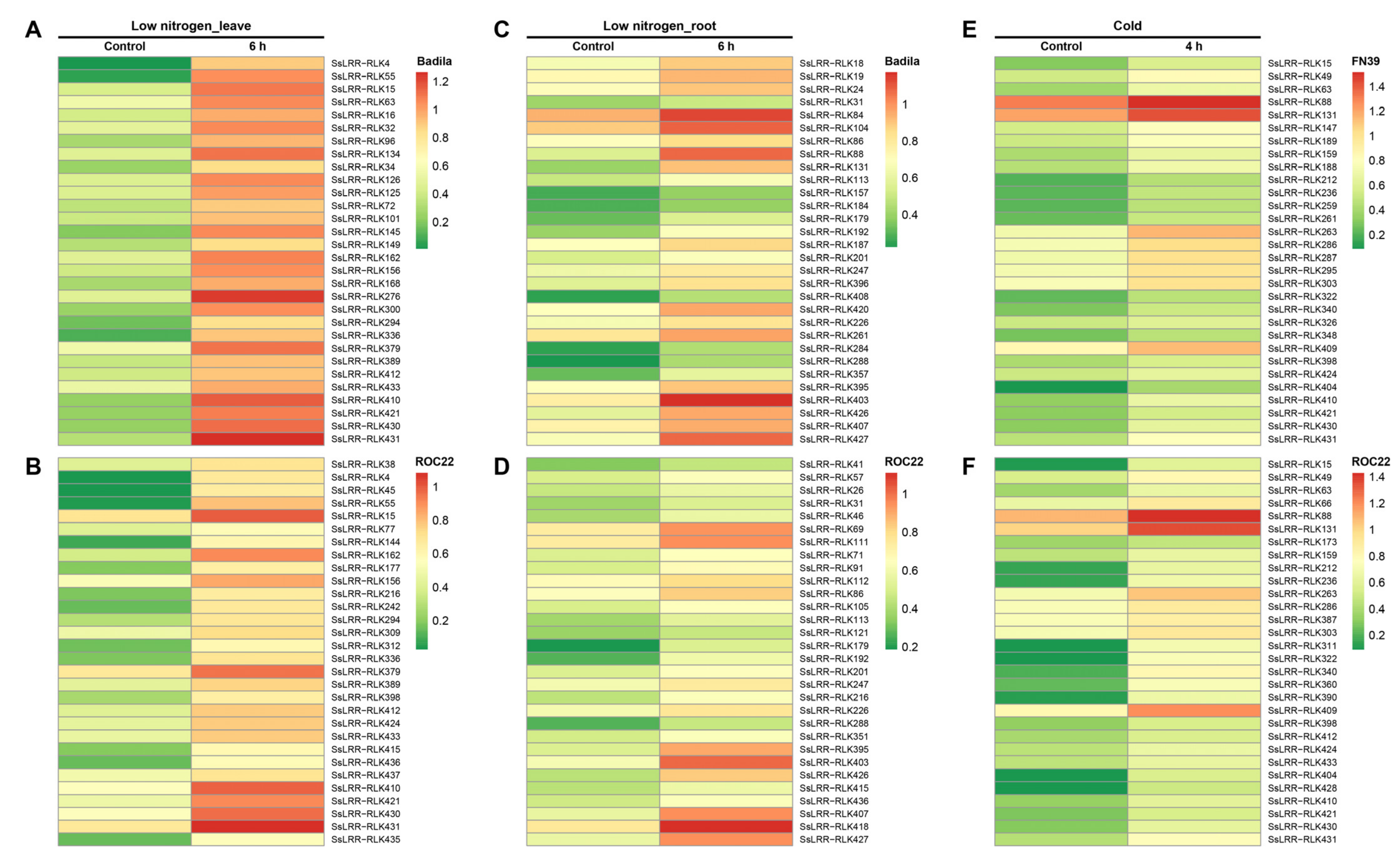
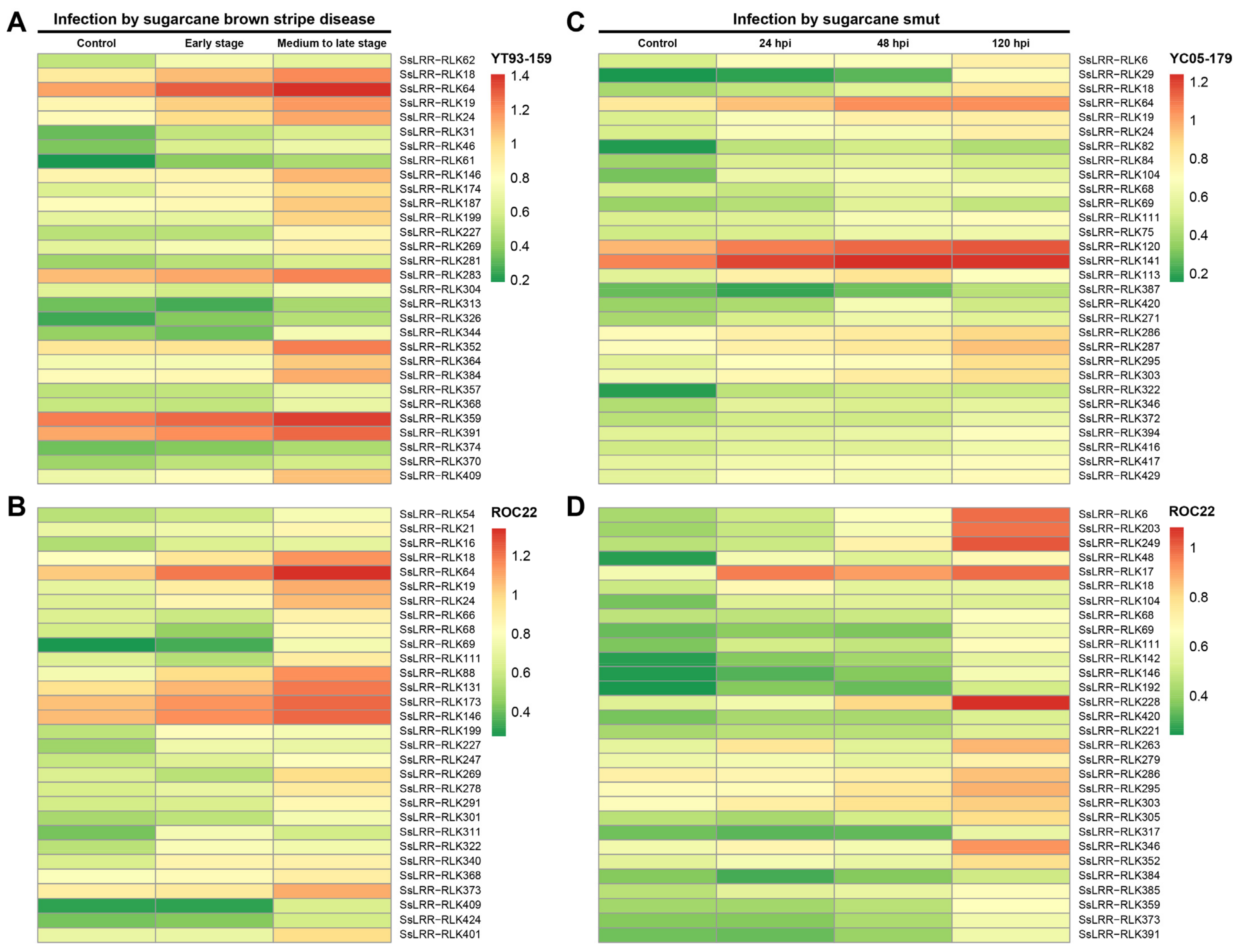
3.6. Expression Profiling of SsLRR-RLK Genes Based on RNA-seq Datasets
3.7. Expression Analysis of SsLRR-RLK Genes by Quantitative Real-Time PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Walker, J.C. Structure and function of the receptor-like protein kinases of higher plants. Plant Mol. Biol. 1994, 26, 1599–1609. [Google Scholar] [CrossRef]
- Geer, P.V.; Hunter, T.; Lindberg, R.A. Receptor protein-tyrosine kinases and their signal transduction pathways. Annu. Rev. Cell Biol. 1994, 10, 251–337. [Google Scholar] [CrossRef] [PubMed]
- Afzal, A.J.; Wood, A.J.; Lightfoot, D.A. Plant receptor-like serine threonine kinases: Roles in signaling and plant defense. Mol. Plant Microbe Interact. 2008, 21, 507–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, J.C.; Zhang, R. Relationship of a putative receptor protein kinase from maize to the S-locus glycoproteins of Brassica. Nature 1990, 345, 743–746. [Google Scholar] [CrossRef]
- Xue, H.W. Global analysis of expression profiles of rice receptor-like kinase genes. Mol. Plant 2012, 5, 143–153. [Google Scholar]
- Shiu, S.H. Plant receptor-like kinase gene family: Diversity, function, and signaling. Sci Stke 2001, 113, re22. [Google Scholar] [CrossRef]
- Kobe, B.; Kajava, A.V. The leucine-rich repeat as a protein recognition motif. Curr. Opin. Struct. Biol. 2001, 11, 725–732. [Google Scholar] [CrossRef]
- Liu, P.L.; Du, L.; Huang, Y.; Gao, S.M.; Yu, M. Origin and diversification of leucine-rich repeat receptor-like protein kinase (LRR-RLK) genes in plants. BMC Evol. Biol. 2017, 17, 47. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Li, L.; Wang, P.; Zhang, S.; Wu, J. Genome-wide characterization, evolution, and expression analysis of the leucine-rich repeat receptor-like protein kinase (LRR-RLK) gene family in Rosaceae genomes. BMC Genom. 2017, 18, 763. [Google Scholar] [CrossRef] [Green Version]
- Zan, Y.; Ji, Y.; Zhang, Y.; Yang, S.; Song, Y.; Wang, J. Genome-wide identification, characterization and expression analysis of populus leucine-rich repeat receptor-like protein kinase genes. BMC Genom. 2013, 14, 318. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Guo, Y.; Qiu, L.J. Genome-wide identification and evolutionary analysis of leucine-rich repeat receptor-like protein kinase genes in soybean. BMC Plant Biol. 2016, 16, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, R.; Wang, S.; Ma, D.; Liu, C. Genome-wide analysis of LRR-RLK gene family in four gossypium species and expression analysis during cotton development and stress responses. Genes 2018, 9, 592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Z.; Wang, J.; Yang, S.; Song, Y. Identification and expression analysis of the LRR-RLK gene family in tomato (Solanum lycopersicum) Heinz 1706. Genome 2015, 58, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Gou, X.; He, K.; Yang, H.; Yuan, T.; Lin, H.; Clouse, S.D.; Li, J. Genome-wide cloning and sequence analysis of leucine-rich repeat receptor-like protein kinase genes in Arabidopsis thaliana. BMC Genom. 2010, 11, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Zhu, Y.F.; Cui, Y.W.; Cheng, K.L.; Liang, W.; Wei, Z.Y.; Zhu, M.S.; Yin, H.J.; Zeng, L.; Xiao, Y.; et al. A group of receptor kinases are essential for CLAVATA signalling to maintain stem cell homeostasis. Nat. Plants 2018, 4, 205–211. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Seto, H.; Fujioka, S.; Yoshida, S.; Chory, J. BRI1 is a critical component of a plasma-membrane receptor for plant steroids. Nature 2001, 410, 380–383. [Google Scholar] [CrossRef]
- Somssich, M.; Je, B.I.; Simon, R.; Jackson, D. CLAVATA-WUSCHEL signaling in the shoot meristem. Development 2016, 143, 3238–3248. [Google Scholar] [CrossRef] [Green Version]
- Nam, K.H.; Jianming, L. BRI1/BAK1, a receptor kinase pair mediating brassinosteroid signaling. Cell 2002, 110, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Jiangqi, W.; Kevin, A.L.; Jason, T.D.; Frans, E.T.; John, C.W. BAK1, an Arabidopsis LRR receptor-like protein kinase, interacts with BRI1 and modulates brassinosteroid signaling. Cell 2002, 110, 213–222. [Google Scholar] [CrossRef] [Green Version]
- Canales, C.; Bhatt, A.M.; Scott, R.; Dickinson, H. EXS, a putative LRR receptor kinase, regulates male germline cell number and tapetal identity and promotes seed development in Arabidopsis. Curr. Biol. 2002, 12, 1718–1727. [Google Scholar] [CrossRef]
- Jinn, T.L.; Stone, J.M.; Walker, J.C. HAESA, an Arabidopsis leucine-rich repeat receptor kinase, controls floral organ abscission. Genes Dev. 2000, 14, 108–117. [Google Scholar]
- Demko, V.; Ako, E.; Perroud, P.F.; Quatrano, R.; Olsen, O.A. The phenotype of the CRINKLY4 deletion mutant of Physcomitrella patens suggests a broad role in developmental regulation in early land plants. Planta 2016, 244, 275–284. [Google Scholar] [CrossRef]
- Hirakawa, Y.; Kondo, Y.; Fukuda, H. TDIF peptide signaling regulates vascular stem cell proliferation via the WOX4 homeobox gene in arabidopsis. Plant Cell 2010, 22, 2618–2629. [Google Scholar] [CrossRef] [Green Version]
- Song, W.Y.; Wang, G.L.; Chen, L.L.; Kim, H.S.; Pi, L.Y.; Holsten, T.; Gardner, J.; Wang, B.; Zhai, W.X.; Zhu, L.H. A receptor kinase-like protein encoded by the rice disease resistance gene, Xa21. Science 1995, 270, 1804–1806. [Google Scholar] [CrossRef] [Green Version]
- Osakabe, Y.; Maruyama, K.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Leucine-rich repeat receptor-like kinase1 is a key membrane-bound regulator of abscisic acid early signaling in arabidopsis. Plant Cell 2005, 17, 1105–1119. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.C.; Hong, S.W.; Whang, S.S.; Lim, P.O.; Nam, H.G.; Koo, J.C. Age-dependent action of an ABA-inducible receptor kinase, RPK1, as a positive regulator of senescence in Arabidopsis leaves. Plant Cell Physiol. 2011, 52, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Godiard, L.; Sauviac, L.; Torii, K.U.; Grenon, O.; Marco, Y. ERECTA, an LRR receptor-like kinase protein controlling development pleiotropically affects resistance to bacterial wilt. Plant J. 2003, 36, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Torii, K.U.; Mitsukawa, N.; Oosumi, T.; Matsuura, Y.; Yokoyama, R.; Whittier, R.F.; Komeda, Y. The Arabidopsis ERECTA gene encodes a putative receptor protein kinase with extracellular leucine-rich repeats. Plant Cell 1996, 8, 735–746. [Google Scholar] [PubMed] [Green Version]
- Chinchilla, D.; Libo, S.; Ping, H.; Sacco de, V.; Birgit, K. One for all: The receptor-associated kinase BAK1. Trends Plant Sci. 2009, 14, 535–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Wang, G.L. Genome-wide identification, characterization and phylogenetic analysis of the rice LRR-kinases. PLoS ONE 2011, 6, e16079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rameneni, J.J.; Lee, Y.; Dhandapani, V.; Yu, X.; Su, R.C.; Oh, M.H.; Yong, P.L. Genomic and post-translational modification analysis of leucine-rich-repeat receptor-like kinases in brassica rapa. PLoS ONE 2015, 10, e0142255. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.; Tang, H.; Zhang, Q.; Hua, X.; Ma, X.; Zhu, F.; Jones, T.; Zhu, X.; Bowers, J.; et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. 2018, 50, 1565–1573. [Google Scholar] [CrossRef] [Green Version]
- Shiu, S.H.; Karlowski, W.M.; Pan, R.; Tzeng, Y.H.; Mayer, K.F.; Li, W.H. Comparative analysis of the receptor-like kinase family in Arabidopsis and rice. Plant Cell 2004, 16, 1220–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.D.; Penelope, C.; Eberhardt, R.Y.; Eddy, S.R.; Jaina, M.; Mitchell, A.L.; Potter, S.C.; Marco, P.; Matloob, Q.; Amaia, S.V.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Doerks, T.; Bork, P. SMART: Recent updates, new developments and status in 2015. Nucleic Acids Res. 2014, 43, D257–D260. [Google Scholar] [CrossRef]
- Viklund, H.; Elofsson, A. Best alpha-helical transmembrane protein topology predictions are achieved using hidden Markov models and evolutionary information. Protein Sci. 2004, 13, 1908–1917. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPASy Server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar]
- Long, R.; Wang, H.; Shen, Y.; Kang, J.; Zhang, T.; Sun, Y.; Zhang, Y.; Li, M.; Yang, Q. Molecular cloning and functional analysis of a salt-induced gene encoding an RNA-binding protein in alfalfa. Mol. Breed. 2014, 34, 1465–1473. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; Heijne, G.V.; Nielsen, H.H. SIGNALP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2-approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Kong, Y.; Wang, Q.; Sun, Y.; Gong, D.; Lv, J.; Liu, G. MapGene2Chrom, a tool to draw gene physical map based on Perl and SVG languages. Hereditas 2015, 37, 91–97. [Google Scholar]
- Hu, B.; Jin, J.; Guo, A.Y.; Zhang, H.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Nadya, W.; Chris, M.; Li, W.W. MEME: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006, 34, W369–W373. [Google Scholar] [CrossRef]
- Lescot, M. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Gao, S.; Shumway, S.Y.; Lin, Z.; Guo, J.; Li, M.; Wang, Z.; Que, Y.; Xu, L. Transcripts and low nitrogen tolerance: Regulatory and metabolic pathways in sugarcane under low nitrogen stress. Environ. Exp. Bot. 2019, 163, 97–111. [Google Scholar] [CrossRef]
- Que, Y.; Su, Y.; Guo, J.; Wu, Q.; Xu, L. A global view of transcriptome dynamics during Sporisorium scitamineum challenge in sugarcane by RNA-seq. PLoS ONE 2014, 9, e106476. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Hou, M.; Liu, Y.; Pei, Y.; Ye, M.; Zhou, Y.; Huang, C.; Zhao, Y.; Ma, H. Genome-wide identification, characterization and expression analysis of the non-specific lipid transfer proteins in potato. BMC Genom. 2019, 20, 375. [Google Scholar] [CrossRef]
- Brooks, M.J.; Rajasimha, H.K.; Roger, J.E.; Swaroop, A. Next-generation sequencing facilitates quantitative analysis of wild-type and Nrl−/− retinal transcriptomes. Mol. Vis. 2011, 17, 3034–3054. [Google Scholar]
- Galili, T.; O’Callaghan, A.; Sidi, J.; Sievert, C. Heatmaply: An R package for creating interactive cluster heatmaps for online publishing. Bioinformatics 2018, 34, 1600–1602. [Google Scholar] [CrossRef]
- Wang, Z.; Ren, H.; Xu, F.; Lu, G.; Cheng, W.; Que, Y.; Xu, L. Genome-wide characterization of lectin receptor kinases in Saccharum spontaneum L. and their responses to Stagonospora tainanensis infection. Plants 2021, 10, 322. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta CT) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, C.; Zhang, M.; Yang, C.; Bao, Y.; Wang, D.; Chen, Y.; Chen, Q. Genome-wide analysis and expression profiling of the Phospholipase D gene family in Solanum tuberosum. Biology 2021, 10, 741. [Google Scholar] [CrossRef] [PubMed]
- Shiu, S.H.; Bleecker, A.B. Receptor-like kinases from Arabidopsis form a monophyletic gene family related to animal receptor kinases. Proc. Natl. Acad. Sci. USA 2001, 98, 10763–10768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.; Xu, L.; Wang, Z.; Peng, Q.; Yang, Y.; Chen, Y.; Que, Y. Comparative proteomics reveals that central metabolism changes are associated with resistance against Sporisorium scitamineum in sugarcane. BMC Genom. 2016, 17, 800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karve, R.; Liu, W.; Willet, S.G.; Torii, K.U.; Shpak, E.D. The presence of multiple introns is essential for ERECTA expression in Arabidopsis. RNA 2011, 17, 1907–1921. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Washida, H.; Onodera, Y.; Harada, K.; Takaiwa, F. Quantitative nature of the Prolamin-box, ACGT and AACA motifs in a rice glutelin gene promoter: Minimal cis-element requirements for endosperm-specific gene expression. Plant J. 2000, 23, 415–421. [Google Scholar] [CrossRef]
- Narusaka, Y.; Nakashima, K.; Shinwari, Z.K.; Sakuma, Y.; Furihata, T.; Abe, H.; Narusaka, M.; Shinozaki, K.; Yamaguchishinozaki, K. Interaction between two cis-acting elements, ABRE and DRE, in ABA-dependent expression of Arabidopsis rd29A gene in response to dehydration and high-salinity stresses. Plant J. 2003, 34, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Ora, H.; Hardtke, C.S. CLAVATA 1-type receptors in plant development. J. Exp. Bot. 2016, 16, 4827–4833. [Google Scholar]
- Zipfel, C.; Kunze, G.; Chinchilla, D.; Canirrd, A.; Jones, J.D.G.; Boller, T.; Felix, G. Perception of the bacterial PAMP EF-Tu by the receptor EFR restricts agrobacterium-mediated transformation. Cell 2006, 125, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Owens, D.K.; Alerding, A.B.; Crosby, K.C.; Bandara, A.B.; Westwood, J.H.; Winkel, B.S.J. Functional analysis of a predicted flavonol synthase gene family in Arabidopsis. Plant Physiol. 2008, 147, 1046–1061. [Google Scholar] [CrossRef] [Green Version]
- Gómezgómez, L.; Boller, T. FLS2: An LRR receptor-like kinase involved in the perception of the bacterial elicitor flagellin in Arabidopsis. Mol. Cell 2000, 5, 1003–1011. [Google Scholar] [CrossRef]
- Hu, H.; Xiong, L.; Yang, Y. Rice SERK1 gene positively regulates somatic embryogenesis of cultured cell and host defense response against fungal infection. Planta 2005, 222, 107–117. [Google Scholar] [CrossRef]
- Gou, X.; Yin, H.; He, K.; Du, J.; Yi, J.; Xu, S.; Lin, H.; Clouse, S.; Li, J. Genetic evidence for an indispensable role of somatic embryogenesis receptor kinases in brassinosteroid signaling. PLoS Genet. 2012, 8, e1002452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albrecht, C.; Russinova, E.; Kemmerling, B.; Kwaaitaal, M.; de Vries, S.C. Arabidopsis SOMATIC EMBRYOGENESIS RECEPTOR KINASE proteins serve brassinosteroid-dependent and -independent signaling pathways. Plant Physiol. 2008, 148, 611–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Subgroups | No. of Genes | Amino Acid Length | With Signal PEPTIDE (%) | Molecular Weight (kDa) | Isoelectric Point |
|---|---|---|---|---|---|---|
| I | a | 12 | 521–1081 | 58.3 | 53.79–117.29 | 5.70–8.57 |
| b | 0 | - | - | - | - | |
| II | 13 | 473–982 | 69.2 | 52.04–105.55 | 5.45–10.63 | |
| III | 75 | 358–1775 | 84.0 | 39.00–189.90 | 5.55–10.49 | |
| IV | 10 | 529–977 | 80.0 | 55.54–108.99 | 6.83–9.07 | |
| V | 15 | 553–1403 | 80.0 | 59.12–153.82 | 5.22–8.69 | |
| VI | a | 8 | 692–771 | 100 | 75.81–82.38 | 6.40–8.87 |
| b | 8 | 572–1070 | 87.5 | 63.67–118.29 | 5.16–6.66 | |
| VII | a | 14 | 925–1930 | 92.9 | 98.09–209.91 | 5.21–7.14 |
| b | 5 | 649–714 | 100 | 69.08–76.10 | 7.25–9.56 | |
| c | 6 | 436–1095 | 33.3 | 48.33–115.15 | 5.54–8.97 | |
| VIII | 6 | 549–1000 | 50.0 | 60.55–110.28 | 5.67–8.77 | |
| IX | 0 | - | - | - | - | |
| X | a | 5 | 560–849 | 80.0 | 61.52–91.70 | 6.44–9.90 |
| b | 59 | 474–1830 | 59.3 | 52.62–197.90 | 5.06–7.25 | |
| c | 3 | 973–983 | 100 | 104.62–106.10 | 5.74–6.18 | |
| XI | a | 124 | 471–2678 | 74.2 | 51.03–292.02 | 5.17–9.50 |
| b | 1 | 3171 | 100 | 349.15 | 5.78 | |
| XII | 51 | 566–2005 | 56.9 | 61.30–213.89 | 5.26–9.56 | |
| XIII | a | 1 | 508 | 100 | 56.19 | 5.60 |
| b | 2 | 958–986 | 100 | 103.67–106.54 | 5.66–5.96 | |
| XIV | 6 | 661–992 | 83.3 | 70.94–107.52 | 8.27–9.78 | |
| XV | 13 | 713–1209 | 92.3 | 76.83–128.91 | 6.02–9.09 |
| Types | Functional Classification | Element Species (ID of PlantCARE) | No. of Elements |
|---|---|---|---|
| 1 | Light responsive elements | GATT-motif, MRE, 3-AF1 binding site, Sp1, CAG-motif, GA-motif, 3-AF3 binding site, chs-CMA1a/2a/2b, Gap-box, LS7, I-box, 4cl-CMA1b/2b, AAAC-motif, ACA-motif, ACE, GT1-motif, AE-box, TCCC-motif, AT1-motif, Pc-CMA2a/2c, ATC-motif, ATCT-motif, Box4, Box II, chs-Unit, LAMP-element, GATA-motif, G-Box, GTGGC-motif, L-box, sbp-SMA1c, TCT-motif | 36 |
| 2 | Hormone responsive elements | AuxRR-core, P-box, TGA-box, ERE, ABRE, ABRE2, JERE, TCA-element, ABRE3a, TATC-box, ABRE4, AT-ABRE, AuxRE, CARE, CGTCA-motif, SARE, TGACG-motif, TGA-element, GARE-motif | 19 |
| 3 | Environmental stress-related elements | MBS, ACTCATCCT-sequence, LTR, AP-1, STRE, MYB recognition site, ARE, as-1, box-S, DRE, MYC, DRE core, DRE1, W box, GC-motif, MYB-like sequence, MYB, WRE3, WUN-motif, TC-rich repeats | 20 |
| 4 | Development-related elements | GCN4_motif, AACA_motif, CCGTCC-motif, AC-I, CCGTCC-box, circadian, dOCT, E2Fb, HD-Zip 1, MSA-like, motif I, Myb-binding site, NON, NON-box, AC-II, O2-site, OCT, RY-element, re2f-1, CAT-box, telo-box | 21 |
| 5 | Promoter-related elements | A-box, AT-TATA-box, Box II-like sequence, CAAT-box, HMG-TATA-region, TATA, TATA-box | 7 |
| 6 | Binding site elements | AT-rich element, HD-Zip 3, AT-rich sequence, MBSI, BOX III, CCAAT-box | 6 |
| 7 | Other no functional description elements | AAGAA-motif, CTAG-motif, F-box, GC-repeat, GRA, H-box, plant_AP-2-like, TCA, Y-box, Unnamed_1/2/3/5/6/8/10/12/14/16 | 19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, W.; Wang, Z.; Xu, F.; Ahmad, W.; Lu, G.; Su, Y.; Xu, L. Genome-Wide Identification of LRR-RLK Family in Saccharum and Expression Analysis in Response to Biotic and Abiotic Stress. Curr. Issues Mol. Biol. 2021, 43, 1632-1651. https://doi.org/10.3390/cimb43030116
Cheng W, Wang Z, Xu F, Ahmad W, Lu G, Su Y, Xu L. Genome-Wide Identification of LRR-RLK Family in Saccharum and Expression Analysis in Response to Biotic and Abiotic Stress. Current Issues in Molecular Biology. 2021; 43(3):1632-1651. https://doi.org/10.3390/cimb43030116
Chicago/Turabian StyleCheng, Wei, Zhoutao Wang, Fu Xu, Waqar Ahmad, Guilong Lu, Yachun Su, and Liping Xu. 2021. "Genome-Wide Identification of LRR-RLK Family in Saccharum and Expression Analysis in Response to Biotic and Abiotic Stress" Current Issues in Molecular Biology 43, no. 3: 1632-1651. https://doi.org/10.3390/cimb43030116
APA StyleCheng, W., Wang, Z., Xu, F., Ahmad, W., Lu, G., Su, Y., & Xu, L. (2021). Genome-Wide Identification of LRR-RLK Family in Saccharum and Expression Analysis in Response to Biotic and Abiotic Stress. Current Issues in Molecular Biology, 43(3), 1632-1651. https://doi.org/10.3390/cimb43030116