
Ketogenic Diet in Cancer Prevention and Therapy: Molecular Targets and Therapeutic Opportunities

 ,
,  , ,
, , 
Abstract
1. Introduction
2. The History of Ketogenic Diet
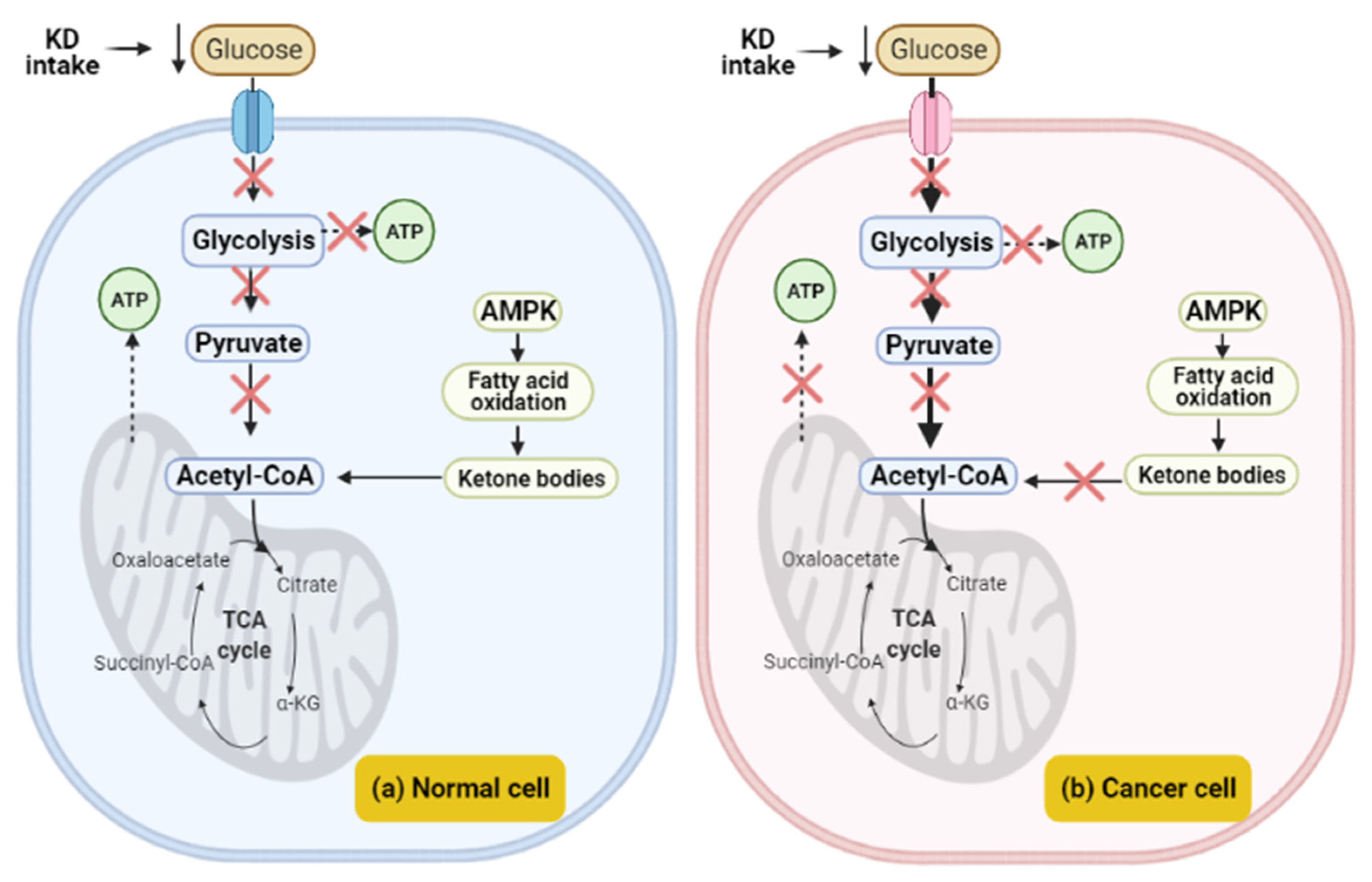
3. Cancer Metabolism and Warburg Effect
3.1. Glutamines
3.2. Serine and One Carbon
3.3. Leucine
3.4. Warburg Effect
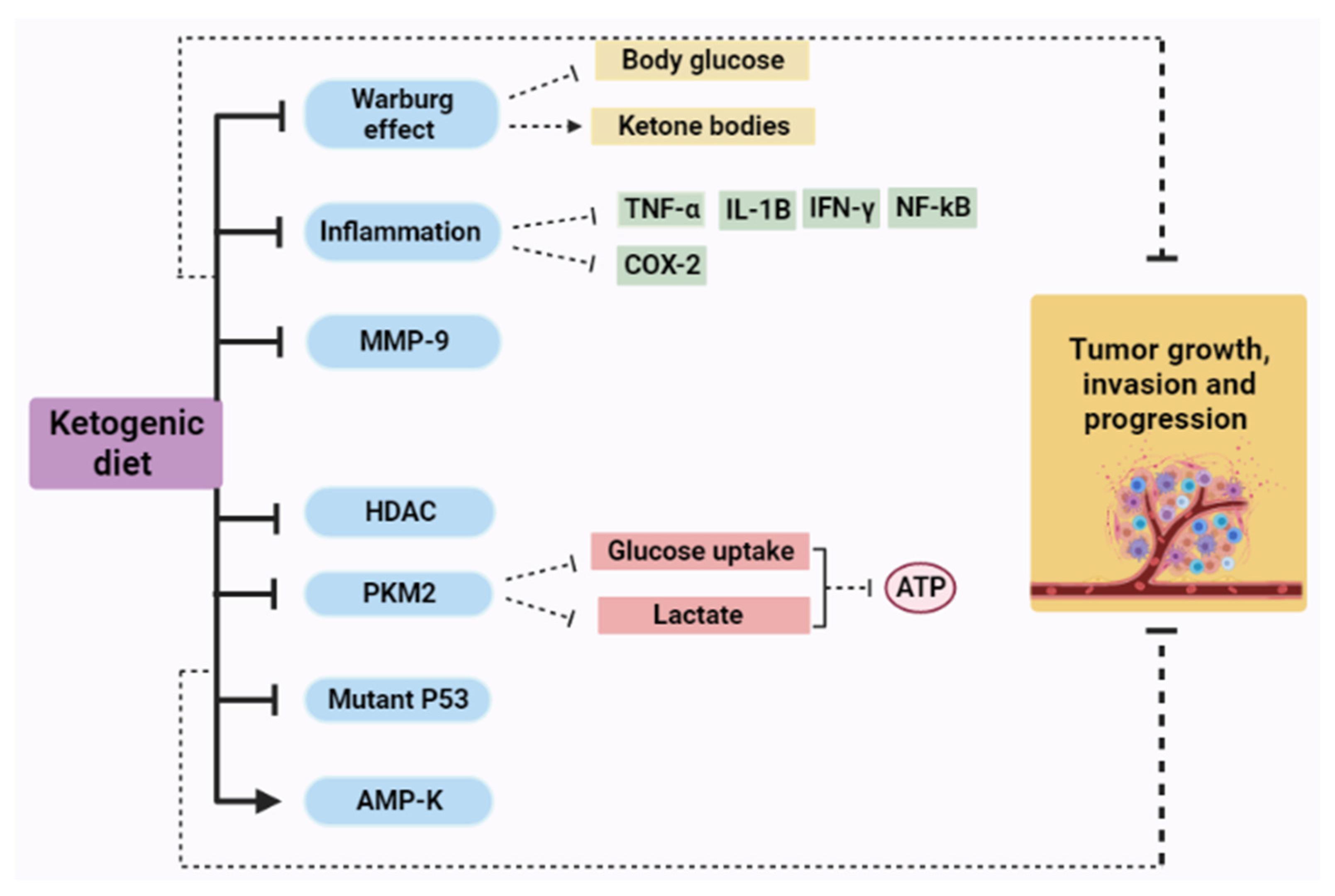
4. Ketogenic Diet as Cancer Therapy: Mechanisms of Action
4.1. The Effect of a Ketogenic Diet in Warburg Effect
4.2. Ketogenic Diet and Inflammation
4.2.1. Tumor Necrosis Factor Alpha (TNF-α)
4.2.2. Cyclooxygenase (COX)
4.3. Ketogenic Diet and Matrix Metalloproteinases (MMPs)
4.4. Ketogenic Diet and Histone Deacetylases (HDAC)
4.5. Ketogenic Diet and Pyruvate Kinase (PK)
4.6. Ketogenic Diet and P53
4.7. Ketogenic Diet and AMP-Activated Protein Kinase (AMPK)
5. Ketogenic Diet as a Prevention of Cancer
6. Ketogenic Diet as an Epigenetic Modifier in Cancer
7. Ketogenic Diet in Experimental and Clinical Anticancer Studies
7.1. Preclinical Studies
Ketogenic Diet as an Experimental Glioma Therapy
7.2. Clinical Studies
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- National Nutrition Council Institute. Cancer Facts & Figures 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar]
- Miller, K.D.; Fidler-Benaoudia, M.; Keegan, T.H.; Hipp, H.S.; Jemal, A.; Siegel, R.L. Cancer statistics for adolescents and young adults, 2020. CA Cancer J. Clin. 2020, 70, 443–459. [Google Scholar] [CrossRef]
- Damyanov, C.; Maslev, I.; Pavlov, V.; Avramov, L. Conventional treatment of cancer realities and problems. Ann. Complement. Altern. Med. 2018, 1, 1–9. [Google Scholar]
- Warburg, O. Uber den stoffwechsel der carcinomzelle. Biochem. Z. 1924, 152, 309–344. [Google Scholar] [CrossRef]
- Warburg, O.; Minami, S. Versuche an überlebendem carcinom-gewebe. J. Mol. Med. 1923, 2, 776–777. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- House, S.W.; Warburg, O.; Burk, D.; Schade, A.L. On respiratory impairment in cancer cells. Science 1956, 124, 267–272. [Google Scholar]
- Seyfried, T.N.; Flores, R.E.; Poff, A.M.; D’Agostino, D.P. Cancer as a metabolic disease: Implications for novel therapeutics. Carcinogenesis 2014, 35, 515–527. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Mukherjee, P.; Iyikesici, M.S.; Slocum, A.; Kalamian, M.; Spinosa, J.-P.; Chinopoulos, C. Consideration of ketogenic metabolic therapy as a complementary or alternative approach for managing breast cancer. Front. Nutr. 2020, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Shelton, L.M. Cancer as a metabolic disease. Nutr. Metab. 2010, 7, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Gillies, R.J.; Robey, I.; Gatenby, R.A. Causes and consequences of increased glucose metabolism of cancers. J. Nucl. Med. 2008, 49, 24S–42S. [Google Scholar] [CrossRef]
- Sattler, U.G.A.; Mueller-Klieser, W. The anti-oxidant capacity of tumour glycolysis. Int. J. Radiat. Biol. 2009, 85, 963–971. [Google Scholar] [CrossRef]
- Alidadi, M.; Banach, M.; Guest, P.C.; Bo, S.; Jamialahmadi, T.; Sahebkar, A. The Effect of Caloric Restriction and Fasting on Cancer. Semin. Cancer Biol. 2021, 73, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Antunes, F.; Erustes, A.G.; Costa, A.J.; Nascimento, A.C.; Bincoletto, C.; Ureshino, R.P.; Pereira, G.J.S.; Smaili, S.S. Autophagy and intermittent fasting: The connection for cancer therapy? Clinics 2018, 73. [Google Scholar] [CrossRef]
- Di Tano, M.; Longo, V.D. A fasting-mimicking diet and vitamin C: Turning anti-aging strategies against cancer. Mol. Cell. Oncol. 2020, 7, 1791671. [Google Scholar] [CrossRef]
- Ibrahim, E.M.; Al-Foheidi, M.H.; Al-Mansour, M.M. Energy and caloric restriction, and fasting and cancer: A narrative review. Support. Care Cancer 2021, 29, 2299–2304. [Google Scholar] [CrossRef] [PubMed]
- Klement, R.J. Fasting, fats, and physics: Combining ketogenic and radiation therapy against cancer. Complement. Med. Res. 2018, 25, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Nencioni, A.; Caffa, I.; Cortellino, S.; Longo, V.D. Fasting and cancer: Molecular mechanisms and clinical application. Nat. Rev. Cancer 2018, 18, 707–719. [Google Scholar] [CrossRef]
- Plotti, F.; Terranova, C.; Luvero, D.; Bartolone, M.; Messina, G.; Feole, L.; Cianci, S.; Scaletta, G.; Marchetti, C.; Di Donato, V. Diet and Chemotherapy: The Effects of Fasting and Ketogenic Diet on Cancer Treatment. Chemotherapy 2020, 65, 77–84. [Google Scholar] [CrossRef]
- Allen, B.G.; Bhatia, S.K.; Anderson, C.M.; Eichenberger-Gilmore, J.M.; Sibenaller, Z.A.; Mapuskar, K.A.; Schoenfeld, J.D.; Buatti, J.M.; Spitz, D.R.; Fath, M.A. Ketogenic diets as an adjuvant cancer therapy: History and potential mechanism. Redox Biol. 2014, 2, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.D.; Aminzadeh-Gohari, S.; Tulipan, J.; Catalano, L.; Feichtinger, R.G.; Kofler, B. Ketogenic diet in the treatment of cancer—Where do we stand? Mol. Metab. 2020, 33, 102–121. [Google Scholar] [CrossRef]
- Abdelwahab, M.G.; Fenton, K.E.; Preul, M.C.; Rho, J.M.; Lynch, A.; Stafford, P.; Scheck, A.C. The ketogenic diet is an effective adjuvant to radiation therapy for the treatment of malignant glioma. PLoS ONE 2012, 7, e36197. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Zhu, X.; Wang, H.; Wang, F.; Guan, W. Roles of caloric restriction, ketogenic diet and intermittent fasting during initiation, progression and metastasis of cancer in animal models: A systematic review and meta-analysis. PLoS ONE 2014, 9, e115147. [Google Scholar] [CrossRef]
- Stafford, P.; Abdelwahab, M.G.; Kim, D.Y.; Preul, M.C.; Rho, J.M.; Scheck, A.C. The ketogenic diet reverses gene expression patterns and reduces reactive oxygen species levels when used as an adjuvant therapy for glioma. Nutr. Metab. 2010, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wheatley, K.E.; Williams, E.A.; Smith, N.C.; Dillard, A.; Park, E.Y.; Nunez, N.P.; Hursting, S.D.; Lane, M.A. Low-carbohydrate diet versus caloric restriction: Effects on weight loss, hormones, and colon tumor growth in obese mice. Nutr. Cancer 2007, 60, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Nebeling, L.C.; Miraldi, F.; Shurin, S.B.; Lerner, E. Effects of a ketogenic diet on tumor metabolism and nutritional status in pediatric oncology patients: Two case reports. J. Am. Coll. Nutr. 1995, 14, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, B.D.; Pauli, C.; Du, X.; Wang, D.G.; Li, X.; Wu, D.; Amadiume, S.C.; Goncalves, M.D.; Hodakoski, C.; Lundquist, M.R. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018, 560, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Temkin, O. The Falling Sickness: A History of Epilepsy from the Greeks to the Beginnings of Modern Neurology; JHU Press: Baltimore, MD, USA, 1994; Volume 4. [Google Scholar]
- Guelpa, G. La lutte contre l′epiepsie par la desintoxication et par la reeducation alimentaire. Rev. Ther. Med. Chir. 1911, 78, 8. [Google Scholar]
- Freeman, J.M.; Kelly, M.T.; Freeman, J.B. The Epilepsy Diet Treatment: An Introduction to the Ketogenic Diet; Demos Vermande: New York, NY, USA, 1996. [Google Scholar]
- Hendricks, M. High fat and seizure free. Johns Hopkins Magazine, April 1995; 14–20. [Google Scholar]
- Wilkinson, J.F. Look at Me: No stunt was too much for millionaire fitness guru Bernarr Macfadden-until he tried to muscle his way into politics. Smithsonian 1997, 28, 136–151. [Google Scholar]
- Lennox, W.G.; Cobb, S. Studies in epilepsy: VIII. The clinical effect of fasting. Arch. Neurol. Psychiatry 1928, 20, 771–779. [Google Scholar] [CrossRef]
- Geyelin, H.R. Fasting as a method for treating epilepsy. Med. Rec. 1921, 99, 1037–1039. [Google Scholar]
- Geyelin, H.R. The relation of chemical influences, including diet and endocrine disturbances, to epilepsy. Ann. Intern. Med. 1929, 2, 678–681. [Google Scholar]
- Pulsifer, M.B.; Gordon, J.M.; Brandt, J.; Vining, E.P.; Freeman, J.M. Effects of ketogenic diet on development and behavior: Preliminary report of a prospective study. Dev. Med. Child. Neurol. 2001, 43, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Wheless, J.W. History and origin of the ketogenic diet. In Epilepsy and the Ketogenic Diet; Humana Press: Totowa, NJ, USA, 2004; pp. 31–50. [Google Scholar]
- Penfield, W.; Erickson, T.C. Epilepsy and Cerebral Localization; Charles C Thomas Publisher: Springfield, IL, USA, 1941. [Google Scholar]
- Swink, T.D.; Vining, E.P.; Freeman, J.M. The ketogenic diet: 1997. Adv. Pediatr. 1997, 44, 297–329. [Google Scholar] [PubMed]
- Welch, H.W.; Goodnow, F.J.; Flexner, S. Memorial meeting for Dr. John Howland. Bull. Johns Hopkins Hosp. 1927, 41, 311–321. [Google Scholar]
- Wilkins, L. Epilepsy in childhood. 3. Results with the ketogenic diet. J. Pediatr. 1937, 10, 341–357. [Google Scholar] [CrossRef]
- Gamble, J.L.; Ross, G.S.; Tisdall, F.F. The metabolism of fixed base during fasting. J. Biol. Chem. 1923, 57, 633–695. [Google Scholar] [CrossRef]
- McQuarrie, I. Epilepsy in children: The relationship of water balance to the occurrence of seizures. Am. J. Dis. Child. 1929, 38, 451–467. [Google Scholar] [CrossRef]
- Helmholz, H.F. The treatment of epilepsy in childhood: Five years’experience with the ketogenic diet. J. Am. Med. Assoc. 1927, 88, 2028–2032. [Google Scholar] [CrossRef]
- Talbot, F.B.; Metcalf, K.M.; Moriarty, M.E. Epilepsy: Chemical investigations of rational treatment by production of ketosis. Am. J. Dis. Child. 1927, 33, 218–225. [Google Scholar] [CrossRef]
- Wilder, R.M. The effects of ketonemia on the course of epilepsy. Mayo Clin. Proc. 1921, 2, 307–308. [Google Scholar]
- Bridge, E.M.; Iob, L.V. The mechanism of the ketogenic diet in epilepsy. Bull. Johns Hopkins Hosp. 1931, 48, 373–389. [Google Scholar] [CrossRef]
- Lennox, W.G.; Cobb, S. Epilepsy: From the standpoint of physiology and treatment. Medicine 1928, 7, 105–290. [Google Scholar] [CrossRef]
- Woodyatt, R. Objects and method of diet adjustment in diabetes. Arch. Intern. Med. 1921, 28, 125–141. [Google Scholar] [CrossRef]
- Peterman, M. The ketogenic diet in the treatment of epilepsy: A preliminary report. Am. J. Dis. Child. 1924, 28, 28–33. [Google Scholar] [CrossRef]
- Peterman, M.G. The ketogenic diet in epilepsy. J. Am. Med. Assoc. 1925, 84, 1979–1983. [Google Scholar] [CrossRef]
- Talbot, F.B. The treatment of epilepsy of childhood by the ketogenic diet. RI Med. J. 1927, 10, 159–162. [Google Scholar]
- Talbot, F.B.; Metcalf, K.; Moriarty, M. The ketogenic diet in the treatment of idiopathic epilepsy. Am. J. Dis. Child. 1926, 32, 316–318. [Google Scholar]
- Talbot, F.B.; Metcalf, K.M.; Moriarty, M.E. A clinical study of epileptic children treated by ketogenic diet. Boston Med. Surg. J. 1927, 196, 89–96. [Google Scholar] [CrossRef]
- McQuarrie, I.; Keith, H.M. Epilepsy in children: Relationship of variations in the degree of ketonuria to occurrence of convulsions in epileptic children on ketogenic diets. Am. J. Dis. Child. 1927, 34, 1013–1029. [Google Scholar] [CrossRef]
- Ford, F. The Epilepsies and Paroxysmal Disorders of the Nervous System; Charles C Thomas Publisher: Springfield, IL, USA, 1937; p. 888. [Google Scholar]
- Aicardi, J. Epilepsy in children. Int. Rev. Child Neurol. Ser. 1994, 138–146. [Google Scholar]
- Dateline, N.B.C. The ketogenic diet. J. Child Nurol. 1995, 10, 419–423. [Google Scholar]
- Wheless, J.W. The Ketogenic Diet: Fa(c)t or Fiction; Sage Publications Sage CA: Thousand Oaks, CA, USA, 1995. [Google Scholar]
- Freeman, J.M. Seizures and Epilepsy in Childhood: A Guide for Parents; Johns Hopkins University Press: Baltimore, MD, USA, 1997. [Google Scholar]
- Barborka, C.J. Ketogenic diet treatment of epilepsy in adults. J. Am. Med. Assoc. 1928, 91, 73–78. [Google Scholar] [CrossRef]
- Barborka, C.J. The ketogenic diet and its use. J. Am. Diet. Assoc. 1933, 8, 471–481. [Google Scholar]
- Fischer, L. Epilepsy: Its treatment by the use of the ketogenic diet versus drugs. Arch. Pediatr. 1935, 52, 131–136. [Google Scholar]
- Helmholz, H.F.; Keith, H.M. Eight years’experience with the ketogenic diet in the treatment of epilepsy. J. Am. Med. Assoc. 1930, 95, 707–709. [Google Scholar] [CrossRef]
- Helmholz, H.F.; Keith, H.M. Ten years’experience in the treatment of epilepsy with ketogenic diet. Arch. Neurol. Psychiatry 1933, 29, 808–812. [Google Scholar] [CrossRef]
- Keeton, R.W.; Mackenzie, H. The Principles Under-Lying the Calculation of Flexible Diabetic and Ketogenic Diets. Ann. Intern. Med. 1929, 3, 546–556. [Google Scholar]
- Pulford, D.S. The present status of the ketogenic diet. Ann. Intern. Med. 1932, 6, 795–801. [Google Scholar] [CrossRef]
- Wilder, R.M.; Pollack, H. Ketosis and the ketogenic diet: Their application to treatment of epilepsy and infections of the urinary tract. Int. Clin. 1935, 1, 1. [Google Scholar]
- Sirven, J.; Whedon, B.; Caplan, D.; Liporace, J.; Glosser, D.; O′Dwyer, J.; Sperling, M.R. The ketogenic diet for intractable epilepsy in adults: Preliminary results. Epilepsia 1999, 40, 1721–1726. [Google Scholar] [CrossRef]
- Kinsman, S.L.; Vining, E.P.G.; Quaskey, S.A.; Mellits, D.; Freeman, J.M. Efficacy of the ketogenic diet for intractable seizure disorders: Review of 58 cases. Epilepsia 1992, 33, 1132–1136. [Google Scholar] [CrossRef]
- Barañano, K.W.; Hartman, A.L. The ketogenic diet: Uses in epilepsy and other neurologic illnesses. Curr. Treat. Options Neurol. 2008, 10, 410–419. [Google Scholar] [CrossRef]
- Vining, E.P.G.; Freeman, J.M.; Ballaban-Gil, K.; Camfield, C.S.; Camfield, P.R.; Holmes, G.L.; Shinnar, S.; Shuman, R.; Trevathan, E.; Wheless, J.W. A multicenter study of the efficacy of the ketogenic diet. Arch. Neurol. 1998, 55, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Courtnay, R.; Ngo, D.C.; Malik, N.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Cancer metabolism and the Warburg effect: The role of HIF-1 and PI3K. Mol. Biol. Rep. 2015, 42, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, M.J.; Brennan, R.A.; Fearon, K.C. Reduction of weight loss and tumour size in a cachexia model by a high fat diet. Br. J. Cancer 1987, 56, 39–43. [Google Scholar] [CrossRef]
- Maurer, G.D.; Brucker, D.P.; Bähr, O.; Harter, P.N.; Hattingen, E.; Walenta, S.; Mueller-Klieser, W.; Steinbach, J.P.; Rieger, J. Differential utilization of ketone bodies by neurons and glioma cell lines: A rationale for ketogenic diet as experimental glioma therapy. BMC Cancer 2011, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Sanderson, T.M.; El-Abbadi, M.M.; McGowan, R.; Mukherjee, P. Role of glucose and ketone bodies in the metabolic control of experimental brain cancer. Br. J. Cancer 2003, 89, 1375–1382. [Google Scholar] [CrossRef]
- Beck, S.A.; Tisdale, M.J. Nitrogen excretion in cancer cachexia and its modification by a high fat diet in mice. Cancer Res. 1989, 49, 3800–3804. [Google Scholar]
- Otto, C.; Kaemmerer, U.; Illert, B.; Muehling, B.; Pfetzer, N.; Wittig, R.; Voelker, H.U.; Thiede, A.; Coy, J.F. Growth of human gastric cancer cells in nude mice is delayed by a ketogenic diet supplemented with omega-3 fatty acids and medium-chain triglycerides. BMC Cancer 2008, 8, 122. [Google Scholar] [CrossRef]
- Freedland, S.J.; Mavropoulos, J.; Wang, A.; Darshan, M.; Demark-Wahnefried, W.; Aronson, W.J.; Cohen, P.; Hwang, D.; Peterson, B.; Fields, T.; et al. Carbohydrate restriction, prostate cancer growth, and the insulin-like growth factor axis. Prostate 2008, 68, 11–19. [Google Scholar] [CrossRef]
- Masko, E.M.; Thomas, J.A.; Antonelli, J.A.; Lloyd, J.C.; Phillips, T.E.; Poulton, S.H.; Dewhirst, M.W.; Pizzo, S.V.; Freedland, S.J. Low-Carbohydrate Diets and Prostate Cancer: How Low Is “Low Enough”? Cancer Prev. Res. 2010, 3, 1124. [Google Scholar] [CrossRef]
- Mavropoulos, J.C.; Buschemeyer, W.C.; Tewari, A.K.; Rokhfeld, D.; Pollak, M.; Zhao, Y.; Febbo, P.G.; Cohen, P.; Hwang, D.; Devi, G.; et al. The Effects of Varying Dietary Carbohydrate and Fat Content on Survival in a Murine LNCaP Prostate Cancer Xenograft Model. Cancer Prev. Res. 2009, 2, 557. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.A.; Sanderson, S.M.; Locasale, J.W. 9—Cancer Metabolism. In Abeloff’s Clinical Oncology, 6th ed.; Niederhuber, J.E., Armitage, J.O., Kastan, M.B., Doroshow, J.H., Tepper, J.E., Eds.; Elsevier: Philadelphia, PA, USA, 2020; pp. 127–138.e124. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Van Dang, C. Cancer Metabolism: The Known, Unknowns. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 1. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C. Metabolism and Cancer: The Future Is Now; Nature Publishing Group: Berlin, Germany, 2020. [Google Scholar]
- Ghaffari, P.; Mardinoglu, A.; Nielsen, J. Cancer metabolism: A modeling perspective. Front. Physiol. 2015, 6, 382. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Maeda, T.; Suzuki, A.; Baba, Y. Cancer metabolism: New insights into classic characteristics. Jpn. Dent. Sci. Rev. 2018, 54, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Pinedo, C.; El Mjiyad, N.; Ricci, J.E. Cancer metabolism: Current perspectives and future directions. Cell Death Dis. 2012, 3, e248. [Google Scholar] [CrossRef]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine metabolism in cancer: Understanding the heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Le, A. Glutamine metabolism in cancer. Heterog. Cancer Metab. 2018, 13–32. [Google Scholar]
- Oizel, K.; Chauvin, C.; Oliver, L.; Gratas, C.; Geraldo, F.; Jarry, U.; Scotet, E.; Rabe, M.; Alves-Guerra, M.-C.; Teusan, R. Efficient mitochondrial glutamine targeting prevails over glioblastoma metabolic plasticity. Clin. Cancer Res. 2017, 23, 6292–6304. [Google Scholar] [CrossRef]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef]
- Reina-Campos, M.; Diaz-Meco, M.T.; Moscat, J. The complexity of the serine glycine one-carbon pathway in cancer. J. Cell Biol. 2019, 219. [Google Scholar] [CrossRef]
- Gorissen, S.H.M.; Crombag, J.J.R.; Senden, J.M.G.; Waterval, W.A.H.; Bierau, J.; Verdijk, L.B.; van Loon, L.J.C. Protein content and amino acid composition of commercially available plant-based protein isolates. Am. Acids 2018, 50, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Vianna, D.; Teodoro, G.F.R.; Torres-Leal, F.L.; Tirapegui, J. Protein synthesis regulation by leucine. Braz. J. Pharm. Sci. 2010, 46, 29–36. [Google Scholar] [CrossRef]
- Katsanos, C.S.; Kobayashi, H.; Sheffield-Moore, M.; Aarsland, A.; Wolfe, R.R. A high proportion of leucine is required for optimal stimulation of the rate of muscle protein synthesis by essential amino acids in the elderly. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E381–E387. [Google Scholar] [CrossRef] [PubMed]
- Masino, S.A.; Rho, J.M. Mechanisms of ketogenic diet action. In Jasper’s Basic Mechanisms of the Epilepsies [Internet], 4th ed.; National Center for Biotechnology Information: Bethesda, MD, USA, 2012. [Google Scholar]
- Li, F.; Yin, Y.; Tan, B.; Kong, X.; Wu, G. Leucine nutrition in animals and humans: mTOR signaling and beyond. Am. Acids 2011, 41, 1185. [Google Scholar] [CrossRef] [PubMed]
- Viana, L.R.; Tobar, N.; Busanello, E.N.B.; Marques, A.C.; de Oliveira, A.G.; Lima, T.I.; Machado, G.; Castelucci, B.G.; Ramos, C.D.; Brunetto, S.Q.; et al. Leucine-rich diet induces a shift in tumour metabolism from glycolytic towards oxidative phosphorylation, reducing glucose consumption and metastasis in Walker-256 tumour-bearing rats. Sci. Rep. 2019, 9, 15529. [Google Scholar] [CrossRef]
- Keenan, M.; Chi, J.-T. Alternative fuels for cancer cells. Cancer J. 2015, 21, 49. [Google Scholar] [CrossRef]
- Mayers, J.R.; Wu, C.; Clish, C.B.; Kraft, P.; Torrence, M.E.; Fiske, B.P.; Yuan, C.; Bao, Y.; Townsend, M.K.; Tworoger, S.S. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat. Med. 2014, 20, 1193–1198. [Google Scholar] [CrossRef]
- Sheen, J.-H.; Zoncu, R.; Kim, D.; Sabatini, D.M. Defective regulation of autophagy upon leucine deprivation reveals a targetable liability of human melanoma cells in vitro and in vivo. Cancer Cell 2011, 19, 613–628. [Google Scholar] [CrossRef]
- Son, S.M.; Park, S.J.; Lee, H.; Siddiqi, F.; Lee, J.E.; Menzies, F.M.; Rubinsztein, D.C. Leucine signals to mTORC1 via its metabolite acetyl-coenzyme A. Cell Metab. 2019, 29, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Xiao, F.; Wang, C.; Yin, H.; Yu, J.; Chen, S.; Fang, J.; Guo, F. Leucine deprivation inhibits proliferation and induces apoptosis of human breast cancer cells via fatty acid synthase. Oncotarget 2016, 7, 63679–63689. [Google Scholar] [CrossRef]
- Jewell, J.L.; Kim, Y.C.; Russell, R.C.; Yu, F.-X.; Park, H.W.; Plouffe, S.W.; Tagliabracci, V.S.; Guan, K.-L. Differential regulation of mTORC1 by leucine and glutamine. Science 2015, 347, 194–198. [Google Scholar] [CrossRef]
- Miko, E.; Margitai, Z.; Czimmerer, Z.; Várkonyi, I.; Dezső, B.; Lányi, Á.; Bacsó, Z.; Scholtz, B. miR-126 inhibits proliferation of small cell lung cancer cells by targeting SLC7A5. FEBS Lett. 2011, 585, 1191–1196. [Google Scholar] [CrossRef] [PubMed]
- Cruz, B.; Oliveira, A.; Viana, L.R.; Lopes-Aguiar, L.; Canevarolo, R.; Colombera, M.C.; Valentim, R.R.; Garcia-Fóssa, F.; de Sousa, L.M.; Castelucci, B.G.; et al. Leucine-Rich Diet Modulates the Metabolomic and Proteomic Profile of Skeletal Muscle during Cancer Cachexia. Cancers 2020, 12, 1880. [Google Scholar] [CrossRef]
- Storck, L.J.; Ruehlin, M.; Gaeumann, S.; Gisi, D.; Schmocker, M.; Meffert, P.J.; Imoberdorf, R.; Pless, M.; Ballmer, P.E. Effect of a leucine-rich supplement in combination with nutrition and physical exercise in advanced cancer patients: A randomized controlled intervention trial. Clin. Nutr. 2020, 39, 3637–3644. [Google Scholar] [CrossRef] [PubMed]
- Viana, L.R.; Canevarolo, R.; Luiz, A.C.P.; Soares, R.F.; Lubaczeuski, C.; de Mattos Zeri, A.C.; Gomes-Marcondes, M.C.C. Leucine-rich diet alters the 1 H-NMR based metabolomic profile without changing the Walker-256 tumour mass in rats. BMC Cancer 2016, 16, 1–14. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Unterlass, J.E.; Curtin, N.J. Warburg and Krebs and related effects in cancer. Expert Rev. Mol. Med. 2019, 21. [Google Scholar] [CrossRef]
- Xu, X.D.; Shao, S.X.; Jiang, H.P.; Cao, Y.W.; Wang, Y.H.; Yang, X.C.; Wang, Y.L.; Wang, X.S.; Niu, H.T. Warburg effect or reverse Warburg effect? A review of cancer metabolism. Oncol. Res. Treat. 2015, 38, 117–122. [Google Scholar] [CrossRef]
- Gray, A.; Dang, B.N.; Moore, T.B.; Clemens, R.; Pressman, P. A review of nutrition and dietary interventions in oncology. SAGE Open Med. 2020, 8. [Google Scholar] [CrossRef]
- Kobliakov, V.A. The mechanisms of regulation of aerobic glycolysis (Warburg Effect) by oncoproteins in carcinogenesis. Biochemistry 2019, 84, 1117–1128. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Shishodia, S.; Sandur, S.K.; Pandey, M.K.; Sethi, G. Inflammation and cancer: How hot is the link? Biochem. Pharmacol. 2006, 72, 1605–1621. [Google Scholar] [CrossRef]
- Klement, R.J. The emerging role of ketogenic diets in cancer treatment. Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.; Zhao, J.; Liu, X.-j.; Yang, W.-r.; Yuan, F. Significance of calorie-restricted ketogenic diet for lung cancer with brain metastases and hepatoma with pulmonary metastases: Report of two cases. Res. Square 2020. [Google Scholar] [CrossRef]
- Tran, Q.; Lee, H.; Kim, C.; Kong, G.; Gong, N.; Kwon, S.H.; Park, J.; Kim, S.-H.; Park, J. Revisiting the Warburg effect: Diet-based strategies for cancer prevention. BioMed Res. Int. 2020, 2020, 8105735. [Google Scholar] [CrossRef] [PubMed]
- Wallis, J. The Ketogenic Diet for Cancer Patients: A Narrative Review. Master’s Thesis, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA, 2018. [Google Scholar] [CrossRef]
- Woolf, E.C.; Scheck, A.C. The ketogenic diet for the treatment of malignant glioma. J. Lipid Res. 2015, 56, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Yang, Y.-Y.; Zhou, M.-W.; Liu, N.; Xing, H.-Y.; Liu, X.-X.; Li, F. Ketogenic diet attenuates oxidative stress and inflammation after spinal cord injury by activating Nrf2 and suppressing the NF-κB signaling pathways. Neurosci. Lett. 2018, 683, 13–18. [Google Scholar] [CrossRef]
- Schetter, A.J.; Heegaard, N.H.H.; Harris, C.C. Inflammation and cancer: Interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis 2010, 31, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R. Tumour necrosis factor and cancer. Prog. Growth Factor Res. 1992, 4, 121–137. [Google Scholar] [CrossRef]
- Crusz, S.M.; Balkwill, F.R. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Shishodia, S.; Sandur, S.K.; Pandey, M.K.; Sethi, G. Inflamación y cáncer:¿ qué tan caliente es el vínculo? Biochem. Pharmacol. 2006, 72, 1605–1621. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Elsakka, A.; Bary, M.A.; Abdelzaher, E.; Elnaggar, M.; Kalamian, M.; Mukherjee, P.; Seyfried, T.N. Management of glioblastoma multiforme in a patient treated with ketogenic metabolic therapy and modified standard of care: A 24-month follow-up. Front. Nutr. 2018, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Sudderth, J.; Dang, T.; Bachoo, R.G.; McDonald, J.G.; DeBerardinis, R.J. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res. 2009, 69, 7986–7993. [Google Scholar] [CrossRef]
- Mukherjee, P.; Augur, Z.M.; Li, M.; Hill, C.; Greenwood, B.; Domin, M.A.; Kondakci, G.; Narain, N.R.; Kiebish, M.A.; Bronson, R.T. Therapeutic benefit of combining calorie-restricted ketogenic diet and glutamine targeting in late-stage experimental glioblastoma. Commun. Biol. 2019, 2, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Cruceriu, D.; Baldasici, O.; Balacescu, O.; Berindan-Neagoe, I. The dual role of tumor necrosis factor-alpha (TNF-α) in breast cancer: Molecular insights and therapeutic approaches. Cell. Oncol. 2020, 43, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Ren, Y.; Dai, Z.-J.; Wu, C.-J.; Ji, Y.-H.; Xu, J. IL-6, IL-8 and TNF-α levels correlate with disease stage in breast cancer patients. Adv. Clin. Exp. Med. 2017, 26, 421–426. [Google Scholar] [CrossRef]
- Zhou, X.-L.; Fan, W.; Yang, G.; Yu, M.-X. The clinical significance of PR, ER, NF-κB, and TNF-α in breast cancer. Dis. Markers 2014, 2014. [Google Scholar] [CrossRef]
- Khodabakhshi, A.; Akbari, M.E.; Mirzaei, H.R.; Mehrad-Majd, H.; Kalamian, M.; Davoodi, S.H. Feasibility, safety, and beneficial effects of MCT-based ketogenic diet for breast cancer treatment: A randomized controlled trial study. Nutr. Cancer 2020, 72, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Khodabakhshi, A.; Akbari, M.E.; Mirzaei, H.R.; Seyfried, T.N.; Kalamian, M.; Davoodi, S.H. Effects of Ketogenic metabolic therapy on patients with breast Cancer: A randomized controlled clinical trial. Clin. Nutr. 2021, 40, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reza, I.; Díaz, L.; García-Becerra, R. Preclinical and clinical aspects of TNF-α and its receptors TNFR1 and TNFR2 in breast cancer. J. Biomed. Sci. 2017, 24, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Karpisheh, V.; Nikkhoo, A.; Hojjat-Farsangi, M.; Namdar, A.; Azizi, G.; Ghalamfarsa, G.; Sabz, G.; Yousefi, M.; Yousefi, B.; Jadidi-Niaragh, F. Prostaglandin E2 as a potent therapeutic target for treatment of colon cancer. Prostaglandins Other Lipid Mediat. 2019, 144, 106338. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; DuBois, R.N. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene 2010, 29, 781–788. [Google Scholar] [CrossRef] [PubMed]
- de Souza, C.P.; Alves, B.; Waisberg, J.; Fonseca, F.; de Oliveira Carmo, A.; Gehrke, F. Detection of COX-2 in liquid biopsy in patients with breast cancer. J. Clin. Pathol. 2020, 73, 826–829. [Google Scholar] [CrossRef]
- Kochel, T.J.; Reader, J.C.; Ma, X.; Kundu, N.; Fulton, A.M. Multiple drug resistance-associated protein (MRP4) exports prostaglandin E2 (PGE2) and contributes to metastasis in basal/triple negative breast cancer. Oncotarget 2017, 8, 6540. [Google Scholar] [CrossRef] [PubMed]
- Edelman, M.J.; Wang, X.; Hodgson, L.; Cheney, R.T.; Baggstrom, M.Q.; Thomas, S.P.; Gajra, A.; Bertino, E.; Reckamp, K.L.; Molina, J. Phase III randomized, placebo-controlled, double-blind trial of celecoxib in addition to standard chemotherapy for advanced non–small-cell lung cancer with cyclooxygenase-2 overexpression: CALGB 30801 (Alliance). J. Clin. Oncol. 2017, 35, 2184. [Google Scholar] [CrossRef]
- Woolf, E.C.; Curley, K.L.; Liu, Q.; Turner, G.H.; Charlton, J.A.; Preul, M.C.; Scheck, A.C. The ketogenic diet alters the hypoxic response and affects expression of proteins associated with angiogenesis, invasive potential and vascular permeability in a mouse glioma model. PLoS ONE 2015, 10, e0130357. [Google Scholar] [CrossRef] [PubMed]
- Laronha, H.; Carpinteiro, I.; Portugal, J.; Azul, A.; Polido, M.; Petrova, K.T.; Salema-Oom, M.; Caldeira, J. Challenges in matrix metalloproteinases inhibition. Biomolecules 2020, 10, 717. [Google Scholar] [CrossRef]
- Said, A.H.; Raufman, J.-P.; Xie, G. The role of matrix metalloproteinases in colorectal cancer. Cancers 2014, 6, 366–375. [Google Scholar] [CrossRef]
- Sampieri, C.L.; De La Peña, S.; Ochoa-Lara, M.; Zenteno-Cuevas, R.; León-Córdoba, K. Expression of matrix metalloproteinases 2 and 9 in human gastric cancer and superficial gastritis. World J. Gastroenterol. WJG 2010, 16, 1500. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mondal, S.; Adhikari, N.; Banerjee, S.; Amin, S.A.; Jha, T. Matrix metalloproteinase-9 (MMP-9) and its inhibitors in cancer: A minireview. Eur. J. Med. Chem. 2020, 194, 112260. [Google Scholar] [CrossRef]
- Zhang, N.; Liu, C.; Jin, L.; Zhang, R.; Wang, T.; Wang, Q.; Chen, J.; Yang, F.; Siebert, H.-C.; Zheng, X. Ketogenic diet elicits antitumor properties through inducing oxidative stress, inhibiting MMP-9 expression, and rebalancing M1/M2 tumor-associated macrophage phenotype in a mouse model of colon cancer. J. Agric. Food Chem. 2020, 68, 11182–11196. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Shang, Y.P.; Chen, H.y.; Li, J. Histone deacetylases function as novel potential therapeutic targets for cancer. Hepatol. Res. 2017, 47, 149–159. [Google Scholar] [CrossRef]
- Sabari, B.R.; Zhang, D.; Allis, C.D.; Zhao, Y. Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 2017, 18, 90. [Google Scholar] [CrossRef] [PubMed]
- West, A.C. Johnstone. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Tan-Shalaby, J. Ketogenic diets and cancer: Emerging evidence. Fed. Pract. 2017, 34, 37S. [Google Scholar]
- Wong, N.; Ojo, D.; Yan, J.; Tang, D. PKM2 contributes to cancer metabolism. Cancer Lett. 2015, 356, 184–191. [Google Scholar] [CrossRef]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Lüftner, D.; Jung, A.; Schmid, P.; Geppert, R.; Kienle, E.; Wernecke, K.D.; Possinger, K. Upregulation of HER-2/neu by ovarian ablation: Results of a randomized trial comparing leuprorelin to CMF as adjuvant therapy in node-positive breast cancer patients. Breast Cancer Res. Treat. 2003, 80, 245–255. [Google Scholar] [CrossRef]
- Brinck, U.; Fischer, G.; Eigenbrodt, E.; Oehmke, M.; Mazurek, S. L-and M 2-pyruvate kinase expression in renal cell carcinomas and their metastases. Virchows Arch. 1994, 424, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.C.; Hu, Y.Y.; Cheng, G.; Liang, L.; Gao, B.; Ren, Y.P.; Liu, J.T.; Cao, X.L.; Zheng, M.H.; Li, S.Z. A ketogenic diet attenuates proliferation and stemness of glioma stem-like cells by altering metabolism resulting in increased ROS production. Int. J. Oncol. 2020, 56, 606–617. [Google Scholar] [CrossRef]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef]
- Ozaki, T.; Nakagawara, A. Role of p53 in cell death and human cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef]
- Li, R.J.; Liu, Y.; Liu, H.Q.; Li, J. Ketogenic diets and protective mechanisms in epilepsy, metabolic disorders, cancer, neuronal loss, and muscle and nerve degeneration. J. Food Biochem. 2020, 44, e13140. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Li, F.; Sun, Q.; Lin, N.; Han, H.; You, K.; Tian, F.; Mao, Z.; Li, T.; Tong, T. p53 β-hydroxybutyrylation attenuates p53 activity. Cell Death Dis. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Motoshima, H.; Goldstein, B.J.; Igata, M.; Araki, E. AMPK and cell proliferation–AMPK as a therapeutic target for atherosclerosis and cancer. J. Physiol. 2006, 574, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Saud, S.M.; Young, M.R.; Chen, G.; Hua, B. Targeting AMPK for cancer prevention and treatment. Oncotarget 2015, 6, 7365. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, N.; Liu, P.; Xie, X. AMPK and Cancer. In AMP-Activated Protein Kinase; Springer: Cham, Switzerland, 2016; pp. 203–226. [Google Scholar]
- Lee, Y.-K.; Park, S.Y.; Kim, Y.-M.; Lee, W.S.; Park, O.J. AMP kinase/cyclooxygenase-2 pathway regulates proliferation and apoptosis of cancer cells treated with quercetin. Exp. Mol. Med. 2009, 41, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, S.; Higurashi, T.; Nakajima, A. AMPK: Therapeutic target for diabetes and cancer prevention. Curr. Pharm. Des. 2017, 23, 3629–3644. [Google Scholar] [CrossRef]
- Bose, S.; Allen, A.E.; Locasale, J.W. The molecular link from diet to Cancer cell metabolism. Mol. Cell 2020, 78, 1034–1044. [Google Scholar] [CrossRef]
- Fine, E.J.; Feinman, R.D. Insulin, carbohydrate restriction, metabolic syndrome and cancer. Expert Rev. Endocrinol. Metab. 2015, 10, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Klement, R.J.; Kämmerer, U. Is there a role for carbohydrate restriction in the treatment and prevention of cancer? Nutr. Metab. 2011, 8, 1–16. [Google Scholar] [CrossRef]
- Choi, J.-W.; Hua, T.N.M. Impact of Lifestyle Behaviors on Cancer Risk and Prevention. J. Lifestyle Med. 2021, 11, 1. [Google Scholar] [CrossRef]
- Hursting, S.D.; Ford, N.A.; Dunlap, S.M.; Hursting, M.J.; Lashinger, L.M. Calorie Restriction and Cancer Prevention: Established and Emerging Mechanisms. In Obesity, Inflammation and Cancer; Springer: New York, NY, USA, 2013; pp. 363–379. [Google Scholar]
- Casari, I.; Falasca, M. Diet and pancreatic cancer prevention. Cancers 2015, 7, 2309–2317. [Google Scholar] [CrossRef]
- Elisia, I.; Krystal, G. The Pros and Cons of Low Carbohydrate and Ketogenic Diets in the Prevention and Treatment of Cancer. Front. Nutr. 2021, 8, 57. [Google Scholar] [CrossRef]
- Goncalves, M.D.; Hopkins, B.D.; Cantley, L.C. Phosphatidylinositol 3-kinase, growth disorders, and cancer. N. Engl. J. Med. 2018, 379, 2052–2062. [Google Scholar] [CrossRef]
- Chan, J.M.; Stampfer, M.J.; Giovannucci, E.; Gann, P.H.; Ma, J.; Wilkinson, P.; Hennekens, C.H.; Pollak, M. Plasma insulin-like growth factor-I and prostate cancer risk: A prospective study. Science 1998, 279, 563–566. [Google Scholar] [CrossRef]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009, 325, 201–204. [Google Scholar] [CrossRef]
- Klement, R.J. Beneficial effects of ketogenic diets for cancer patients: A realist review with focus on evidence and confirmation. Med. Oncol. 2017, 34, 1–15. [Google Scholar] [CrossRef]
- Mavropoulos, J.C.; Isaacs, W.B.; Pizzo, S.V.; Freedland, S.J. Is there a role for a low-carbohydrate ketogenic diet in the management of prostate cancer? Urology 2006, 68, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Maxmen, A. Calorie restriction falters in the long run. Nat. News 2012, 488, 569. [Google Scholar] [CrossRef]
- Meynet, O.; Ricci, J.-E. Caloric restriction and cancer: Molecular mechanisms and clinical implications. Trends Mol. Med. 2014, 20, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, K.L. The science of fatty acids and inflammation. Adv. Nutr. 2015, 6, 293S–301S. [Google Scholar] [CrossRef]
- Calder, P.C. Omega-3 polyunsaturated yağ turşuları və iltihabi proseslər: Bəslənmə və farmakologiya. İngilis Klinik Farmakologiya Jurnalı 2013, 75, 645–662. [Google Scholar]
- Harvey, A.E.; Lashinger, L.M.; Hursting, S.D. The growing challenge of obesity and cancer: An inflammatory issue. Ann. N. Y. Acad. Sci. 2011, 1229, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef]
- Ferrere, G.; Alou, M.T.; Liu, P.; Goubet, A.-G.; Fidelle, M.; Kepp, O.; Durand, S.; Iebba, V.; Fluckiger, A.; Daillère, R. Ketogenic diet and ketone bodies enhance the anticancer effects of PD-1 blockade. JCI Insight 2021, 6, e145207. [Google Scholar] [CrossRef]
- Parida, S.; Siddharth, S.; Sharma, D. Adiponectin, obesity, and cancer: Clash of the bigwigs in health and disease. Int. J. Mol. Sci. 2019, 20, 2519. [Google Scholar] [CrossRef]
- Bandera-Merchan, B.; Boughanem, H.; Crujeiras, A.B.; Macias-Gonzalez, M.; Tinahones, F.J. Ketotherapy as an epigenetic modifier in cancer. Rev. Endocr. Metab. Disord. 2020, 21, 509–519. [Google Scholar] [CrossRef]
- Diaz-Lagares, A.; Crujeiras, A.B.; Lopez-Serra, P.; Soler, M.; Setien, F.; Goyal, A.; Sandoval, J.; Hashimoto, Y.; Martinez-Cardús, A.; Gomez, A.; et al. Epigenetic inactivation of the p53-induced long noncoding RNA TP53 target 1 in human cancer. Proc. Natl. Acad. Sci. USA 2016, 113, E7535. [Google Scholar] [CrossRef]
- Dabek, A.; Wojtala, M.; Pirola, L.; Balcerczyk, A. Modulation of Cellular Biochemistry, Epigenetics and Metabolomics by Ketone Bodies. Implications of the Ketogenic Diet in the Physiology of the Organism and Pathological States. Nutrients 2020, 12, 788. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.-B.; Crawford, P.A. Ketone bodies as epigenetic modifiers. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 260–266. [Google Scholar] [CrossRef]
- Boison, D. New insights into the mechanisms of the ketogenic diet. Curr. Opin. Neurol. 2017, 30, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Masino, S.A.; Li, T.; Theofilas, P.; Sandau, U.S.; Ruskin, D.N.; Fredholm, B.B.; Geiger, J.D.; Aronica, E.; Boison, D. A ketogenic diet suppresses seizures in mice through adenosine A1 receptors. J. Clin. Investig. 2011, 121, 2679–2683. [Google Scholar] [CrossRef]
- Benjamin, J.S.; Pilarowski, G.O.; Carosso, G.A.; Zhang, L.; Huso, D.L.; Goff, L.A.; Vernon, H.J.; Hansen, K.D.; Bjornsson, H.T. A ketogenic diet rescues hippocampal memory defects in a mouse model of Kabuki syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, 125–130. [Google Scholar] [CrossRef]
- Jaworski, D.M.; Namboodiri, A.M.A.; Moffett, J.R. Acetate as a Metabolic and Epigenetic Modifier of Cancer Therapy. J. Cell. Biochem. 2016, 117, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Shirahata, M.; Tang, W.-Y.; Kostuk, E.W. A Short-Term Fasting in Neonates Induces Breathing Instability and Epigenetic Modification in the Carotid Body. In Arterial Chemoreceptors in Physiology and Pathophysiology; Peers, C., Kumar, P., Wyatt, C., Gauda, E., Nurse, C.A., Prabhakar, N., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 187–193. [Google Scholar] [CrossRef]
- Bao, B.; Wang, Z.; Li, Y.; Kong, D.; Ali, S.; Banerjee, S.; Ahmad, A.; Sarkar, F.H. The complexities of obesity and diabetes with the development and progression of pancreatic cancer. Biochim. Biophys. Acta Rev. Cancer 2011, 1815, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Parasramka, M.A.; Ho, E.; Williams, D.E.; Dashwood, R.H. MicroRNAs, diet, and cancer: New mechanistic insights on the epigenetic actions of phytochemicals. Mol. Carcinog. 2012, 51, 213–230. [Google Scholar] [CrossRef] [PubMed]
- Schnekenburger, M.; Diederich, M. Epigenetics offer new horizons for colorectal cancer prevention. Curr. Colorectal Cancer Rep. 2012, 8, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Cannataro, R.; Perri, M.; Gallelli, L.; Caroleo, M.C.; De Sarro, G.; Cione, E. Ketogenic diet acts on body remodeling and microRNAs expression profile. Microrna 2019, 8, 116–126. [Google Scholar] [CrossRef]
- Gulyaeva, L.F.; Kushlinskiy, N.E. Regulatory mechanisms of microRNA expression. J. Transl. Med. 2016, 14, 1–10. [Google Scholar] [CrossRef]
- Ramalingam, S.; Subramaniam, D.; Anant, S. Manipulating miRNA expression: A novel approach for colon cancer prevention and chemotherapy. Curr. Pharmacol. Rep. 2015, 1, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Woolf, E.C. Ketogenic Therapy as an Adjuvant for Malignant Glioma: Impacts on Anti-Tumor Immunity. Ph.D. Dissertation, Arizona State University, Tempe, AZ, USA, 2018. [Google Scholar]
- DeSano, J.T.; Xu, L. MicroRNA regulation of cancer stem cells and therapeutic implications. AAPS J. 2009, 11, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.J.; Ray, A. MicroRNAs in the search for understanding human diseases. BioDrugs 2007, 21, 97–104. [Google Scholar] [CrossRef]
- Florean, C.; Schnekenburger, M.; Grandjenette, C.; Dicato, M.; Diederich, M. Epigenomics of leukemia: From mechanisms to therapeutic applications. Epigenomics 2011, 3, 581–609. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. MicroRNA-cancer connection: The beginning of a new tale. Cancer Res. 2006, 66, 7390–7394. [Google Scholar] [CrossRef]
- Tokarz, P.; Blasiak, J. The role of microRNA in metastatic colorectal cancer and its significance in cancer prognosis and treatment. Acta Biochim. Pol. 2012, 59. [Google Scholar] [CrossRef]
- Ludwig, N.; Leidinger, P.; Becker, K.; Backes, C.; Fehlmann, T.; Pallasch, C.; Rheinheimer, S.; Meder, B.; Stähler, C.; Meese, E. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016, 44, 3865–3877. [Google Scholar] [CrossRef]
- Kumarswamy, R.; Volkmann, I.; Thum, T. Regulation and function of miRNA-21 in health and disease. RNA Biol. 2011, 8, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-Y.; Yeh, K.-Y.; Lin, C.-Y.; Hsieh, Y.-W.; Lai, H.-H.; Chen, J.-R.; Hsu, C.-C.; Her, G.M. MicroRNA-21 Plays Multiple Oncometabolic Roles in the Process of NAFLD-Related Hepatocellular Carcinoma via PI3K/AKT, TGF-β, and STAT3 Signaling. Cancers 2021, 13, 940. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yu, J.; Yu, S.; Lavker, R.M.; Cai, L.; Liu, W.; Yang, K.; He, X.; Chen, S. MicroRNA-21 acts as an oncomir through multiple targets in human hepatocellular carcinoma. J. Hepatol. 2010, 53, 98–107. [Google Scholar] [CrossRef]
- Tomimaru, Y.; Eguchi, H.; Nagano, H.; Wada, H.; Kobayashi, S.; Marubashi, S.; Tanemura, M.; Tomokuni, A.; Takemasa, I.; Umeshita, K. Circulating microRNA-21 as a novel biomarker for hepatocellular carcinoma. J. Hepatol. 2012, 56, 167–175. [Google Scholar] [CrossRef]
- Zhang, T.; Yang, Z.; Kusumanchi, P.; Han, S.; Liangpunsakul, S. Critical role of microRNA-21 in the pathogenesis of liver diseases. Front. Med. 2020, 7, 7. [Google Scholar] [CrossRef]
- Varkaris, A.; Katsiampoura, A.; Davis, J.S.; Shah, N.; Lam, M.; Frias, R.L.; Ivan, C.; Shimizu, M.; Morris, J.; Menter, D. Circulating inflammation signature predicts overall survival and relapse-free survival in metastatic colorectal cancer. Br. J. Cancer 2019, 120, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Nf, H.G.K.M. KB and STAT3—Key players in 1iver inflammation and cancer. Cell Res. 2011, 21, 159. [Google Scholar]
- Loboda, A.; Sobczak, M.; Jozkowicz, A.; Dulak, J. TGF-β1/Smads and miR-21 in renal fibrosis and inflammation. Mediat. Inflamm. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Si, M.-L.; Wu, H.; Mo, Y.-Y. MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1). J. Biol. Chem. 2007, 282, 14328–14336. [Google Scholar] [CrossRef]
- Chang, K.H.; Miller, N.; Kheirelseid, E.A.H.; Ingoldsby, H.; Hennessy, E.; Curran, C.E.; Curran, S.; Smith, M.J.; Regan, M.; McAnena, O.J. MicroRNA-21 and PDCD4 expression in colorectal cancer. Eur. J. Surg. Oncol. 2011, 37, 597–603. [Google Scholar] [CrossRef]
- Liu, C.-Z.; Liu, W.; Zheng, Y.; Su, J.-M.; Li, J.-J.; Yu, L.; He, X.-D.; Chen, S.-S. PTEN and PDCD4 are Bona Fide Targets of microRNA-21 in Human Cholangiocarcinoma△. Chin. Med. Sci. J. 2012, 27, 65–72. [Google Scholar]
- Carpi, S.; Polini, B.; Fogli, S.; Podestà, A.; Ylösmäki, E.; Cerullo, V.; Romanini, A.; Nieri, P. Circulating microRNAs as biomarkers for early diagnosis of cutaneous melanoma. Expert Rev. Mol. Diagn. 2020, 20, 19–30. [Google Scholar] [CrossRef]
- Ferracin, M.; Lupini, L.; Salamon, I.; Saccenti, E.; Zanzi, M.V.; Rocchi, A.; Da Ros, L.; Zagatti, B.; Musa, G.; Bassi, C. Absolute quantification of cell-free microRNAs in cancer patients. Oncotarget 2015, 6, 14545. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.C.; John, S.M.; Carrera-Bastos, P.; Schmitz, G. MicroRNA-21-enriched exosomes as epigenetic regulators in melanomagenesis and melanoma progression: The impact of western lifestyle factors. Cancers 2020, 12, 2111. [Google Scholar] [CrossRef]
- Neagu, M.; Constantin, C.; Cretoiu, S.M.; Zurac, S. miRNAs in the Diagnosis and Prognosis of Skin Cancer. Front. Cell Dev. Biol. 2020, 8, 71. [Google Scholar] [CrossRef] [PubMed]
- Satzger, I.; Mattern, A.; Kuettler, U.; Weinspach, D.; Niebuhr, M.; Kapp, A.; Gutzmer, R. micro RNA-21 is upregulated in malignant melanoma and influences apoptosis of melanocytic cells. Exp. Dermatol. 2012, 21, 509–514. [Google Scholar] [CrossRef]
- Yang, Z.; Liao, B.; Xiang, X.; Ke, S. miR-21-5p promotes cell proliferation and G1/S transition in melanoma by targeting CDKN2C. FEBS Open Bio 2020, 10, 752–760. [Google Scholar] [CrossRef]
- Woolf, E.C.; Syed, N.; Scheck, A.C. Tumor metabolism, the ketogenic diet and β-hydroxybutyrate: Novel approaches to adjuvant brain tumor therapy. Front. Mol. Neurosci. 2016, 9, 122. [Google Scholar] [CrossRef] [PubMed]
- Condrat, C.E.; Thompson, D.C.; Barbu, M.G.; Bugnar, O.L.; Boboc, A.; Cretoiu, D.; Suciu, N.; Cretoiu, S.M.; Voinea, S.C. miRNAs as biomarkers in disease: Latest findings regarding their role in diagnosis and prognosis. Cells 2020, 9, 276. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.; Priyanka, K.; Tuli, H.S. Emergence of circulating microRNAs in breast cancer as diagnostic and therapeutic efficacy biomarkers. Mol. Diagn. Ther. 2020, 24, 153–173. [Google Scholar] [CrossRef] [PubMed]
- Petri, B.J.; Klinge, C.M. Regulation of breast cancer metastasis signaling by miRNAs. Cancer Metastasis Rev. 2020, 39, 837–886. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.A.; Jeon, B.T.; Shin, H.J.; Kim, N.; Lee, D.H.; Kim, H.J.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Roh, G.S. Ketogenic diet-induced peroxisome proliferator-activated receptor-γ activation decreases neuroinflammation in the mouse hippocampus after kainic acid-induced seizures. Exp. Neurol. 2011, 232, 195–202. [Google Scholar] [CrossRef]
- Arndt, G.M.; Dossey, L.; Cullen, L.M.; Lai, A.; Druker, R.; Eisbacher, M.; Zhang, C.; Tran, N.; Fan, H.; Retzlaff, K. Characterization of global microRNA expression reveals oncogenic potential of miR-145 in metastatic colorectal cancer. BMC Cancer 2009, 9, 1–17. [Google Scholar] [CrossRef]
- Earle, J.S.L.; Luthra, R.; Romans, A.; Abraham, R.; Ensor, J.; Yao, H.; Hamilton, S.R. Association of microRNA expression with microsatellite instability status in colorectal adenocarcinoma. J. Mol. Diagn. 2010, 12, 433–440. [Google Scholar] [CrossRef]
- Kondo, T.; Oka, T.; Sato, H.; Shinnou, Y.; Washio, K.; Takano, M.; Morito, T.; Takata, K.; Ohara, N.; Ouchida, M. Accumulation of aberrant CpG hypermethylation by Helicobacter pylori infection promotes development and progression of gastric MALT lymphoma. Int. J. Oncol. 2009, 35, 547–557. [Google Scholar] [PubMed]
- Akao, Y.; Nakagawa, Y.; Naoe, T. let-7 microRNA functions as a potential growth suppressor in human colon cancer cells. Biol. Pharm. Bull. 2006, 29, 903–906. [Google Scholar] [CrossRef]
- King, C.E.; Wang, L.; Winograd, R.; Madison, B.B.; Mongroo, P.S.; Johnstone, C.N.; Rustgi, A.K. LIN28B fosters colon cancer migration, invasion and transformation through let-7-dependent and-independent mechanisms. Oncogene 2011, 30, 4185–4193. [Google Scholar] [CrossRef]
- Sha, D.; Lee, A.M.; Shi, Q.; Alberts, S.R.; Sargent, D.J.; Sinicrope, F.A.; Diasio, R.B. Association study of the let-7 miRNA-complementary site variant in the 3′ untranslated region of the KRAS gene in stage III colon cancer (NCCTG N0147 Clinical Trial). Clin. Cancer Res. 2014, 20, 3319–3327. [Google Scholar] [CrossRef]
- Deng, J.; Lei, W.; Fu, J.-C.; Zhang, L.; Li, J.-H.; Xiong, J.-P. Targeting miR-21 enhances the sensitivity of human colon cancer HT-29 cells to chemoradiotherapy in vitro. Biochem. Biophys. Res. Commun. 2014, 443, 789–795. [Google Scholar] [CrossRef]
- Oue, N.; Anami, K.; Schetter, A.J.; Moehler, M.; Okayama, H.; Khan, M.A.; Bowman, E.D.; Mueller, A.; Schad, A.; Shimomura, M. High miR-21 expression from FFPE tissues is associated with poor survival and response to adjuvant chemotherapy in colon cancer. Int. J. Cancer 2014, 134, 1926–1934. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Yu, Y.; Padhye, S.B.; Sarkar, F.H.; Majumdar, A.P.N. Difluorinated-curcumin (CDF) restores PTEN expression in colon cancer cells by down-regulating miR-21. PLoS ONE 2013, 8, e68543. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guo, H.; Zhang, H.; Wang, H.; Qian, G.; Fan, X.; Hoffman, A.R.; Hu, J.F.; Ge, S. Putative tumor suppressor miR-145 inhibits colon cancer cell growth by targeting oncogene friend leukemia virus integration 1 gene. Cancer 2011, 117, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Lee, C.Y.; Johnson, R.L.; Wichterman, J.; Huang, R.; DePamphilis, M.L. An image-based, high-throughput screening assay for molecules that induce excess DNA replication in human cancer cells. Mol. Cancer Res. 2011, 9, 294–310. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.-Y.; Park, Y.K. Rationale, Feasibility and Acceptability of Ketogenic Diet for Cancer Treatment. J. Cancer Prev. 2017, 22, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Shea, A.; Harish, V.; Afzal, Z.; Chijioke, J.; Kedir, H.; Dusmatova, S.; Roy, A.; Ramalinga, M.; Harris, B.; Blancato, J.; et al. MicroRNAs in glioblastoma multiforme pathogenesis and therapeutics. Cancer Med. 2016, 5, 1917–1946. [Google Scholar] [CrossRef]
- Beck, S.A.; Tisdale, M.J. Effect of insulin on weight loss and tumour growth in a cachexia model. Br. J. Cancer 1989, 59, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Tonouchi, H.; Sasayama, A.; Ashida, K. A ketogenic formula prevents tumor progression and cancer cachexia by attenuating systemic inflammation in colon 26 tumor-bearing mice. Nutrients 2018, 10, 206. [Google Scholar] [CrossRef]
- Kasumi, E.; Sato, N. A ketogenic diet improves the prognosis in a mouse model of peritoneal dissemination without tumor regression. J. Clin. Biochem. Nutr. 2019, 64, 201–208. [Google Scholar] [CrossRef]
- Shukla, S.K.; Gebregiworgis, T.; Purohit, V.; Chaika, N.V.; Gunda, V.; Radhakrishnan, P.; Mehla, K.; Pipinos, I.I.; Powers, R.; Yu, F. Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer Metab. 2014, 2, 1–19. [Google Scholar]
- Zhang, J.; Jia, P.-P.; Liu, Q.-L.; Cong, M.-H.; Gao, Y.; Shi, H.-P.; Yu, W.-N.; Miao, M.-Y. Low ketolytic enzyme levels in tumors predict ketogenic diet responses in cancer cell lines in vitro and in vivo. J. Lipid Res. 2018, 59, 625–634. [Google Scholar] [CrossRef]
- Zahra, A.; Fath, M.A.; Opat, E.; Mapuskar, K.A.; Bhatia, S.K.; Ma, D.C.; Snyders, T.P.; Chenard, C.A.; Eichenberger-Gilmore, J.M.; Bodeker, K.L. Consuming a ketogenic diet while receiving radiation and chemotherapy for locally advanced lung cancer and pancreatic cancer: The University of Iowa experience of two phase 1 clinical trials. Radiat. Res. 2017, 187, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Gluschnaider, U.; Hertz, R.; Ohayon, S.; Smeir, E.; Smets, M.; Pikarsky, E.; Bar-Tana, J. Long-chain fatty acid analogues suppress breast tumorigenesis and progression. Cancer Res. 2014, 74, 6991–7002. [Google Scholar] [CrossRef] [PubMed]
- Stemmer, K.; Zani, F.; Habegger, K.M.; Neff, C.; Kotzbeck, P.; Bauer, M.; Yalamanchilli, S.; Azad, A.; Lehti, M.; Martins, P.J.F. FGF21 is not required for glucose homeostasis, ketosis or tumour suppression associated with ketogenic diets in mice. Diabetologia 2015, 58, 2414–2423. [Google Scholar] [CrossRef]
- Rieger, J.; Bähr, O.; Maurer, G.D.; Hattingen, E.; Franz, K.; Brucker, D.; Walenta, S.; Kämmerer, U.; Coy, J.F.; Weller, M. ERGO: A pilot study of ketogenic diet in recurrent glioblastoma Erratum in/ijo/45/6/2605. Int. J. Oncol. 2014, 44, 1843–1852. [Google Scholar] [CrossRef] [PubMed]
- Dang, M.T.; Wehrli, S.; Dang, C.V.; Curran, T. The ketogenic diet does not affect growth of hedgehog pathway medulloblastoma in mice. PLoS ONE 2015, 10, e0133633. [Google Scholar] [CrossRef]
- Kim, H.S.; Masko, E.M.; Poulton, S.L.; Kennedy, K.M.; Pizzo, S.V.; Dewhirst, M.W.; Freedland, S.J. Carbohydrate restriction and lactate transporter inhibition in a mouse xenograft model of human prostate cancer. BJU Int. 2012, 110, 1062. [Google Scholar] [CrossRef]
- Allott, E.H.; Macias, E.; Sanders, S.; Knudsen, B.S.; Thomas, G.V.; Hursting, S.D.; Freedland, S.J. Impact of carbohydrate restriction in the context of obesity on prostate tumor growth in the Hi-Myc transgenic mouse model. Prostate Cancer Prostatic Dis. 2017, 20, 165–171. [Google Scholar] [CrossRef]
- Zhuang, Y.; Chan, D.K.; Haugrud, A.B.; Miskimins, W.K. Mechanisms by which low glucose enhances the cytotoxicity of metformin to cancer cells both in vitro and in vivo. PLoS ONE 2014, 9, e108444. [Google Scholar] [CrossRef]
- Allen, B.G.; Bhatia, S.K.; Buatti, J.M.; Brandt, K.E.; Lindholm, K.E.; Button, A.M.; Szweda, L.I.; Smith, B.J.; Spitz, D.R.; Fath, M.A. Ketogenic diets enhance oxidative stress and radio-chemo-therapy responses in lung cancer xenografts. Clin. Cancer Res. 2013, 19, 3905–3913. [Google Scholar] [CrossRef]
- Xia, S.; Lin, R.; Jin, L.; Zhao, L.; Kang, H.-B.; Pan, Y.; Liu, S.; Qian, G.; Qian, Z.; Konstantakou, E. Prevention of dietary-fat-fueled ketogenesis attenuates BRAF V600E tumor growth. Cell Metab. 2017, 25, 358–373. [Google Scholar] [CrossRef]
- Vidali, S.; Aminzadeh-Gohari, S.; Feichtinger, R.G.; Vatrinet, R.; Koller, A.; Locker, F.; Rutherford, T.; O’Donnell, M.; Stöger-Kleiber, A.; Lambert, B. The ketogenic diet is not feasible as a therapy in a CD-1 nu/nu mouse model of renal cell carcinoma with features of Stauffer’s syndrome. Oncotarget 2017, 8, 57201. [Google Scholar] [CrossRef] [PubMed]
- Liśkiewicz, A.D.; Kasprowska, D.; Wojakowska, A.; Polański, K.; Lewin–Kowalik, J.; Kotulska, K.; Jędrzejowska–Szypułka, H. Long-term High Fat Ketogenic Diet Promotes Renal Tumor Growth in a Rat Model of Tuberous Sclerosis. Sci. Rep. 2016, 6, 21807. [Google Scholar] [CrossRef]
- Byrne, F.L.; Hargett, S.R.; Lahiri, S.; Roy, R.J.; Berr, S.S.; Caldwell, S.H.; Hoehn, K.L. Serial MRI Imaging Reveals Minimal Impact of Ketogenic Diet on Established Liver Tumor Growth. Cancers 2018, 10, 312. [Google Scholar] [CrossRef]
- Healy, M.E.; Chow, J.D.Y.; Byrne, F.L.; Breen, D.S.; Leitinger, N.; Li, C.; Lackner, C.; Caldwell, S.H.; Hoehn, K.L. Dietary effects on liver tumor burden in mice treated with the hepatocellular carcinogen diethylnitrosamine. J. Hepatol. 2015, 62, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Magee, B.A.; Potezny, N.; Rofe, A.M.; Conyers, R.A. The inhibition of malignant cell growth by ketone bodies. Aust. J. Exp. Biol. Med. Sci. 1979, 57, 529–539. [Google Scholar] [CrossRef]
- Ludwig, D.S. The ketogenic diet: Evidence for optimism but high-quality research needed. J. Nutr. 2020, 150, 1354–1359. [Google Scholar] [CrossRef] [PubMed]
- Champ, C.E.; Palmer, J.D.; Volek, J.S.; Werner-Wasik, M.; Andrews, D.W.; Evans, J.J.; Glass, J.; Kim, L.; Shi, W. Targeting metabolism with a ketogenic diet during the treatment of glioblastoma multiforme. J. Neurooncol. 2014, 117, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Artzi, M.; Liberman, G.; Vaisman, N.; Bokstein, F.; Vitinshtein, F.; Aizenstein, O.; Ben Bashat, D. Changes in cerebral metabolism during ketogenic diet in patients with primary brain tumors: (1)H-MRS study. J. Neurooncol. 2017, 132, 267–275. [Google Scholar] [CrossRef]
- Martin-McGill, K.J.; Marson, A.G.; Tudur Smith, C.; Jenkinson, M.D. The Modified Ketogenic Diet in Adults with Glioblastoma: An Evaluation of Feasibility and Deliverability within the National Health Service. Nutr. Cancer 2018, 70, 643–649. [Google Scholar] [CrossRef]
- Strowd, R.E.; Cervenka, M.C.; Henry, B.J.; Kossoff, E.H.; Hartman, A.L.; Blakeley, J.O. Glycemic modulation in neuro-oncology: Experience and future directions using a modified Atkins diet for high-grade brain tumors. Neuro Oncol. Pract. 2015, 2, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Porper, K.; Shpatz, Y.; Plotkin, L.; Pechthold, R.G.; Talianski, A.; Hemi, R.; Mardor, Y.; Jan, E.; Genssin, H.; Symon, Z.; et al. DDRE-17. A phase i clinical trial of dose-escalated metabolic therapy combined with concomitant radiation therapy in high-grade glioma. Neurooncol. Adv. 2021, 3, i10. [Google Scholar] [CrossRef]
- Kato, I.; Dyson, G.; Snyder, M.; Kim, H.-R.; Severson, R.K. Differential effects of patient-related factors on the outcome of radiation therapy for rectal cancer. J. Radiat. Oncol. 2016, 5, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Branca, J.J.V.; Pacini, S.; Ruggiero, M. Effects of Pre-surgical Vitamin D Supplementation and Ketogenic Diet in a Patient with Recurrent Breast Cancer. Anticancer Res. 2015, 35, 5525. [Google Scholar]
- Klement, R.J.; Weigel, M.M.; Sweeney, R.A. A ketogenic diet consumed during radiotherapy improves several aspects of quality of life and metabolic health in women with breast cancer. Clin. Nutr. 2021. [Google Scholar] [CrossRef]
- İyikesici, M.S.; Slocum, A.K.; Slocum, A.; Berkarda, F.B.; Kalamian, M.; Seyfried, T.N. Efficacy of Metabolically Supported Chemotherapy Combined with Ketogenic Diet, Hyperthermia, and Hyperbaric Oxygen Therapy for Stage IV Triple-Negative Breast Cancer. Cureus 2017, 9, e1445. [Google Scholar] [CrossRef]
- Iyikesici, M.S. Feasibility study of metabolically supported chemotherapy with weekly carboplatin/paclitaxel combined with ketogenic diet, hyperthermia and hyperbaric oxygen therapy in metastatic non-small cell lung cancer. Int. J. Hyperth. 2019, 36, 446–455. [Google Scholar] [CrossRef]
- Cohen, C.W.; Fontaine, K.R. Favorable Effects of a Ketogenic Diet on Physical Function, Perceived Energy, and Food Cravings in Women with Ovarian or Endometrial Cancer: A Randomized, Controlled Trial. Nutrients 2018, 10, 1187. [Google Scholar] [CrossRef]
- Cohen, C.W.; Fontaine, K.R.; Arend, R.C.; Alvarez, R.D.; Leath, C.A., III; Huh, W.K.; Bevis, K.S.; Kim, K.H.; Straughn, J.M., Jr.; Gower, B.A. A Ketogenic Diet Reduces Central Obesity and Serum Insulin in Women with Ovarian or Endometrial Cancer. J. Nutr. 2018, 148, 1253–1260. [Google Scholar] [CrossRef]
- Schroeder, U.; Himpe, B.; Pries, R.; Vonthein, R.; Nitsch, S.; Wollenberg, B. Decline of lactate in tumor tissue after ketogenic diet: In vivo microdialysis study in patients with head and neck cancer. Nutr. Cancer 2013, 65, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.G.; Da Cruz, W.M.S.; Schönthal, A.H.; Salazar, M.D.; Fontes, C.A.P.; Quirico-Santos, T.; Da Fonseca, C.O. Efficacy of a ketogenic diet with concomitant intranasal perillyl alcohol as a novel strategy for the therapy of recurrent glioblastoma. Oncol. Lett. 2018, 15, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Choi, J. Diet-Based Interventions Against Cancer. Arbutus Rev. 2020, 11, 22–30. [Google Scholar] [CrossRef]
- Schmidt, M.; Pfetzer, N.; Schwab, M.; Strauss, I.; Kämmerer, U. Effects of a ketogenic diet on the quality of life in 16 patients with advanced cancer: A pilot trial. Nutr. Metab. 2011, 8, 54. [Google Scholar] [CrossRef]
- Rossi-Fanelli, F.; Franchi, F.; Mulieri, M.; Cangiano, C.; Cascino, A.; Ceci, F.; Muscaritoli, M.; Seminara, P.; Bonomo, L. Effect of energy substrate manipulation on tumour cell proliferation in parenterally fed cancer patients. Clin. Nutr. 1991, 10, 228–232. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Cancer Type | Cell Line | Animal Model | KD Ratio | Study Group | Mechanism and Results of the Studies Compared with SD | Ref. |
|---|---|---|---|---|---|---|
| Glioblastoma | T98G, U87MG, NIH3T3, A172, LNT-229, U251MG | athymic nude mice | 3:1 | SD, KD | KD resulted in a significant increase in BHB (KB), but it had no effect on glioma cell lines, TP, BG levels, or survival → no effect. | [75] |
| U87MG | athymic nude mice | 3:1 | SD ± CT, KD ± CT | KD alone: increase in KBs but no effect on TP or survival; KD+CT: increase the activity of CT drug → increase survival. | [252] | |
| GL261-Luc2 | albino C57BL/6 mice | 4:1 | SD ± RT, KD, KD ± RT | KD alone: increase in BHB (KB) and survival; KD+RT: enhance antitumor additive effect from RT alone → increase survival. | [22] | |
| GL261-Luc2 | albino C57BL/6 mice | 4:1 | SD, KD | The expression of VEGF receptor, MMP-2 and vimentin were reduced in tumors from animals on KD, and significantly reduced in the peritumoral edema. | [142] | |
| Medullo-blastoma | Spontaneous tumor development | Ptch1/-Trp53/mice | 4:1 | SD, KD | KD reduced the insulin level and increased the KB level in mice but there was no effect on the TP or survival. | [253] |
| Medulloblastoma from Ptch1/-Trp53/mice | NOD SCID mice | 6:1 | SD, KD | KD reduced the insulin level and increased the KB level in mice but there was no effect on the TP or survival. | [253] | |
| Prostate cancer | LAPC-4 | athymic nude mice | 2:1 | SD ± MCT1 inhibitor, KD ± MCT1 inhibitor | MCT1 inhibitor did not affect TP and increased necrotic fraction; KD decreased TP compared to SD and decreased the necrotic fraction. | [254] |
| Spontaneous tumor development | transgenic Hi-Myc mice | 2:1 | SD, KD | KD worked as a protumor (preventive). | [255] | |
| Colon cancer | colon 26 | CDF1 mice | 3:1 | SD, KD | KD increased KB and decreased TP and plasma IL-6 levels compared with tumor-bearing mice taking SD. | [245] |
| colon 26 | BALB/c mice | 4:1 | SD, KD | The KD group showed an increase in survival and better health status, no effect on TP → KD good as a potential preventive therapy. | [246] | |
| Pancreatic cancer | S2-013 | athymic nude mice | 2:1 | SD, KD | KD caused reduced TP and inhibition of muscle and body weight loss by decreasing BG, glycolytic flux in tumor cells and increasing KB, which diminished glutamine uptake, overall ATP content, and survival of the pancreatic cancer cell lines, while inducing apoptosis of it. | [247] |
| PANC-1 | nu/nu mice | 3:1 | SD, KD | KD decreased TP and increased the survival rate by reducing energy supplies to cells, which damage the tumor microenvironment → antitumor effect. | [248] | |
| MIA PaCa-2 | athymic nude mice | 4:1 | SD ± RT, KD ± RT | KD increased radiation sensitivity in a pancreatic cancer compared with radiation alone. | [249] | |
| Breast cancer | Spontaneous tumor development | transgenic FVB MMTV-PyMT mice | 4:1 | SD, KD | KD decreased TP by suppressing tumorigenesis; this may perhaps reflect the inherent tumor-suppressive efficacy of free LCFA or their respective CoA-thioesters by suppressing their esterification into lipids due to limiting insulin and glycerol-3-phosphate. | [250] |
| 4T1 | BALB/c mice | 6:1 | SD ± metformin, CR-KD ± metformin | KD enhanced the cytotoxic effect of metformin on tumor growth by decreasing ATP production and inhibiting survival signaling pathways. | [256] | |
| ES-272 | C57BL/6 mice | 6:1 | SD ± PI3K inhibitors, KD ± PI3K inhibitors | KD enhanced PI3K inhibitors to decrease TP in tumor cell → increased the antitumor effect. | [27] | |
| Lung cancer | LLC1 | C57BL/6 (Fgf21 WT and KO) mice | 3:1, 8:1 | low-fat diet (SD), regular protein KD, low protein KD | Regular protein KD had no effect on TP but low protein KD showed decreased TP, i.e., an antitumor effect by the extreme increase of fibroblast growth factor 21 levels because of protein starvation. | [251] |
| TC-1_luc | BALB/c mice | 4:1 | SD, KD, KD + mABs | KD increased BHB that slowed TP and induced a T cell-dependent anticancer effect. | [185] | |
| NCI-H292, A549 | nu/nu mice | 4:1 | different experiments with different IR doses, but overall: SD ± RT, KD ± RT, SD + RT/CT, KD + RT/CT | KD enhanced the antitumor effect of RT that decreased TP compared with RT alone by a mechanism that may involve increased oxidative stress. | [257] | |
| Melanoma | A375, A2058 (BRAF V600E) | nu/nu mice | 4:1, 6:1 | SD, KD | KD decreased glucose level and increased AcA, leading to increased TP → protumor effect. | [258] |
| RET melanoma | C57BL/6JolaHsd BALB/c mice | 4:1 | SD, KD, KD + mABs | KD increased BHB that slowed TP and induced a T cell-dependent anticancer effect and KD had synergistic antitumor effects when combined with a combination of immunostimulatory mAbs. | [185] | |
| Kidney cancer | 786-O | CD-1 nude mice | 8:1 | SD, LCT-KD, MCT-KDs | KD reduced TP, but mouse survival was dramatically reduced due to massive weight loss in the KD group. | [259] |
| RENCA-luc | BALB/c mice | 4:1 | SD, KD, KD + mABs | KD delayed the progression of TP, preventing tumor outgrowth in some mice and smaller tumors were observed in others. | [185] | |
| Spontaneous tumor development | Eker (Tsc2) rats | 8:1 | SD, KD | KD promoted TP by recruiting ERK1/2 and mTOR, which are correlated with the accumulation of oleic acid and the overproduction of growth hormone. | [260] | |
| Liver cancer | DEN-induced hepatocellularcarcinoma | C57BL/6N mice | 4:1 | SD, KD | KD had no effect on TP. | [261] |
| DEN-induced hepatocellularcarcinoma | C57BL/6N mice | 5:1 | low-fat/low-sucrose diet, KD, western diets, fructose diet | KD and a low-fat/low-sucrose diet feeding decreased TP compared with a high-sucrose diet; this effect correlated with sugar intake and was independent of excess adiposity or insulin resistance. | [262] | |
| Uterus cancer | HeLa | nu/nu mice | 3:1 | SD, KD | KD showed an increase in TP and decreased survival because HeLa tumors actively consumed KB as an energy source. | [248] |
| Patient-derived xenograft | nude mice | 6:1 | SD ± PI3K inhibitors, KD ± PI3K inhibitors | KD had no effect on TP alone but an enhanced antitumor effect of KD+PI3Kinhibitors compared with CD + PI3K. | [27] | |
| Acute myeloid leukemia | MLL-AF9 Ds-Red | C57BL/6 mice | 6:1 | SD ± PI3K inhibitors, KD ± PI3K inhibitors | KD alone decreased the survival → protumor effect, but enhanced survival in KD + PI3K inhibitors group compared with CD + PI3K inhibitor. | [27] |
| Bladder cancer | Patient-derived xenograft | nude mice | 6:1 | SD ± PI3K inhibitors, KD ± PI3K inhibitors | KD alone decreased TP, and with PI3K inhibitors had an additive antitumor effect because the efficacy of PI3K inhibition can be limited in the presence of insulin feedback and in KD reduced levels of phosphorylated insulin receptor, decreasing the levels of tumor proliferation, increasing apoptosis, and enhancing PI3K inhibitors activity | [27] |
| Component | Control, Standard Diet | Ketogenic Diet |
|---|---|---|
| Fat | 6.1 | 35.5 |
| Carbohydrate | 55.6 | 0.2 |
| Protein | 21.8 | 13.0 |
| Fiber | 3.8 | 14.8 |
| Ashes | 5.3 | 2.1 |
| Energy [kJ/g] | 15.8 | 15.4 |
| Ketogenic ratio | 0.08:1 | 2.7:1 |
| Cancer Type | Study Group Size (n) | Dietary Intervention (n) | Combined with Tumor Therapy (n) | Study Duration | Results of the Studies | Effect on QoL | Ref. |
|---|---|---|---|---|---|---|---|
| Glioblastoma | 20 | KD 60 g CHO/day | ST as RT, CT, or antiangiogenic treatment | 6+ weeks | PFS was observed in patient with stable ketosis (8); one with complete response and five with partial response. | Three patients stopped KD because they felt that KD decreased their QoL but there were no serious side effects. | [252] |
| 53 | KD 30–50 g CHO/day (5), CR-KD (1) | RT | 3–12 months | No tumor recurrence was observed on CR and KD patients after 12 months from RT. | Not specified. | [265] | |
| Glioblastoma and gliomatosis cerebri | 9 | KD 4:1 (5), CD (4) | ST | 2–31 months | This study shows the accumulation of Acn and AcAc in the brain in patients with brain tumors on KD; KD may have potential as a treatment given the metabolic changes. | Not specified, but some of the patients stopped KD because they felt that KD decreased their QoL. | [266] |
| Glioma | 172 | modified KD 70% kcal fat, 20 g CHO/day (6) | ST | 3 months | KD appears to be a good adjuvant therapy; no data on TP. | Self-reported good QoL, but two patients reporting constipation, which was resolved through dietary changes. | [267] |
| 8 | MAD 20 g CHO/day (8) | ST | 2–24 months | KD increased the control of seizure in brain tumor patients. Increase in the survival rate. | Not specified. | [268] | |
| 13 6 (newly diagnosed) 7 (recurrent) | KD + MCT + Metformin 850 | RT (60Gy) for recurrent RT (35Gy) for newly | 6 weeks (recurrent) 2 weeks (newly diagnosed) | Increase in survival rate. Synergistic interaction between radiation therapy and KD. Metformin has in-vitro anti-cancer activity through AMPK activation and mTOR inhibition. | Five patients stopped KD + MCT. Metformin 850 mg three-times daily was poorly tolerated. | [269] | |
| Invasive rectal cancer | 359 | KD ≥ 40% kcal fat and <100 g/day glycemic load (48) | RT (18/48) | not specified | KD significantly reduced risk of cancer-specific deaths compared with NSAIDs cancer-specific death, smoking, or other diseases. | Not specified. | [270] |
| Breast cancer | 1 | strict KD + high dose vitamin D3, not further specified (1) | No | 3 weeks | KD + vitamin D caused changes in biological markers of breast cancer (negativization of HER2 expression and increased expression of PgR). | Not specified. | [271] |
| 29 (on KD) 30 (on SD) | KD, SD | RT | 5–6 weeks | The increase of TP was less pronounced in the KD group compared to the SD group (KD enhanced the RT effect). | Improvements in emotional functioning, social functioning, sleep quality, and side effects. | [272] | |
| Triple-negative breast cancer (TNBC) | 1 | KD | MSCT + HT + BHO | 6 months | KD effective in treating advanced TNBC clinical, radiological, and pathological complete with good response. | Self-reported increase in QoL. | [273] |
| Lung and pancreatic cancer | 9 7 (lung cancer) 2 (pancreatic cancer) | KD 4:1 90% of calories from fat, 8% from protein and 2% from carbohydrate with a 4:1 ratio of fat to combined protein and carbohydrate | ST | 5–6 weeks | Four patients were unable to comply with the diet and withdrew, two completed the study and one was withdrawn due to a dose-limiting toxicity. Two pancreatic patients—one completed the study, and the other was withdrawn due to a dose-limiting toxicity. | Difficulty for adults to comply with a ketogenic diet while receiving concurrent RT and CT. | [249] |
| Non-small cell lung cancer | 44 | mild KD, avoidance of high CHO foods | MSCT + HT + HBOT | 6 months | KD with CT and HBOT improved survival outcomes and increased treatment response rates by targeting several corresponding metabolic pathways and weaknesses of cancer cells. | Not specified. | [274] |
| Ovarian and endometrial cancer | 73 | KD 70% kcal fat, 30% kcal CHO + protein (37), CD (36) | ST | 3 months | Increase in ketone bodies with increase in physical function. KD group without chemotherapy reported significant increase in energy at 12 weeks follow up, no data on TP. | KD does not diminish QoL; KD may even increase QoL. | [275,276] |
| Head and neck cancer | 12 | KD, not further specified | not specified | 7 days | Increase in body weight. No data on TP. | Not specified. | [277] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Talib, W.H.; Mahmod, A.I.; Kamal, A.; Rashid, H.M.; Alashqar, A.M.D.; Khater, S.; Jamal, D.; Waly, M. Ketogenic Diet in Cancer Prevention and Therapy: Molecular Targets and Therapeutic Opportunities. Curr. Issues Mol. Biol. 2021, 43, 558-589. https://doi.org/10.3390/cimb43020042
Talib WH, Mahmod AI, Kamal A, Rashid HM, Alashqar AMD, Khater S, Jamal D, Waly M. Ketogenic Diet in Cancer Prevention and Therapy: Molecular Targets and Therapeutic Opportunities. Current Issues in Molecular Biology. 2021; 43(2):558-589. https://doi.org/10.3390/cimb43020042
Chicago/Turabian StyleTalib, Wamidh H., Asma Ismail Mahmod, Ayah Kamal, Hasan M. Rashid, Aya M. D. Alashqar, Samar Khater, Duaa Jamal, and Mostafa Waly. 2021. "Ketogenic Diet in Cancer Prevention and Therapy: Molecular Targets and Therapeutic Opportunities" Current Issues in Molecular Biology 43, no. 2: 558-589. https://doi.org/10.3390/cimb43020042
APA StyleTalib, W. H., Mahmod, A. I., Kamal, A., Rashid, H. M., Alashqar, A. M. D., Khater, S., Jamal, D., & Waly, M. (2021). Ketogenic Diet in Cancer Prevention and Therapy: Molecular Targets and Therapeutic Opportunities. Current Issues in Molecular Biology, 43(2), 558-589. https://doi.org/10.3390/cimb43020042