
Microwave-Assisted Synthesis of a MK2 Inhibitor by Suzuki-Miyaura Coupling for Study in Werner Syndrome Cells

 , and
, and
Abstract
:
{kind=link}
{kind=link}
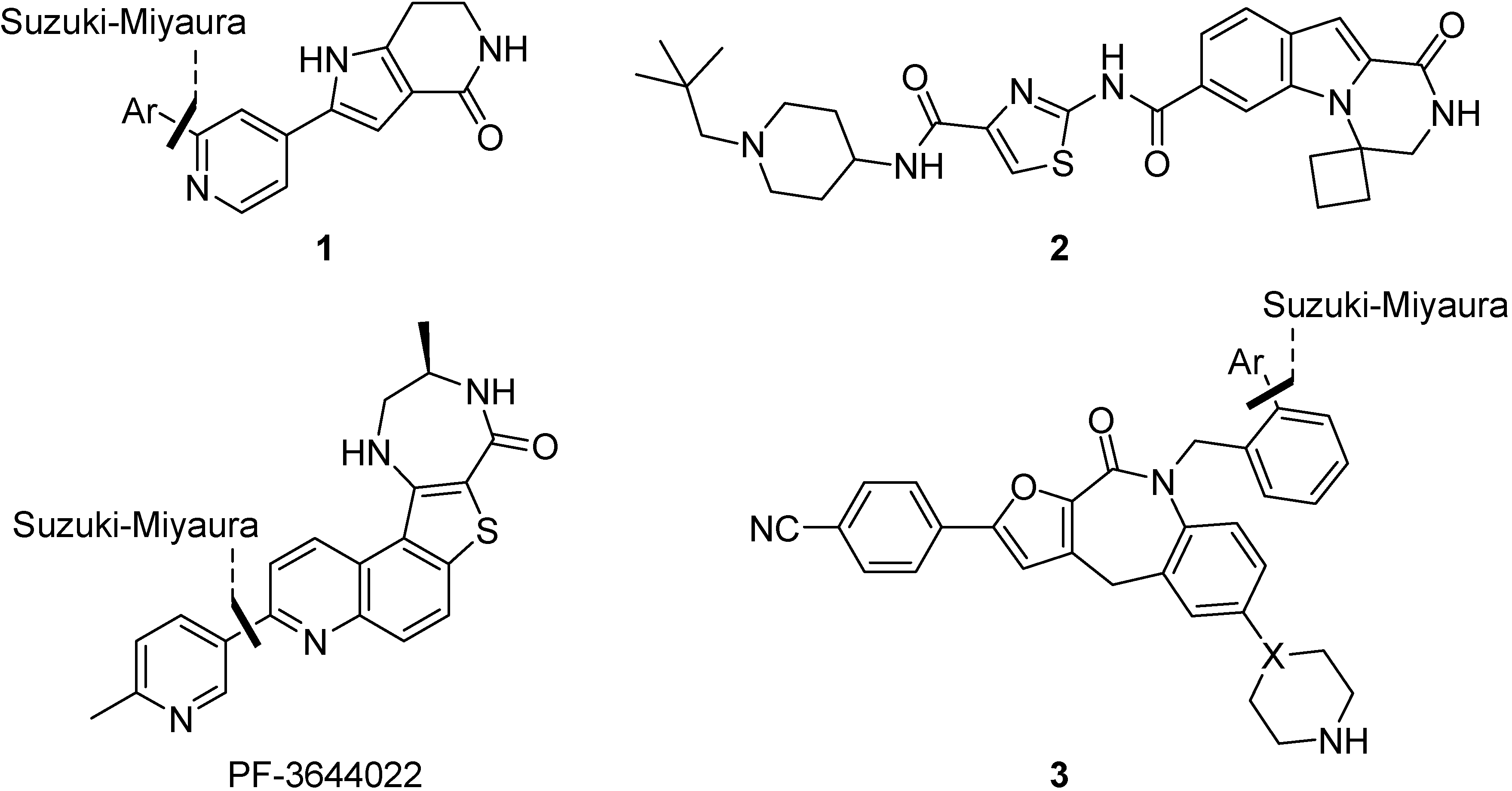{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
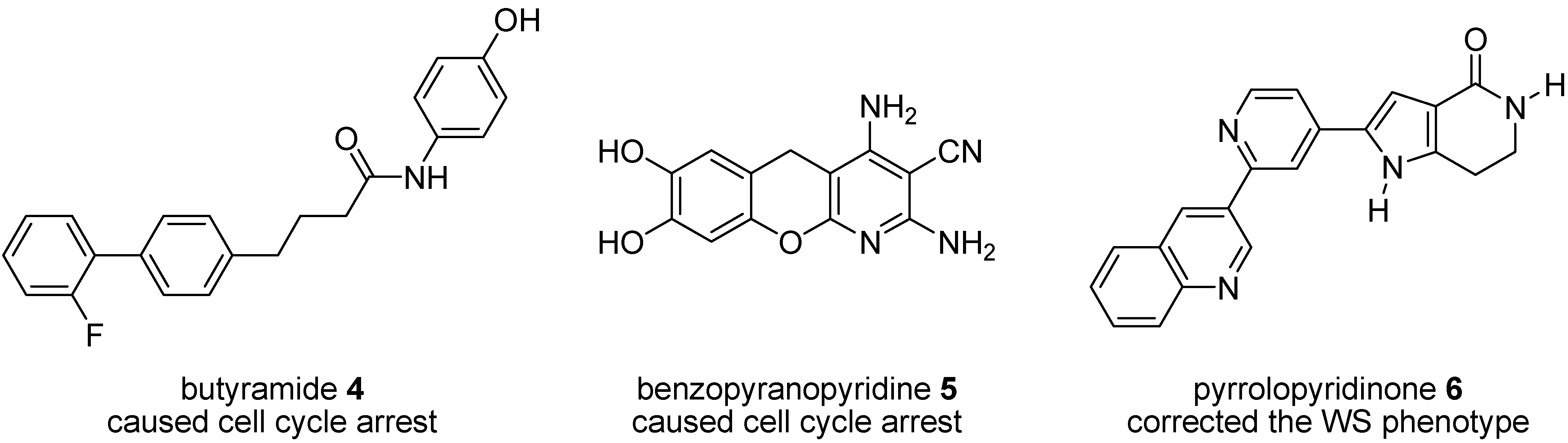
1.1. Studies of P38 Inhibitors in WS Cells
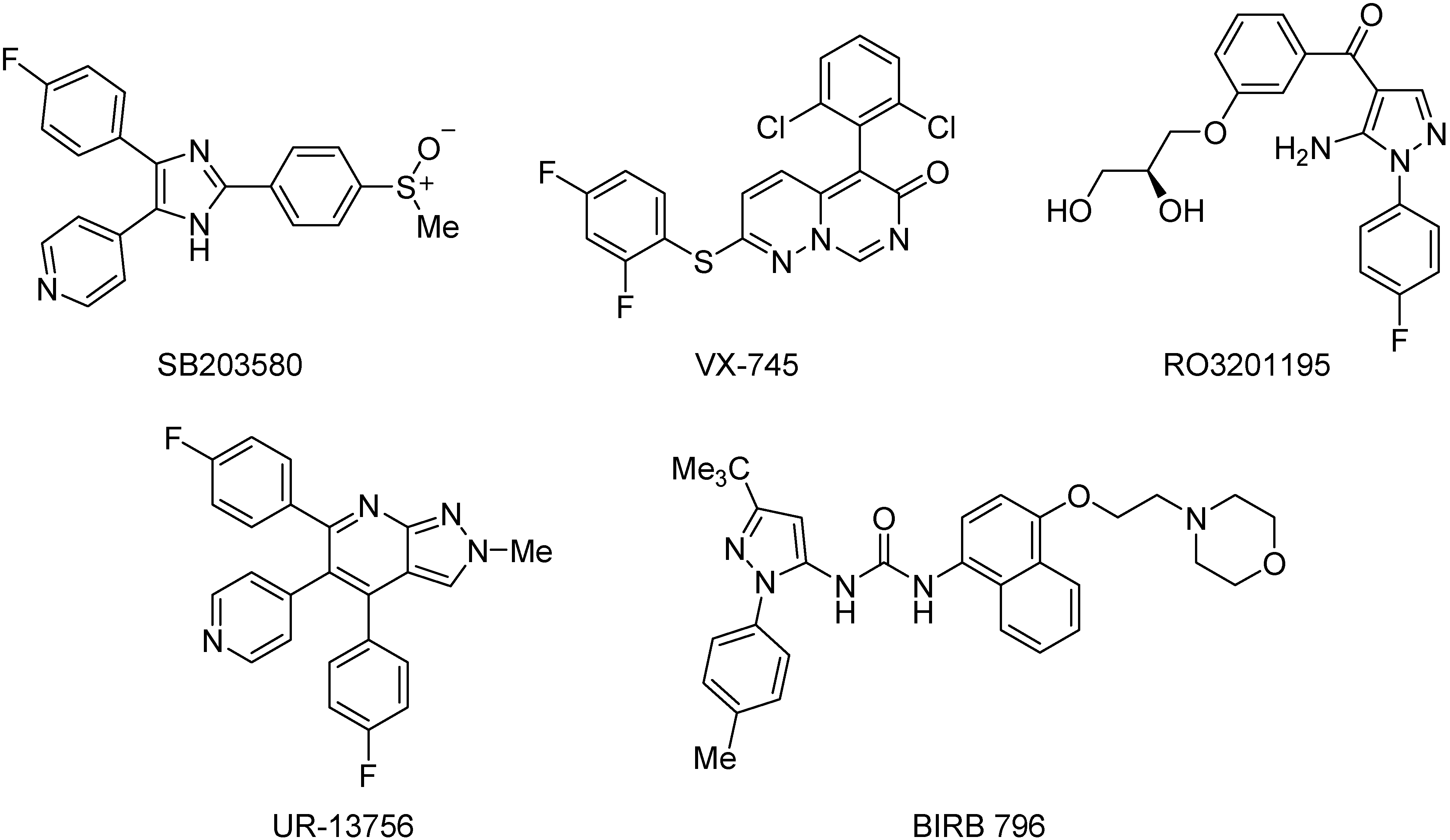
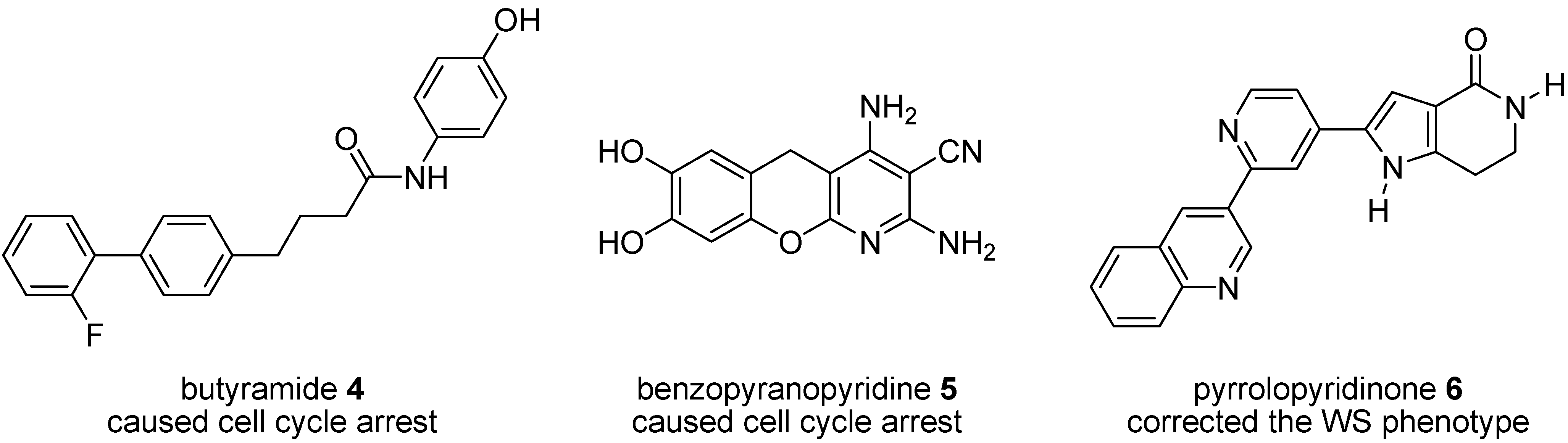
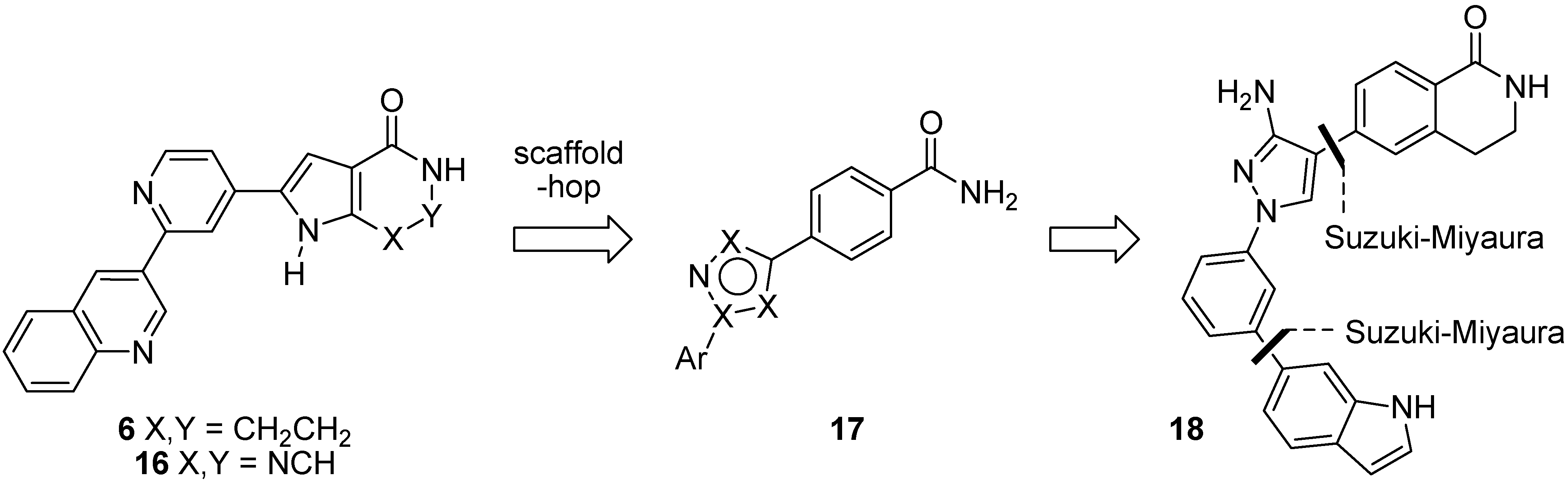
1.2. Synthesis and Properties of MK2 Inhibitors
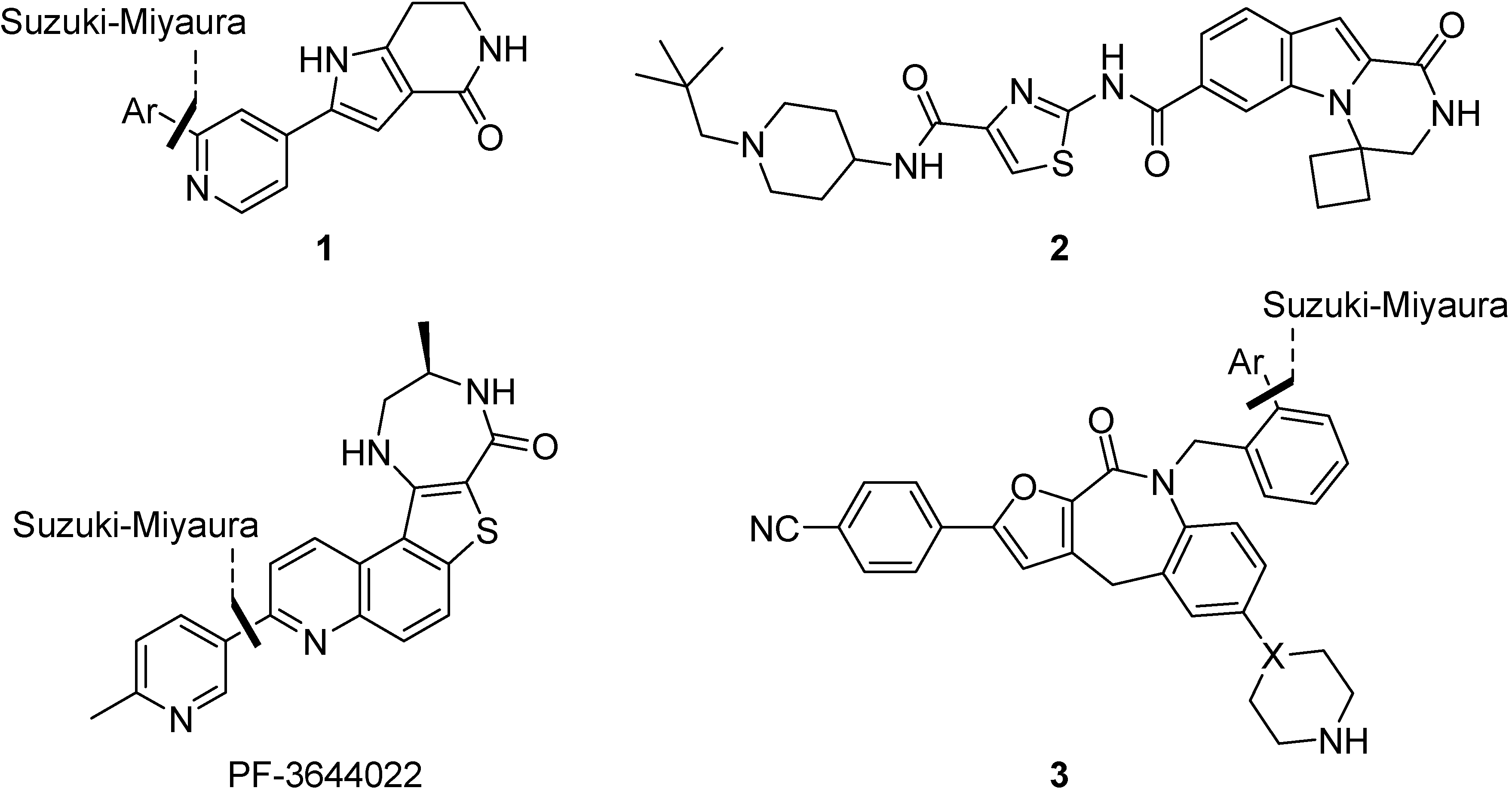
2. Results and Discussion

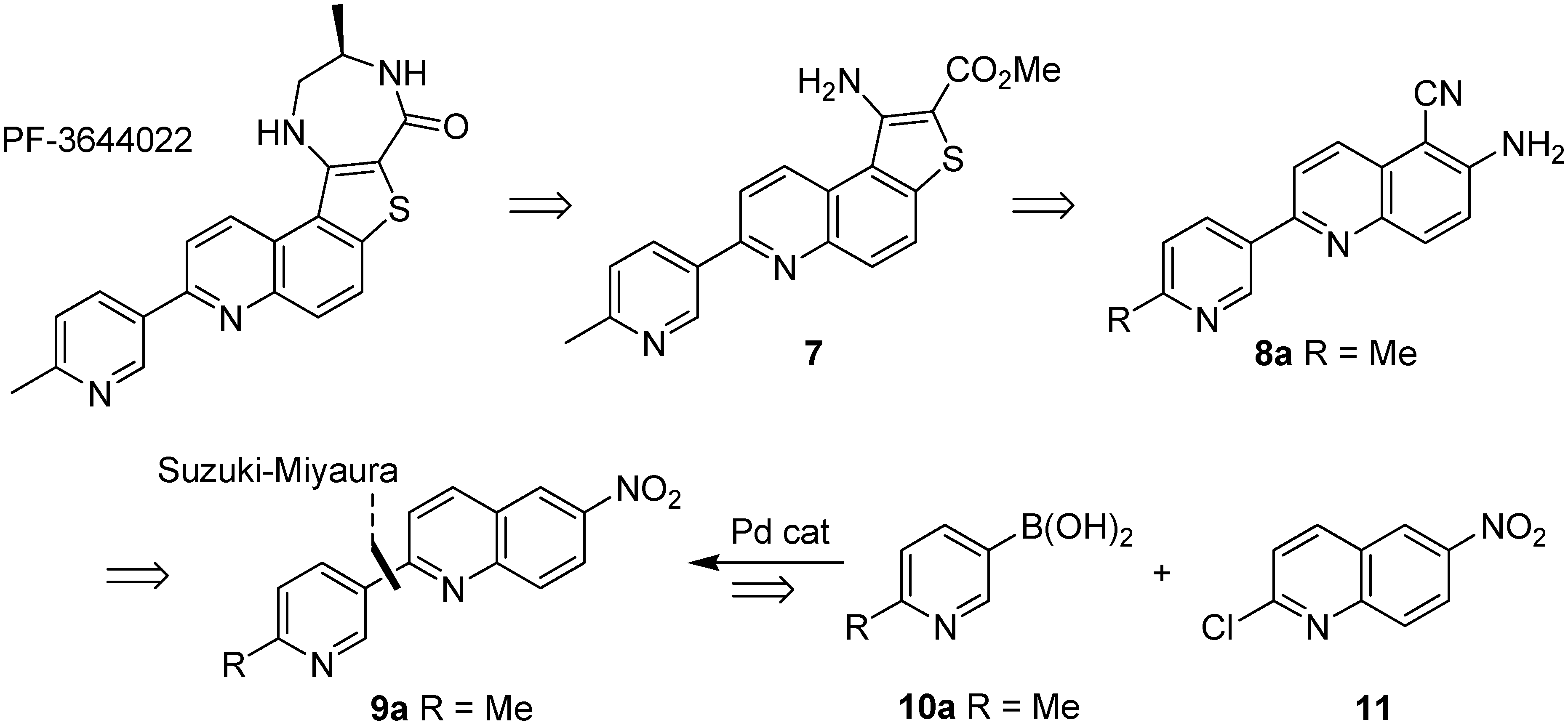
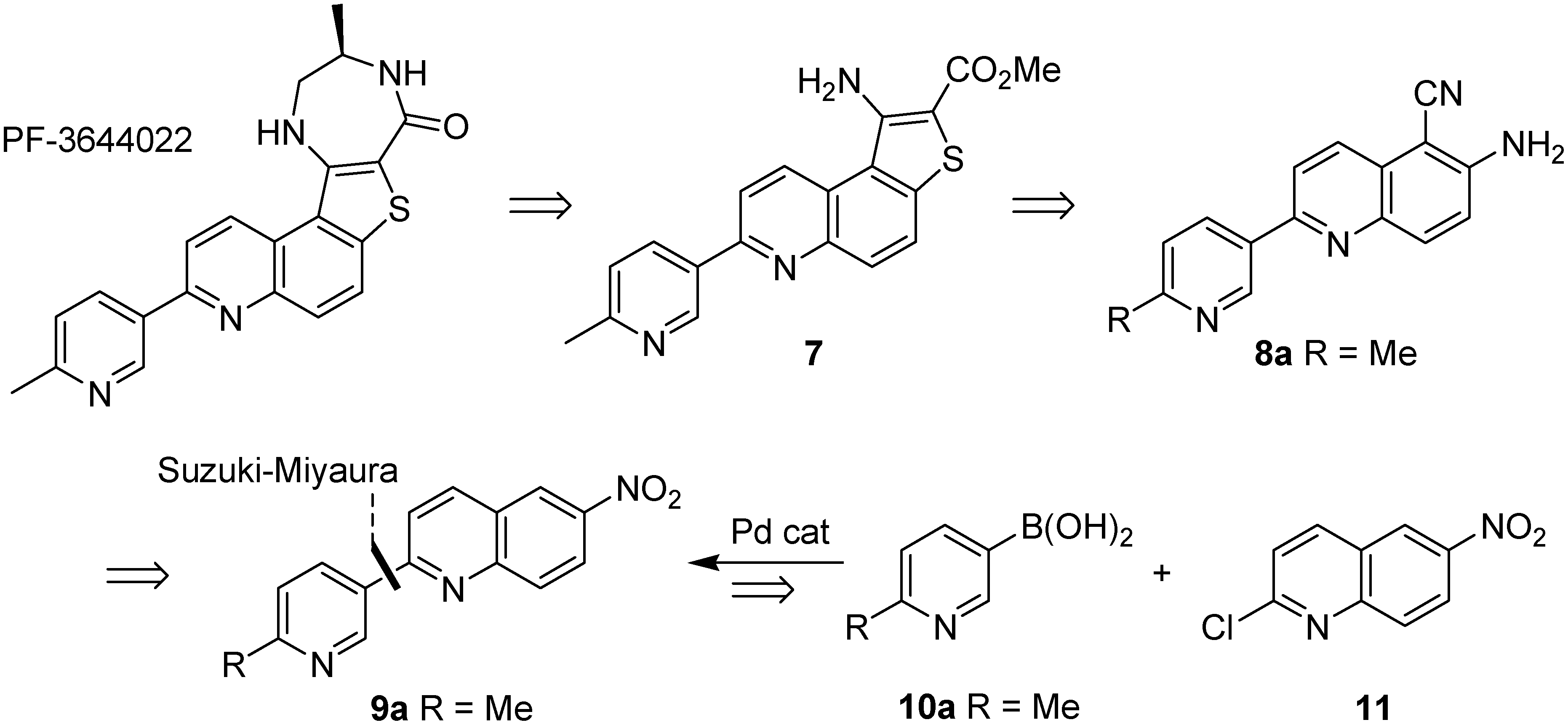
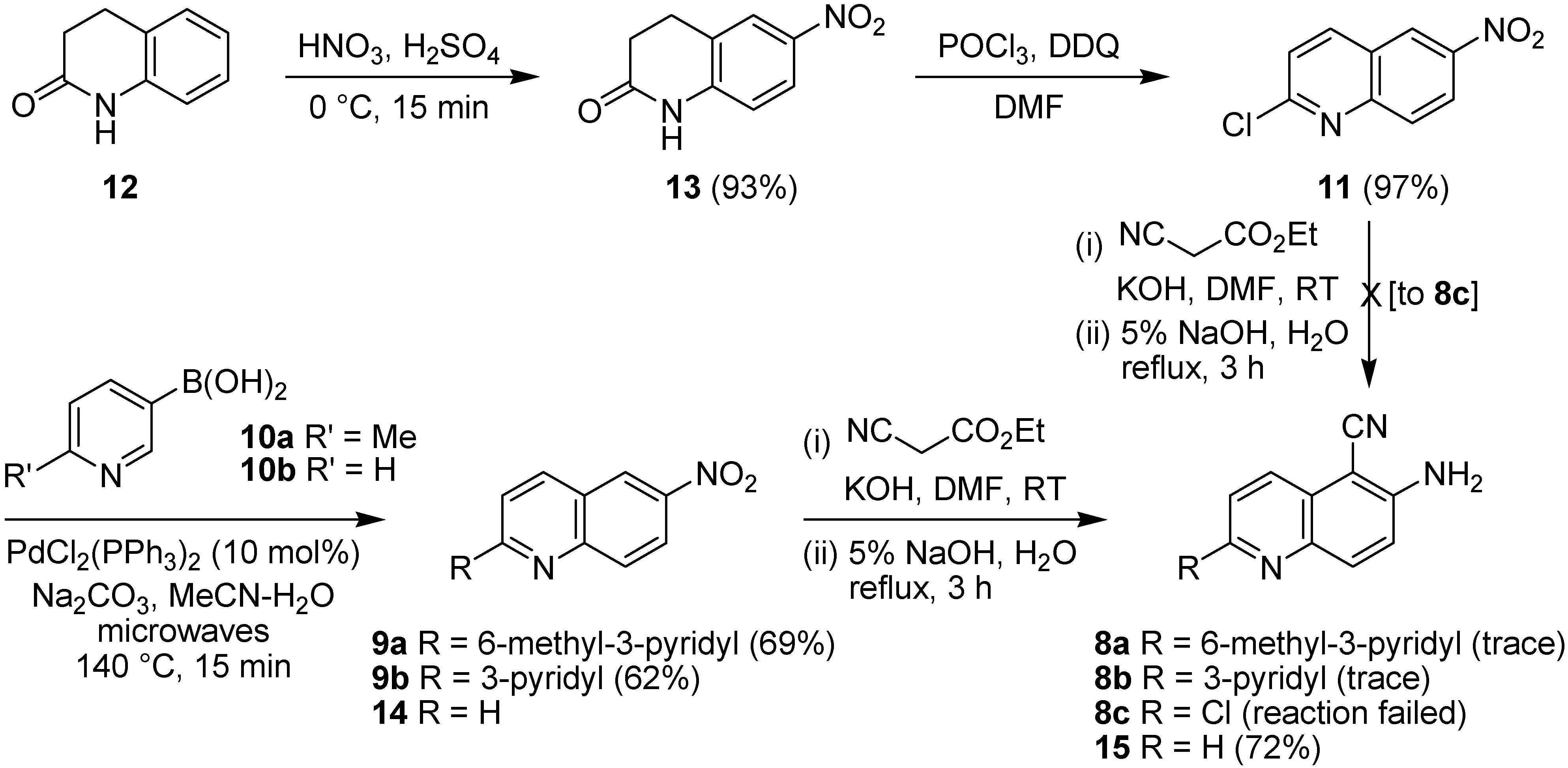
2.1. Early-Stage Suzuki-Miyaura Cross-Coupling Reactions for the Synthesis of PF-3644022
2.2. Suzuki-Miyaura Cross-Coupling Reactions for the Synthesis of (Indolyl)phenylpyrazole MK2 Inhibitor 18
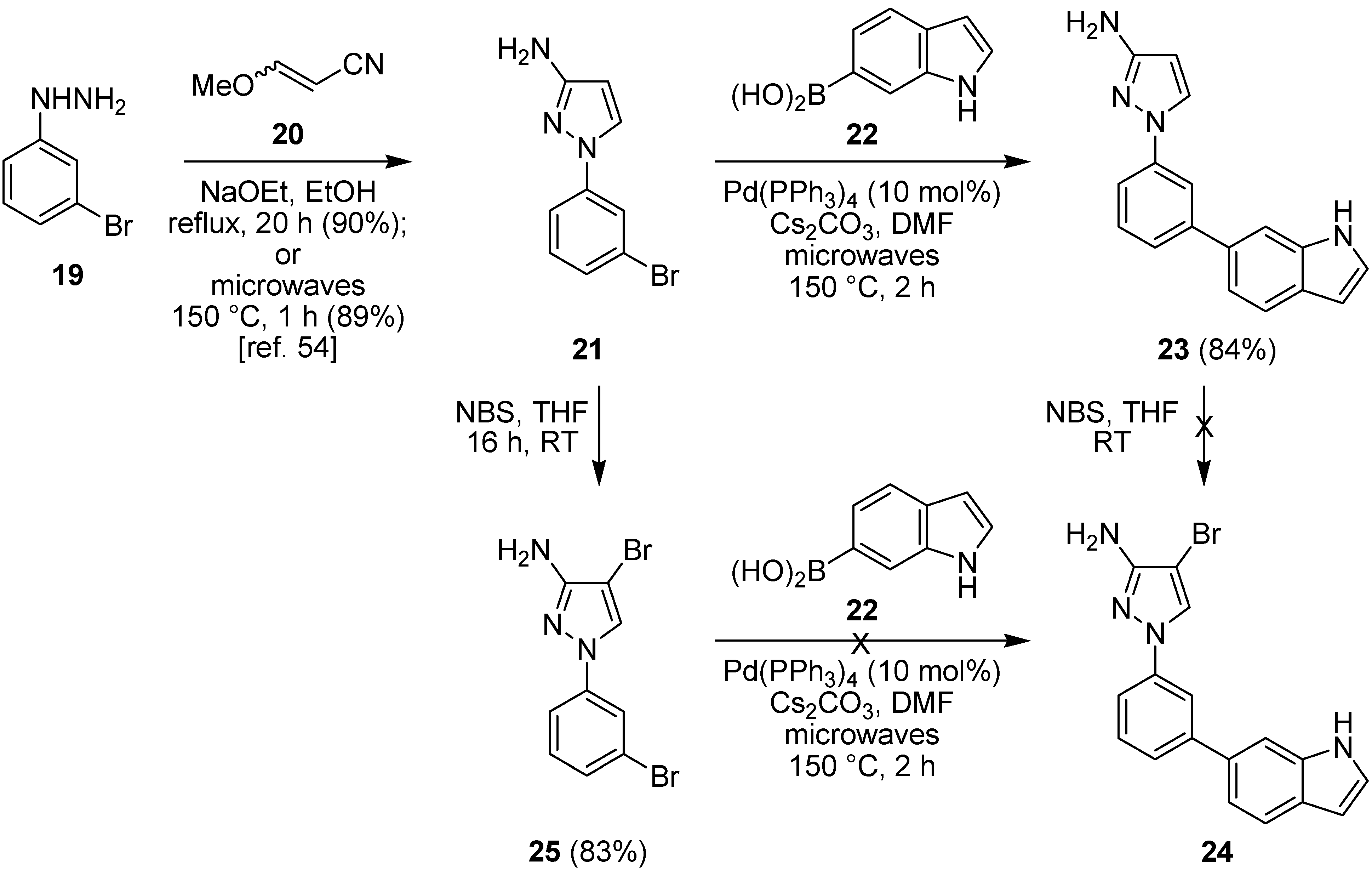
2.3. Suzuki-Miyaura Cross-Coupling Reactions for the Synthesis of Aminopyrazole MK2 Inhibitor 26
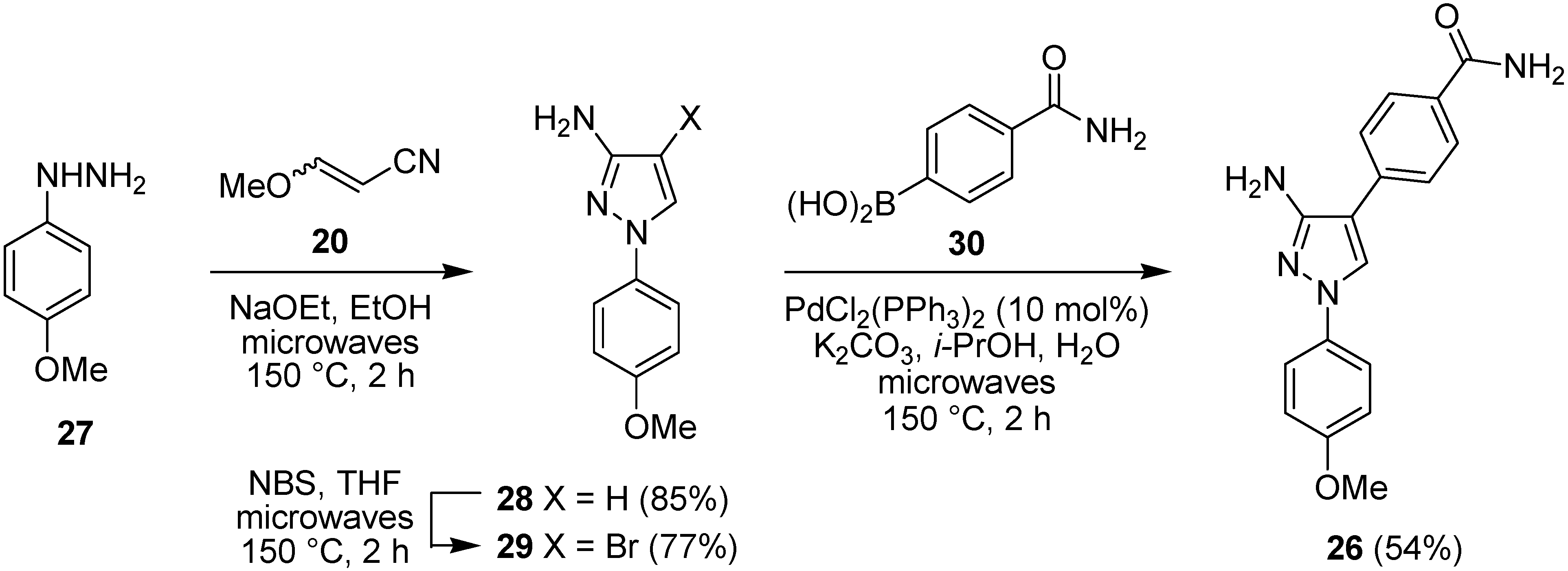
2.4. Biological Evaluation of Aminopyrazole MK2 Inhibitor 26 in WS Cells
2.5. The Role of MK2 in the WS Cell Phenotype and Its Modulation Using Chemical Inhibitors
3. Experimental Section
3.1. Synthetic Materials and Methods
3.2. General Synthetic Procedures
General Procedure for Suzuki-Miyaura Cross-Coupling
3.3. Synthetic Experimental Procedures
3.3.1. 3,4-Dihydro-6-nitroquinolin-2(1H)-one (13)
3.3.2. 2-Chloro-6-nitroquinoline (11)
3.3.3. 2-(6-Methylpyridin-3-yl)-6-nitroquinoline (9a)
3.3.4. 2-(Pyridin-3-yl)-6-nitroquinoline (9b)
3.3.5. 6-Amino-5-cyanoquinoline (15)
3.3.6. 3-Amino-1-(3-bromophenyl)-1H-pyrazole (21)
3.3.7. 3-Amino-1-[3-(1H-indol-6-yl)phenyl]-1H-pyrazole (23)
3.3.8. 3-Amino-4-bromo-1-(3-bromophenyl)-1H-pyrazole (25)
3.3.9. 3-Amino-1-(4-methoxyphenyl)-1H-pyrazole (28)
3.3.10. 3-Amino-4-bromo-1-(4-methoxyphenyl)-1H-pyrazole (29)
3.3.11. 3-Amino-4-(4-aminocarbonylphenyl)-1-(4-methoxyphenyl)-1H-pyrazole (26)
3.4. Biological Procedures
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Martin, G.M.; Oshima, J.; Gray, M.D.; Poot, M. What geriatricians should know about the werner syndrome. J. Am. Geriatr. Soc. 1999, 47, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Kipling, D.; Davis, T.; Ostler, E.L.; Faragher, R.G. What can progeroid syndromes tell us about human aging? Science 2004, 305, 1426–1431. [Google Scholar] [CrossRef] [PubMed]
- Werner, O. On Cataract Associated in Conjunction with Scleroderma. Ph.D. Thesis, Kiel University, Schmidt and Klaunig, Kiel, Germany, 1904. [Google Scholar]
- Thannhauser, S.J. Werner’s syndrome (progeria of the adult) and rothmund’s syndrome: Two types of closely related heredofamilial atrophic dermatosis with juvenile cataracts and endocrine features. A critcal study of five new cases. Ann. Int. Med. 1945, 23, 559–625. [Google Scholar]
- Davis, T.; Kipling, D. Werner syndrome as an example of inflamm-aging: Possible therapeutic opportunities for a progeroid syndrome? Rejuvenation Res. 2006, 9, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Yokote, K.; Hara, K.; Mori, S.; Kadowaki, T.; Saito, Y.; Goto, M. Dysadipocytokinemia in werner syndrome and its recovery by treatment with pioglitazone. Diabetes Care 2004, 27, 2562–2563. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; de Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Vasto, S.; Candore, G.; Balistreri, C.R.; Caruso, M.; Colonna-Romano, G.; Grimaldi, M.P.; Listi, F.; Nuzzo, D.; Lio, D.; Caruso, C. Inflammatory networks in ageing, age-related diseases and longevity. Mech. Ageing Dev. 2007, 128, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Lopez, A.M.; Jackson, D.A.; Iborra, F.; Cox, L.S. Asymmetry of DNA replication fork progression in Werner’s syndrome. Aging Cell 2002, 1, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Meyn, M.S. Chromosome instability syndromes: Lessons for carcinogenesis. Curr. Top. Microbiol. Immunol. 1997, 221, 71–148. [Google Scholar] [PubMed]
- Pagano, G.; Zatterale, A.; Degan, P.; d'Ischia, M.; Kelly, F.J.; Pallardo, F.V.; Kodama, S. Multiple involvement of oxidative stress in werner syndrome phenotype. Biogerontology 2005, 6, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.; Baird, D.M.; Haughton, M.F.; Jones, C.J.; Kipling, D. Prevention of accelerated cell aging in werner syndrome using a p38 mitogen-activated protein kinase inhibitor. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Huot, J.; Houle, F.; Marceau, F.; Landry, J. Oxidative stress-induced actin reorganization mediated by the p38 mitogen-activated protein kinase/heat shock protein 27 pathway in vascular endothelial cells. Circ. Res. 1997, 80, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.Y.; Mercer, S.E.; Ewton, D.Z.; Yan, Z.; Jin, K.; Friedman, E. The stress-activated protein kinases p38 alpha and JNK1 stabilize p21(cip1) by phosphorylation. J. Biol. Chem. 2002, 277, 29792–29802. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, J.X.; Liao, R.; Deng, Q.; Zhou, J.J.; Huang, S.; Sun, P. Sequential activation of the MEK-extracellular signal-regulated kinase and MKK3/6-p38 mitogen-activated protein kinase pathways mediates oncogenic ras-induced premature senescence. Mol. Cell. Biol. 2002, 22, 3389–3403. [Google Scholar] [CrossRef] [PubMed]
- Bagley, M.C.; Davis, T.; Murziani, P.G.S.; Widdowson, C.S.; Kipling, D. Use of p38 MAPK inhibitors for the treatment of werner syndrome. Pharmaceuticals 2010, 3, 1842–1872. [Google Scholar] [CrossRef]
- Davis, T.; Haughton, M.F.; Jones, C.J.; Kipling, D. Prevention of accelerated cell aging in the werner syndrome. Ann. N. Y. Acad. Sci. 2006, 1067, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.; Bachler, M.A.; Wyllie, F.S.; Bagley, M.C.; Kipling, D. Evaluating the role of p38 map kinase in growth of werner syndrome fibroblasts. Ann. N. Y. Acad. Sci. 2010, 1197, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Bagley, M.C.; Davis, T.; Dix, M.C.; Rokicki, M.J.; Kipling, D. Rapid synthesis of VX-745: p38 map kinase inhibition in werner syndrome cells. Bioorg. Med. Chem. Lett. 2007, 17, 5107–5110. [Google Scholar] [CrossRef] [PubMed]
- Bagley, M.C.; Davis, T.; Dix, M.C.; Fusillo, V.; Pigeaux, M.; Rokicki, M.J.; Kipling, D. Microwave-assisted ullmann C-S bond formation: Synthesis of the p38alpha MAPK clinical candidate VX-745. J. Org. Chem. 2009, 74, 8336–8342. [Google Scholar] [CrossRef] [PubMed]
- Bagley, M.C.; Davis, T.; Dix, M.C.; Fusillo, V.; Pigeaux, M.; Rokicki, M.J.; Kipling, D. Gramme-scale synthesis of the p38α MAPK inhibitor VX-745 for pre-clinical studies into werner syndrome. Future Med. Chem. 2010, 2, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Bagley, M.C.; Davis, T.; Dix, M.C.; Murziani, P.G.; Rokicki, M.J.; Kipling, D. Microwave-assisted synthesis of 5-aminopyrazol-4-yl ketones and the p38(MAPK) inhibitor RO3201195 for study in werner syndrome cells. Bioorg. Med. Chem. Lett. 2008, 18, 3745–3748. [Google Scholar] [CrossRef] [PubMed]
- Bagley, M.C.; Davis, T.; Dix, M.C.; Murziani, P.G.; Rokicki, M.J.; Kipling, D. Microwave-assisted synthesis of a pyrazolyl ketone library for evaluation as p38 MAPK inhibitors in werner syndrome cells. Future Med. Chem. 2010, 2, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Bagley, M.C.; Davis, T.; Rokicki, M.J.; Widdowson, C.S.; kipling, D. Synthesis of the highly selective p38 MAPK inhibitor UR-13756 for possible therapeutic use in werner syndrome. Future Med. Chem. 2010, 2, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Bagley, M.C.; Davis, T.; Dix, M.C.; Widdowson, C.S.; Kipling, D. Microwave-assisted synthesis of N-pyrazole ureas and the p38alpha inhibitor BIRB 796 for study into accelerated cell ageing. Org. Biomol. Chem. 2006, 4, 4158–4164. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.; Wyllie, F.S.; Rokicki, M.J.; Bagley, M.C.; Kipling, D. The role of cellular senescence in werner syndrome: Toward therapeutic intervention in human premature aging. Ann. N. Y. Acad. Sci. 2007, 1100, 455–469. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C. Inhibition of p38: Has the fat lady sung? Arthritis Rheum. 2009, 60, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Bagley, M.C.; Baashen, M.; Dwyer, J.; Milbeo, P.; Kipling, D.; Davis, T. Microwave-assisted synthesis of inhibitors of MK2 for targeting p38 map kinase signalling in werner syndrome cells. In Microwaves in Drug Discovery and Development: Recent Advances; Spencer, J., Bagley, M., Eds.; Future Sci Ltd.: London, UK, 2014; pp. 86–104. [Google Scholar]
- Dean, J.L.; Sully, G.; Clark, A.R.; Saklatvala, J. The involvement of AU-rich element-binding proteins in p38 mitogen-activated protein kinase pathway-mediated mrna stabilisation. Cell Signal. 2004, 16, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Manke, I.A.; Nguyen, A.; Lim, D.; Stewart, M.Q.; Elia, A.E.; Yaffe, M.B. Mapkap kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol. Cell 2005, 17, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Hegen, M.; Gaestel, M.; Nickerson-Nutter, C.L.; Lin, L.L.; Telliez, J.B. MAPKAP kinase 2-deficient mice are resistant to collagen-induced arthritis. J. Immunol. 2006, 177, 1913–1917. [Google Scholar] [CrossRef] [PubMed]
- Ronkina, N.; Kotlyarov, A.; Gaestel, M. MK2 and MK3—A pair of isoenzymes? Front. Biosci. 2008, 13, 5511–5521. [Google Scholar] [CrossRef] [PubMed]
- Duraisamy, S.; Bajpai, M.; Bughani, U.; Dastidar, S.G.; Ray, A.; Chopra, P. MK2: A novel molecular target for anti-inflammatory therapy. Expert Opin. Ther. Targets 2008, 12, 921–936. [Google Scholar] [CrossRef] [PubMed]
- Schlapbach, A.; Huppertz, C. Low-molecular-weight MK2 inhibitors: A tough nut to crack! Future Med. Chem. 2009, 1, 1243–1257. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.R.; Meyers, M.J.; Vernier, W.F.; Mahoney, M.W.; Kurumbail, R.G.; Caspers, N.; Poda, G.I.; Schindler, J.F.; Reitz, D.B.; Mourey, R.J. Pyrrolopyridine inhibitors of mitogen-activated protein kinase-activated protein kinase 2 (MK-2). J. Med. Chem. 2007, 50, 2647–2654. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, D.R.; Choi, Y.; Cogan, D.; Corson, M.; DeLeon, R.; Gao, A.; Gruenbaum, L.; Hao, M.H.; Joseph, D.; Kashem, M.A.; et al. Pyrazinoindolone inhibitors of MAPKAP-K2. Bioorg. Med. Chem. Lett. 2008, 18, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Mourey, R.J.; Burnette, B.L.; Brustkern, S.J.; Daniels, J.S.; Hirsch, J.L.; Hood, W.F.; Meyers, M.J.; Mnich, S.J.; Pierce, B.S.; Saabye, M.J.; et al. A benzothiophene inhibitor of mitogen-activated protein kinase-activated protein kinase 2 inhibits tumor necrosis factor alpha production and has oral anti-inflammatory efficacy in acute and chronic models of inflammation. J. Pharmacol. Exp. Ther. 2010, 333, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Palani, A.; Huang, X.; Sofolarides, M.; Zhou, W.; Chen, X.; Aslanian, R.; Guo, Z.; Fossetta, J.; Tian, F.; et al. Conformation constraint of anilides enabling the discovery of tricyclic lactams as potent MK2 non-ATP competitive inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 3262–3266. [Google Scholar] [CrossRef] [PubMed]
- Daniels, J.S.; Lai, Y.; South, S.; Chiang, P.C.; Walker, D.; Feng, B.; Mireles, R.; Whiteley, L.O.; McKenzie, J.W.; Stevens, J.; et al. Inhibition of hepatobiliary transporters by a novel kinase inhibitor contributes to hepatotoxicity in beagle dogs. Drug Metab. Lett. 2013, 7, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Miyaura, N.; Lahiri, K.; Suzuki, A. A new stereospecific cross-coupling by the palladium catalyzed reaction of 1-alkenylboranes with 1-alkenyl or 1-alkynyl halides. Tetrahedron Lett. 1979, 36, 3437–3440. [Google Scholar] [CrossRef]
- Kotha, S.; Lahiri, K.; Kashinath, D. Recent applications of the suzuki-miyaura cross-coupling reaction in organic synthesis. Tetrahedron 2002, 58, 9633–9695. [Google Scholar] [CrossRef]
- Suzuki, A. Recent advances in the cross-coupling reactions of organoboron derivatives with organic electrophiles 1995–1998. J. Organomet. Chem. 1999, 576, 147–168. [Google Scholar] [CrossRef]
- Roughley, S.D.; Jordan, A.M. The medicinal chemist’s toolbox: An analysis of reactions used in the pursuit of drug candidates. J. Med. Chem. 2011, 54, 3451–3479. [Google Scholar] [CrossRef] [PubMed]
- Mavandadi, F.; Pilotti, Å. The impact of microwave-assisted organic synthesis in drug discovery. Drug Discov. Today 2006, 11, 165–174. [Google Scholar] [CrossRef]
- Davis, T.; Rokicki, M.J.; Bagley, M.C.; Kipling, D. The effect of small-molecule inhibition of MAPKAPK2 on cell ageing phenotypes of fibroblasts from human werner syndrome. Chem. Cent. J. 2013, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.; Bagley, M.C.; Dix, M.C.; Murziani, P.G.; Rokicki, M.J.; Widdowson, C.S.; Zayed, J.M.; Bachler, M.A.; Kipling, D. Synthesis and in vivo activity of MK2 and MK2 substrate-selective p38α(MAPK) inhibitors in werner syndrome cells. Bioorg. Med. Chem. Lett. 2007, 17, 6832–6835. [Google Scholar] [CrossRef] [PubMed]
- Davidson, W.; Frego, L.; Peet, G.W.; Kroe, R.R.; Labadia, M.E.; Lukas, S.M.; Snow, R.J.; Jakes, S.; Grygon, C.A.; Pargellis, C.; et al. Discovery and characterization of a substrate selective p38alpha inhibitor. Biochemistry 2004, 43, 11658–11671. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.R.; Hegde, S.; Reinhard, E.; Gomez, L.; Vernier, W.F.; Lee, L.; Liu, S.; Sambandam, A.; Snider, P.A.; Masih, L. Aminocyanopyridine inhibitors of mitogen activated protein kinase-activated protein kinase 2 (MK-2). Bioorg. Med. Chem. Lett. 2005, 15, 1587–1590. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.R.; Meyers, M.J.; Kurumbail, R.G.; Caspers, N.; Poda, G.I.; Long, S.A.; Pierce, B.S.; Mahoney, M.W.; Mourey, R.J. Benzothiophene inhibitors of MK2. Part 1: Structure-activity relationships, assessments of selectivity and cellular potency. Bioorg. Med. Chem. Lett. 2009, 19, 4878–4881. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.R.; Meyers, M.J.; Kurumbail, R.G.; Caspers, N.; Poda, G.I.; Long, S.A.; Pierce, B.S.; Mahoney, M.W.; Mourey, R.J.; Parikh, M.D. Benzothiophene inhibitors of MK2. Part 2: Improvements in kinase selectivity and cell potency. Bioorg. Med. Chem. Lett. 2009, 19, 4882–4884. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Lee, B.C.; Jun, J.-G.; Chi, D.Y. A new efficient synthesis of 6-nitroquipazine. Heterocycles 1998, 48, 2637–2641. [Google Scholar]
- Tomioka, Y.; Ohkubo, K.; Yamazaki, M. Studies on aromatic nitro compounds V. A simple one-pot preparation of o-aminoarylnitriles from some aromatic nitro compounds. Chem. Pharm. Bull. 1985, 33, 1360–1366. [Google Scholar] [CrossRef] [Green Version]
- Velcicky, J.; Feifel, R.; Hawtin, S.; Heng, R.; Huppertz, C.; Koch, G.; Kroemer, M.; Moebitz, H.; Revesz, L.; Scheufler, C.; et al. Novel 3-aminopyrazole inhibitors of MK-2 discovered by scaffold hopping strategy. Bioorg. Med. Chem. Lett. 2010, 20, 1293–1297. [Google Scholar] [CrossRef] [PubMed]
- Bagley, M.C.; Baashen, M.; Paddock, V.L.; Kipling, D.; Davis, T. Microwave-assisted synthesis of 3- and 5-aminopyrazole, pyrazolo[3,4-d]pyrimidine, pyrazolo[3,4-b]pyridine and pyrazolo[3,4-b]quinolin-4-one scaffolds for mapk inhibition. Tetrahedron 2013, 68, 8429–8438. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [PubMed]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Tomioka, Y.; Mochiike, A.; Himeno, J.; Yamazaki, M. Studies on aromatic nitro compounds. I. Reaction of 6-nitroquinoline with active methylene compounds in the presence of bases. Chem. Pharm. Bull. 1981, 29, 1286–1291. [Google Scholar] [CrossRef] [Green Version]
- Bagley, M.C.; Dwyer, J.E.; Molina, M.D.B.; Rand, A.; Rand, H.L.; Tomkinson, N.C.O. Microwave-assisted synthesis of 3-aminobenzo[b]thiophene scaffolds for the preparation of kinase inhibitors. Org. Biomol. Chem. 2015. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bagley, M.C.; Baashen, M.; Chuckowree, I.; Dwyer, J.E.; Kipling, D.; Davis, T. Microwave-Assisted Synthesis of a MK2 Inhibitor by Suzuki-Miyaura Coupling for Study in Werner Syndrome Cells. Pharmaceuticals 2015, 8, 257-276. https://doi.org/10.3390/ph8020257
Bagley MC, Baashen M, Chuckowree I, Dwyer JE, Kipling D, Davis T. Microwave-Assisted Synthesis of a MK2 Inhibitor by Suzuki-Miyaura Coupling for Study in Werner Syndrome Cells. Pharmaceuticals. 2015; 8(2):257-276. https://doi.org/10.3390/ph8020257
Chicago/Turabian StyleBagley, Mark C., Mohammed Baashen, Irina Chuckowree, Jessica E. Dwyer, David Kipling, and Terence Davis. 2015. "Microwave-Assisted Synthesis of a MK2 Inhibitor by Suzuki-Miyaura Coupling for Study in Werner Syndrome Cells" Pharmaceuticals 8, no. 2: 257-276. https://doi.org/10.3390/ph8020257
APA StyleBagley, M. C., Baashen, M., Chuckowree, I., Dwyer, J. E., Kipling, D., & Davis, T. (2015). Microwave-Assisted Synthesis of a MK2 Inhibitor by Suzuki-Miyaura Coupling for Study in Werner Syndrome Cells. Pharmaceuticals, 8(2), 257-276. https://doi.org/10.3390/ph8020257