
Combined Transcriptome and Proteome Analysis of Maize (Zea mays L.) Reveals A Complementary Profile in Response to Phosphate Deficiency

Abstract
:1. Introduction
2. Material and Methods
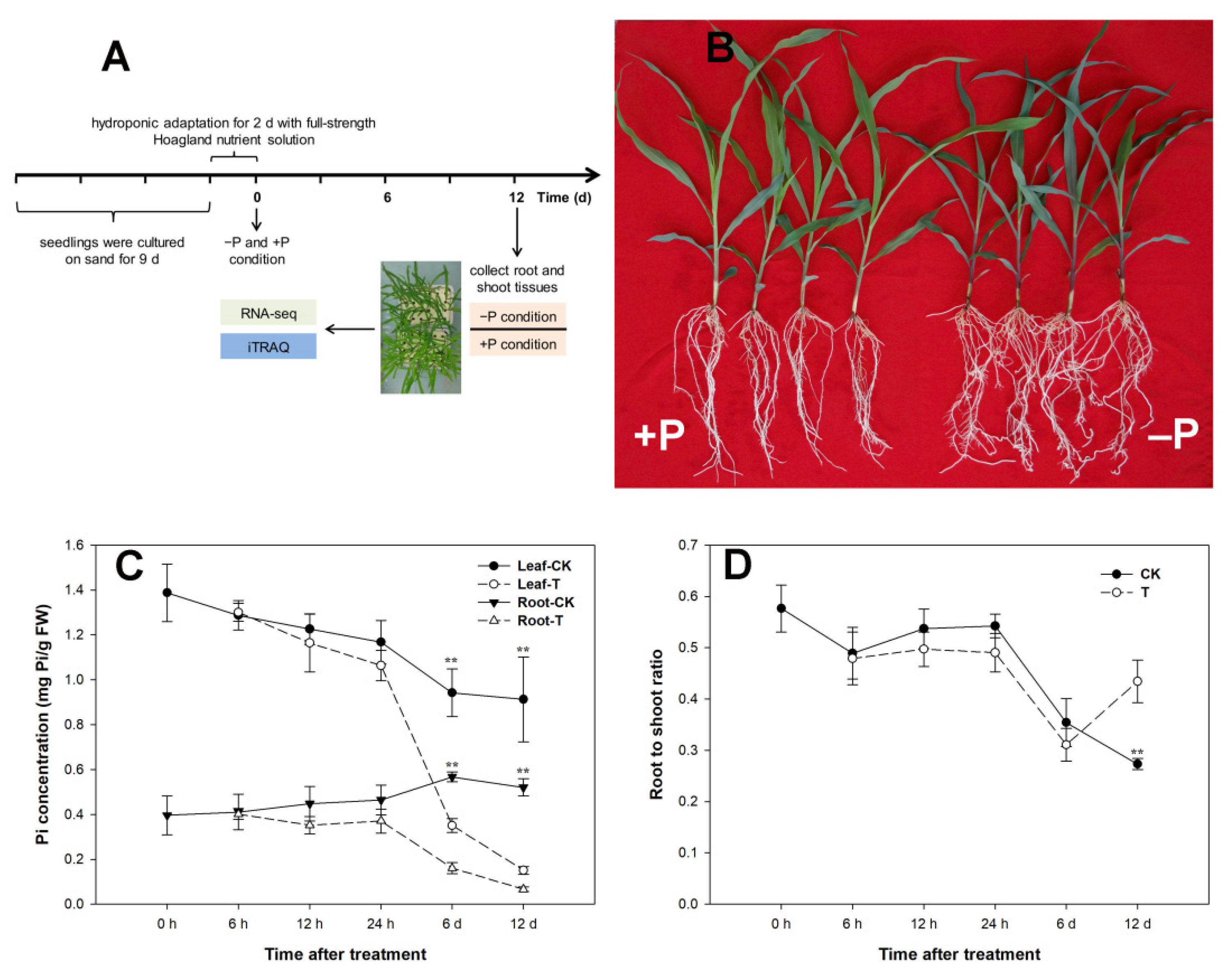
2.1. Plant Materials, Hydroponic Conditions and Measurement of Physiological Indexes
2.2. RNA and Protein Extraction
2.3. RNA-seq Analysis
2.4. iTRAQ Analysis
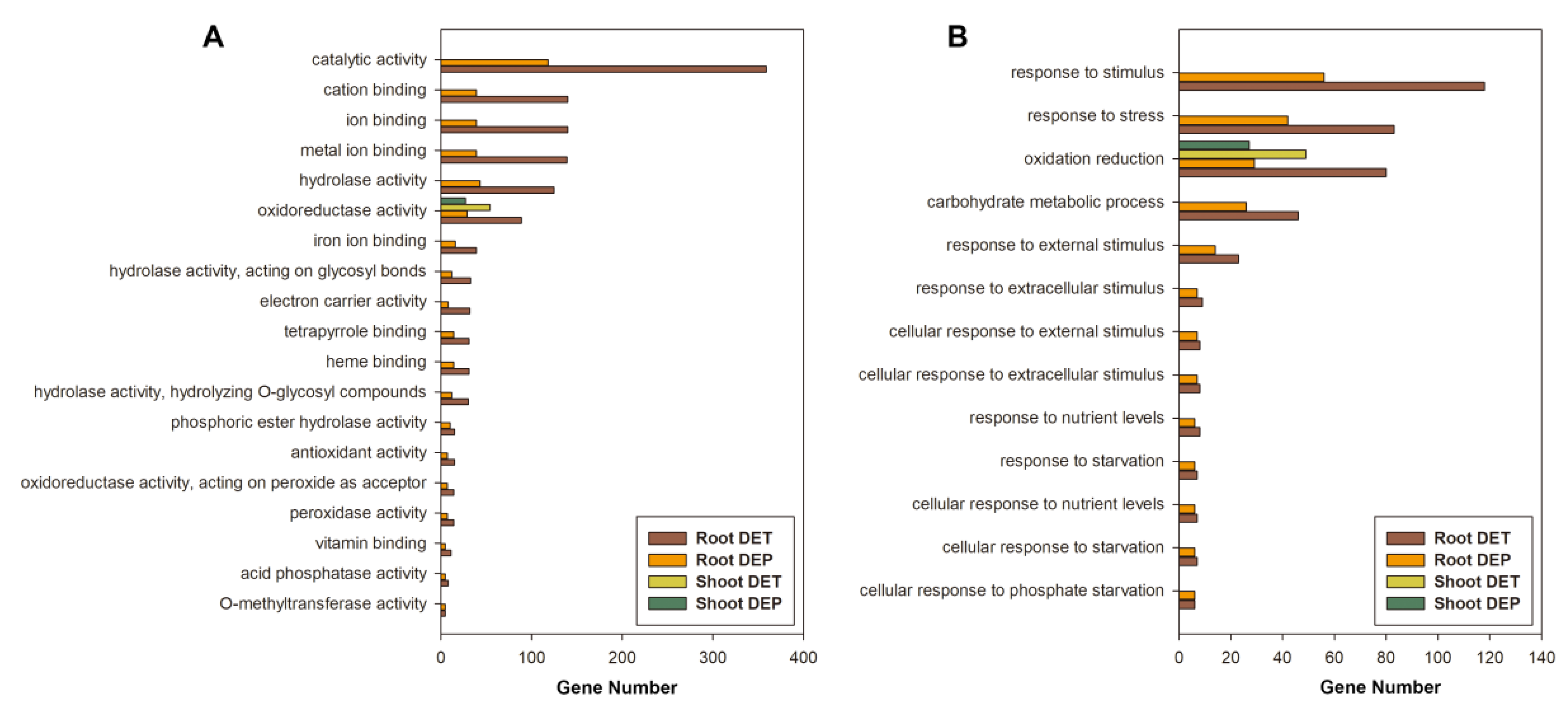
2.5. Enrichment Analysis
2.6. Quantitative Real-Time PCR Validation
2.7. Identification of Pi-Responsive TFs and Analysis of Protein–Protein Interactions
3. Results
3.1. Changes in Phenotypes and Soluble P Concentrations under Pi Starvation
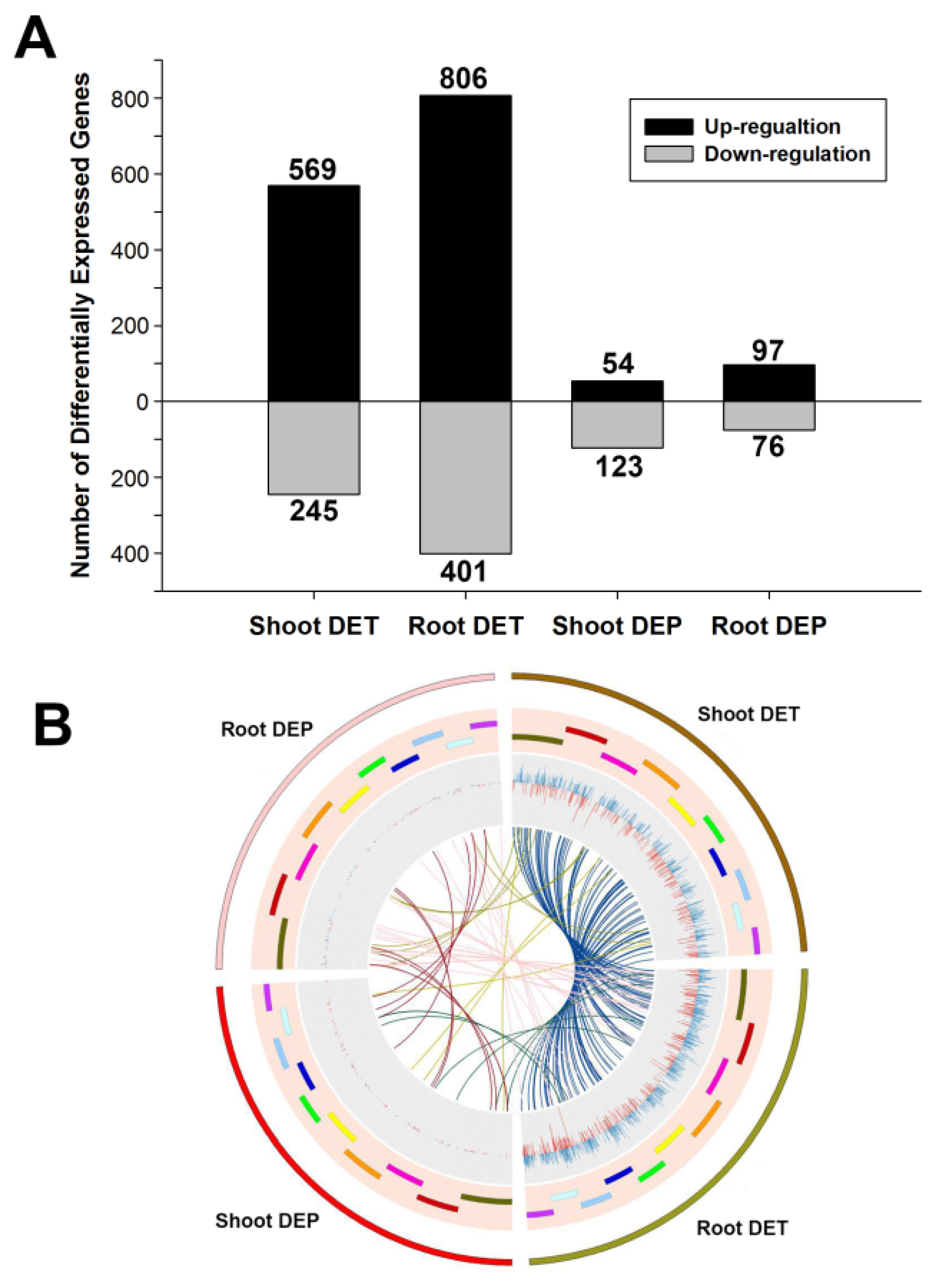
3.2. Differentially Expressed Transcripts under Pi Starvation
3.3. Differentially Expressed Proteins under Pi Starvation
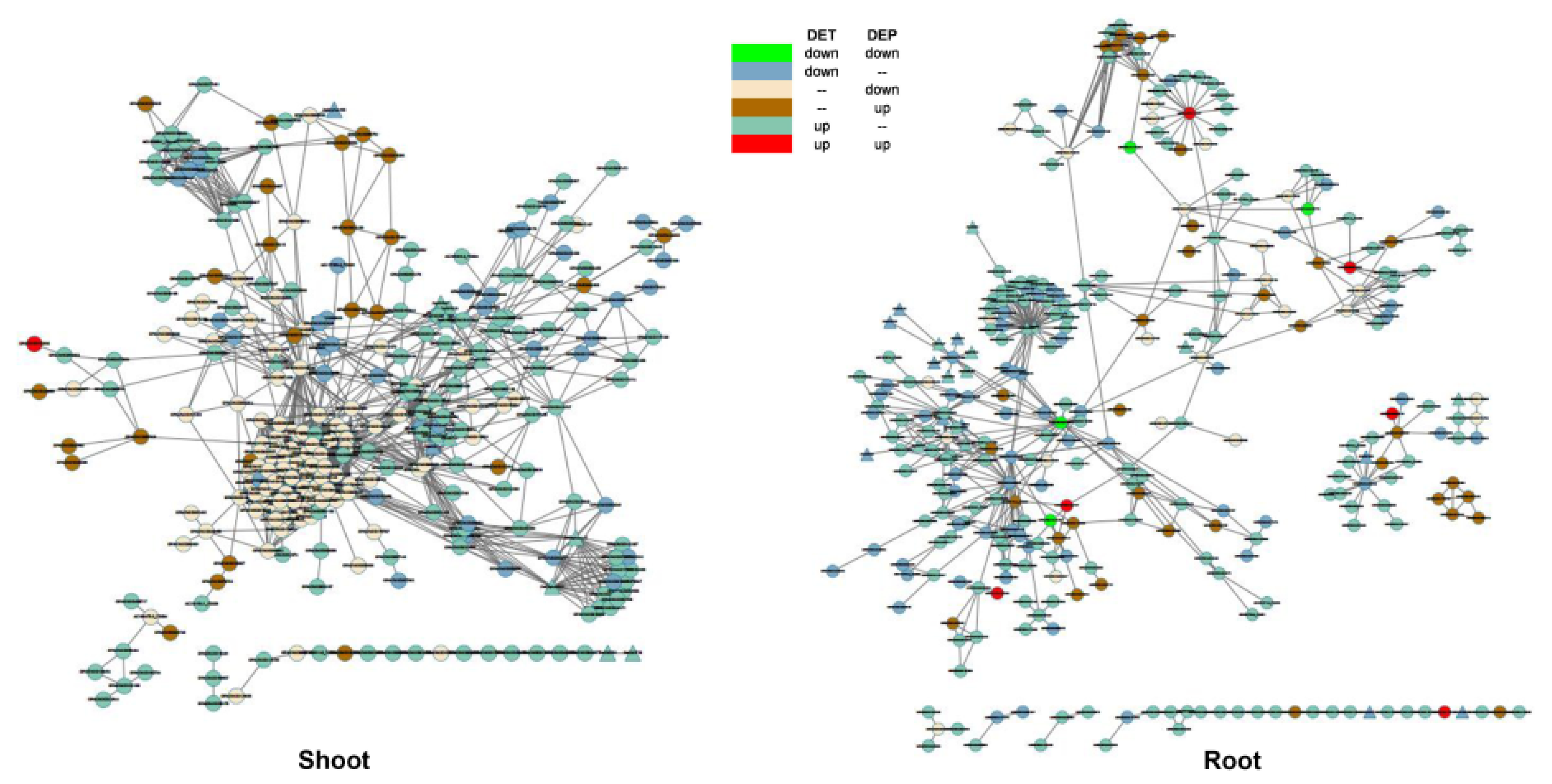
3.4. Relationship between Differentially Expressed Transcripts and Proteins
3.5. Low-Pi Induced Genes
3.6. Landscape of the Interaction between DEGs and Transcription Factors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Puga, M.I.; Rojas-Triana, M.; de Lorenzo, L.; Leyva, A.; Rubio, V.; Paz-Ares, J. Novel signals in the regulation of Pi starvation responses in plants: Facts and promises. Curr. Opin. Plant Biol. 2017, 39, 40–49. [Google Scholar] [CrossRef]
- Vance, C.P.; Uhde-Stone, C.; Allan, D.L. Phosphorus acquisition and use: Critical adaptations by plants for securing a nonrenewable resource. New Phytol. 2003, 157, 423–447. [Google Scholar] [CrossRef] [Green Version]
- López-Arredondo, D.L.; Leyva-González, M.A.; González-Morales, S.I.; López-Bucio, J.; Herrera-Estrella, L. Phosphate Nutrition: Improving Low-Phosphate Tolerance in Crops. Annu. Rev. Plant Biol. 2014, 65, 95–123. [Google Scholar] [CrossRef]
- Cordell, D.; Drangert, J.-O.; White, S. The story of phosphorus: Global food security and food for thought. Glob. Environ. Chang. 2009, 19, 292–305. [Google Scholar] [CrossRef]
- Ai, P.; Sun, S.; Zhao, J.; Fan, X.; Xin, W.; Guo, Q.; Yu, L.; Shen, Q.; Wu, P.; Miller, A.J.; et al. Two rice phosphate transporters, OsPht1;2 and OsPht1;6, have different functions and kinetic properties in uptake and translocation. Plant J. Cell Mol. Biol. 2009, 57, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Shin, H.S.; Dewbre, G.R.; Harrison, M.J. Phosphate transport in Arabidopsis: Pht1;1 and Pht1;4 play a major role in phosphate acquisition from both low- and high-phosphate environments. Plant J. Cell Mol. Biol. 2004, 39, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Jin, Y.; Wussler, C.; Blancaflor, E.B.; Motes, C.M.; Versaw, W.K. Functional analysis of the Arabidopsis PHT4 family of intracellular phosphate transporters. New Phytol. 2008, 177, 889–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Versaw, W.K.; Harrison, M.J. A chlorolast phosphate transporter, PHT2;1, influences allocation of phosphate within the plant and phosphate-starvation responses. Plant Cell 2002, 14, 1751–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cubero, B.; Nakagawa, Y.; Jiang, X.-Y.; Miura, K.-J.; Li, F.; Raghothama, K.G.; Bressan, R.A.; Hasegawa, P.M.; Pardo, J.M. The Phosphate Transporter PHT4;6 Is a Determinant of Salt Tolerance that Is Localized to the Golgi Apparatus of Arabidopsis. Mol. Plant 2009, 2, 535–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.-Y.; Huang, T.-K.; Yang, S.-Y.; Hong, Y.-T.; Huang, S.-M.; Wang, F.-N.; Chiang, S.-F.; Tsai, S.-Y.; Lu, W.-C.; Chiou, T.-J. Identification of plant vacuolar transporters mediating phosphate storage. Nat. Commun. 2016, 7, 11095. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Piñeros, M.A.; Li, X.; Yang, H.; Liu, Y.; Murphy, A.S.; Kochian, L.V.; Liu, D. An Arabidopsis ABC Transporter Mediates Phosphate Deficiency-Induced Remodeling of Root Architecture by Modulating Iron Homeostasis in Roots. Mol. Plant 2017, 10, 244–259. [Google Scholar] [CrossRef] [Green Version]
- Ticconi, C.A.; Lucero, R.D.; Sakhonwasee, S.; Adamson, A.W.; Creff, A.; Nussaume, L.; Desnos, T.; Abel, S. ER-resident proteins PDR2 and LPR1 mediate the developmental response of root meristems to phosphate availability. Proc. Natl. Acad. Sci. USA 2009, 106, 14174–14179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio, V.; Linhares, F.; Solano, R.; Martin, A.C.; Iglesias, J.; Leyva, A.; Paz-Ares, J. A conserved MYB transcription factor involved in phosphate starvation signaling both in vascular plants and in unicellular algae. Genes Dev. 2001, 15, 2122–2133. [Google Scholar] [CrossRef] [Green Version]
- Bari, R.; Pant, B.D.; Stitt, M.; Wolf-Rüdiger, S. PHO2, MicroRNA399, and PHR1 Define a Phosphate-Signaling Pathway in Plants[W][OA]. Plant Physiol. 2006, 141, 988–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Ruan, W.; Shi, J.; Zhang, L.; Xiang, D.; Yang, C.; Li, C.; Wu, Z.; Liu, Y.; Yu, Y.; et al. Rice SPX1 and SPX2 inhibit phosphate starvation responses through interacting with PHR2 in a phosphate-dependent manner. Proc. Natl. Acad. Sci. USA 2014, 111, 14953–14958. [Google Scholar] [CrossRef] [Green Version]
- Puga, M.I.; Mateos, I.; Charukesi, R.; Wang, Z.; Franco-Zorrilla, J.M.; de Lorenzo, L.; Irigoyen, M.L.; Masiero, S.; Bustos, R.; Rodríguez, J.; et al. SPX1 is a phosphate-dependent inhibitor of PHOSPHATE STARVATION RESPONSE 1 in Arabidopsis. Proc. Natl. Acad. Sci. USA 2014, 111, 14947–14952. [Google Scholar] [CrossRef] [Green Version]
- Lv, Q.; Zhong, Y.; Wang, Y.; Wang, Z.; Zhang, L.; Shi, J.; Wu, Z.; Liu, Y.; Mao, C.; Yi, K.; et al. SPX4 Negatively Regulates Phosphate Signaling and Homeostasis through Its Interaction with PHR2 in Rice. Plant Cell 2014, 26, 1586–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, P.; Li, W.; Schmidt, W. Complementary Proteome and Transcriptome Profiling in Phosphate-deficient Arabidopsis Roots Reveals Multiple Levels of Gene Regulation. Mol. Cell. Proteom. 2012, 11, 1156–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, H.; Wang, G.; Zhang, Y.; Hu, X.; Pi, E.; Zhu, Y.; Wang, H.; Du, L. Genome-wide identification of phosphate-deficiency-responsive genes in soybean roots by high-throughput sequencing. Plant Soil 2015, 398, 207–227. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, S.; Yang, K.; Zhang, Z.; Lin, H.; Huang, N.; Zheng, M.; Xu, K.; Chen, Y.; Pan, G. Screening and identification of low-phosphorus-tolerant maize germplasm. J. Plant Genet. Resour. 2008, 9, 335–339. [Google Scholar]
- Nie, Z.; Ren, Z.; Wang, L.; Su, S.; Wei, X.; Zhang, X.; Wu, L.; Liu, D.; Tang, H.; Liu, H.; et al. Genome-wide identification of microRNAs responding to early stages of phosphate deficiency in maize. Physiol. Plant. 2016, 157, 161–174. [Google Scholar] [CrossRef]
- Zhou, J.; Jiao, F.; Wu, Z.; Li, Y.; Wang, X.; He, X.; Zhong, W.; Wu, P. OsPHR2 is involved in phosphate-starvation signaling and excessive phosphate accumulation in shoots of plants. Plant Physiol. 2008, 146, 1673–1686. [Google Scholar] [CrossRef] [Green Version]
- Nanamori, M.; Shinano, T.; Wasaki, J.; Yamamura, T.; Rao, I.M.; Osaki, M. Low Phosphorus Tolerance Mechanisms Phosphorus Recycling and Photosynthate Partitioning in the Tropical Forage Grass, Brachiaria Hybrid Cultivar Mulato Compared with Rice. Plant Cell Physiol. 2004, 45, 460–469. [Google Scholar] [CrossRef] [Green Version]
- Méchin, V.; Damerval, C.; Zivy, M. Total Protein Extraction with TCA-Acetone. In Plant Proteomics: Methods and Protocols; Thiellement, H., Zivy, M., Damerval, C., Méchin, V., Eds.; Humana Press: Totowa, NJ, USA, 2007; pp. 1–8. [Google Scholar]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Xu, Z.; Zhang, Y.; Chai, L.; Yi, H.; Deng, X. An integrative analysis of the transcriptome and proteome of the pulp of a spontaneous late-ripening sweet orange mutant and its wild type improves our understanding of fruit ripening in citrus. J. Exp. Bot. 2014, 65, 1651–1671. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.; Su, Z. agriGO: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, W64–W70. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Paschold, A.; Marcon, C.; Liu, S.; Tai, H.; Nestler, J.; Yeh, C.-T.; Opitz, N.; Lanz, C.; Schnable, P.S.; et al. The Aux/IAA gene rum1 involved in seminal and lateral root formation controls vascular patterning in maize (Zea mays L.) primary roots. J. Exp. Bot. 2014, 65, 4919–4930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Rodríguez, P.; Riaño-Pachón, D.M.; Corrêa, L.G.G.; Rensing, S.A.; Kersten, B.; Mueller-Roeber, B. PlnTFDB: Updated content and new features of the plant transcription factor database. Nucleic Acids Res. 2010, 38, D822–D827. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, A.; Nishiyama, M.Y.; Fuentes, B.G.; Souza, G.M.; Janies, D.; Gray, J.; Grotewold, E. GRASSIUS: A Platform for Comparative Regulatory Genomics across the Grasses. Plant Physiol. 2009, 149, 171–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.; Tian, F.; Yang, D.-C.; Meng, Y.-Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Mollier, A.; Pellerin, S. Maize root system growth and development as influenced by phosphorus deficiency. J. Exp. Bot. 1999, 50, 487–497. [Google Scholar] [CrossRef]
- Williamson, L.C.; Ribrioux, S.P.C.P.; Fitter, A.H.; Leyser, H.M.O. Phosphate Availability Regulates Root System Architecture in Arabidopsis. Plant Physiol. 2001, 126, 875–882. [Google Scholar] [CrossRef] [Green Version]
- Peret, B.; Clement, M.; Nussaume, L.; Desnos, T. Root developmental adaptation to phosphate starvation: Better safe than sorry. Trends Plant Sci. 2011, 16, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Liu, L.; Zhuang, X.; Yu, Y.; Liu, X.; Cui, X.; Ji, L.; Pan, Z.; Cao, X.; Mo, B.; et al. MicroRNAs Inhibit the Translation of Target mRNAs on the Endoplasmic Reticulum in Arabidopsis. Cell 2013, 153, 562–574. [Google Scholar] [CrossRef] [Green Version]
- Di Pietro, M.; Vialaret, J.; Li, G.-W.; Hem, S.; Prado, K.; Rossignol, M.; Maurel, C.; Santoni, V. Coordinated Post-translational Responses of Aquaporins to Abiotic and Nutritional Stimuli in Arabidopsis Roots. Mol. Cell. Proteom. 2013, 12, 3886–3897. [Google Scholar] [CrossRef] [Green Version]
- Hammond, J.P.; Bennett, M.J.; Bowen, H.C.; Broadley, M.R.; Eastwood, D.C.; May, S.T.; Rahn, C.; Swarup, R.; Woolaway, K.E.; White, P.J. Changes in gene expression in Arabidopsis shoots during phosphate starvation and the potential for developing smart plants. Plant Physiol. 2003, 132, 578–596. [Google Scholar] [CrossRef] [Green Version]
- Franco-Zorrilla, J.M.; Martin, A.C.; Leyva, A.; Paz-Ares, J. Interaction between phosphate-starvation, sugar, and cytokinin signaling in Arabidopsis and the roles of cytokinin receptors CRE1/AHK4 and AHK3. Plant Physiol. 2005, 138, 847–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essigmann, B.; Guler, S.; Narang, R.A.; Linke, D.; Benning, C. Phosphate availability affects the thylakoid lipid composition and the expression of SQD1, a gene required for sulfolipid biosynthesis in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 1998, 95, 1950–1955. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Kulkarni, K.; Souret, F.F.; MuthuValliappan, R.; Tej, S.S.; Poethig, R.S.; Henderson, I.R.; Jacobsen, S.E.; Wang, W.; Green, P.J.; et al. MicroRNAs and other small RNAs enriched in the Arabidopsis RNA-dependent RNA polymerase-2 mutant. Genome Res. 2006, 16, 1276–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Yuan, L.; Zhang, J.; Li, H.; Bai, Z.; Chen, X.; Zhang, W.; Zhang, F. Phosphorus Dynamics: From Soil to Plant. Plant Physiol. 2011, 156, 997–1005. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Calderón, L.; López-Bucio, J.; Chacón-López, A.; Cruz-Ramírez, A.; Nieto-Jacobo, F.; Dubrovsky, J.G.; Herrera-Estrella, L. Phosphate Starvation Induces a Determinate Developmental Program in the Roots of Arabidopsis thaliana. Plant Cell Physiol. 2005, 46, 174–184. [Google Scholar] [CrossRef]
- Wei, K.; Pan, S. Maize protein phosphatase gene family: Identification and molecular characterization. BMC Genom. 2014, 15, 773. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Poling, M.D.; Karthikeyan, A.S.; Blakeslee, J.J.; Peer, W.A.; Titapiwatanakun, B.; Murphy, A.S.; Raghothama, K.G. Differential effects of sucrose and auxin on localized phosphate deficiency-induced modulation of different traits of root system architecture in Arabidopsis. Plant Physiol. 2007, 144, 232–247. [Google Scholar] [CrossRef] [Green Version]
- Miura, K.; Lee, J.; Gong, Q.; Ma, S.; Jin, J.B.; Yoo, C.Y.; Miura, T.; Sato, A.; Bohnert, H.J.; Hasegawa, P.M. SIZ1 regulation of phosphate starvation-induced root architecture remodeling involves the control of auxin accumulation. Plant Physiol. 2011, 155, 1000–1012. [Google Scholar] [CrossRef] [Green Version]
- Casimiro, I.; Marchant, A.; Bhalerao, R.P.; Beeckman, T.; Dhooge, S.; Swarup, R.; Graham, N.; Inzé, D.; Sandberg, G.; Casero, P.J.; et al. Auxin Transport Promotes Arabidopsis Lateral Root Initiation. Plant Cell 2001, 13, 843–852. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Pandya-Kumar, N.; Dam, A.; Haor, H.; Mayzlish-Gati, E.; Belausov, E.; Wininger, S.; Abu-Abied, M.; McErlean, C.S.P.; Bromhead, L.J.; et al. Arabidopsis response to low-phosphate conditions includes active changes in actin filaments and PIN2 polarization and is dependent on strigolactone signalling. J. Exp. Bot. 2015, 66, 1499–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barakat, A.; Szick-Miranda, K.; Ing-Feng, C.; Guyot, R.; Delseny, M.; Bailey-Serres, J. The organization of cytoplasmic ribosomal protein genes in the Arabidopsis genome. Plant Physiol. 2001, 127, 398–415. [Google Scholar] [CrossRef] [PubMed]
- Ishige, T.; Krause, M.; Bott, M.; Wendisch, V.F.; Sahm, H. The Phosphate Starvation Stimulon of Corynebacterium glutamicum Determined by DNA Microarray Analyses. J. Bacteriol. 2003, 185, 4519–4529. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene_ID | RNA-seq | iTRAQ | |||
|---|---|---|---|---|---|
| log2_FC (TL/CL) | log2_FC (TR/CR) | log2_FC (TL/CL) | log2_FC (TR/CR) | Description | |
| GRMZM2G009800 | -- | 11.80 | -- | -- | Inorganic phosphate transporter |
| GRMZM2G154090 | 3.62 | -- | -- | 1.88 | Pht1;1 |
| GRMZM2G326707 | -- | -- | -- | 0.93 | Pht1;2 |
| GRMZM2G041595 | -- | 12.38 | -- | -- | Pht1;5 |
| GRMZM5G881088 | -- | 9.79 | -- | -- | Pht1;6 |
| AC202435.3_FG003 | -- | 7.38 | -- | 2.41 | acid phosphatase |
| GRMZM2G014193 | -- | 2.94 | -- | -- | Chloroplast purple acid phosphatase |
| GRMZM2G093101 | 5.25 | 5.20 | -- | 1.56 | acid phosphatase |
| GRMZM2G109071 | 5.97 | -- | -- | -- | acid phosphatase |
| GRMZM2G134054 | -- | 5.54 | -- | -- | acid phosphatase |
| GRMZM2G141584 | -- | 4.79 | -- | -- | acid phosphatase |
| GRMZM2G152447 | -- | 8.50 | -- | 3.49 | acid phosphatase |
| GRMZM2G152477 | 3.10 | -- | -- | 1.13 | Purple acid phosphatase 1 |
| GRMZM2G315848 | -- | 4.67 | 0.68 | 1.46 | Nucleotide pyrophosphatase/phosphodiesterase |
| GRMZM2G015908 | -- | 8.80 | -- | -- | acid phosphatase |
| GRMZM2G100652 | 1.82 | 1.80 | -- | -- | UDP-sulfoquinovose synthase (SQD2) |
| GRMZM2G053322 | -- | 3.54 | 1.05 | 1.38 | UDP-sulfoquinovose synthase |
| GRMZM2G175995 | -- | 2.93 | -- | -- | Auxin regulated gene involved in organ size 5 |
| GRMZM2G354338 | -- | 6.08 | -- | -- | Auxin regulated gene involved in organ size 8 |
| GRMZM2G082943 | -- | 3.52 | -- | -- | Auxin regulated gene involved in organ size 9 |
| GRMZM2G429940 | 2.28 | -- | -- | -- | CBL-interacting serine/threonine-protein kinase 15 |
| GRMZM2G059544 | 8.87 | -- | -- | -- | IAA25-auxin-responsive Aux/IAA family member |
| GRMZM2G126260 | -- | 1.23 | -- | -- | Putative auxin efflux carrier PIN10a |
| GRMZM2G025742 | -- | 7.83 | -- | -- | Putative auxin efflux carrier PIN5a |
| GRMZM2G040911 | 3.90 | -- | -- | -- | Putative auxin efflux carrier PIN5c |
| GRMZM2G074427 | 1.86 | -- | -- | -- | ZmIAA23 |
| GRMZM2G045057 | 6.75 | -- | -- | -- | ZmLAX4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nie, Z.; Luo, B.; Zhang, X.; Wu, L.; Liu, D.; Guo, J.; He, X.; Gao, D.; Gao, S.; Gao, S. Combined Transcriptome and Proteome Analysis of Maize (Zea mays L.) Reveals A Complementary Profile in Response to Phosphate Deficiency. Curr. Issues Mol. Biol. 2021, 43, 1142-1155. https://doi.org/10.3390/cimb43020081
Nie Z, Luo B, Zhang X, Wu L, Liu D, Guo J, He X, Gao D, Gao S, Gao S. Combined Transcriptome and Proteome Analysis of Maize (Zea mays L.) Reveals A Complementary Profile in Response to Phosphate Deficiency. Current Issues in Molecular Biology. 2021; 43(2):1142-1155. https://doi.org/10.3390/cimb43020081
Chicago/Turabian StyleNie, Zhi, Bowen Luo, Xiao Zhang, Ling Wu, Dan Liu, Jialei Guo, Xuan He, Duojiang Gao, Shiqiang Gao, and Shibin Gao. 2021. "Combined Transcriptome and Proteome Analysis of Maize (Zea mays L.) Reveals A Complementary Profile in Response to Phosphate Deficiency" Current Issues in Molecular Biology 43, no. 2: 1142-1155. https://doi.org/10.3390/cimb43020081
APA StyleNie, Z., Luo, B., Zhang, X., Wu, L., Liu, D., Guo, J., He, X., Gao, D., Gao, S., & Gao, S. (2021). Combined Transcriptome and Proteome Analysis of Maize (Zea mays L.) Reveals A Complementary Profile in Response to Phosphate Deficiency. Current Issues in Molecular Biology, 43(2), 1142-1155. https://doi.org/10.3390/cimb43020081