Role of Microglial M1/M2 Polarization in Relapse and Remission of Psychiatric Disorders and Diseases

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
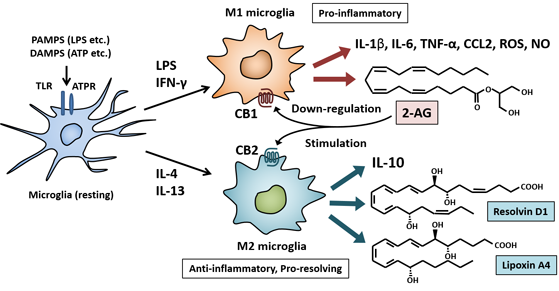
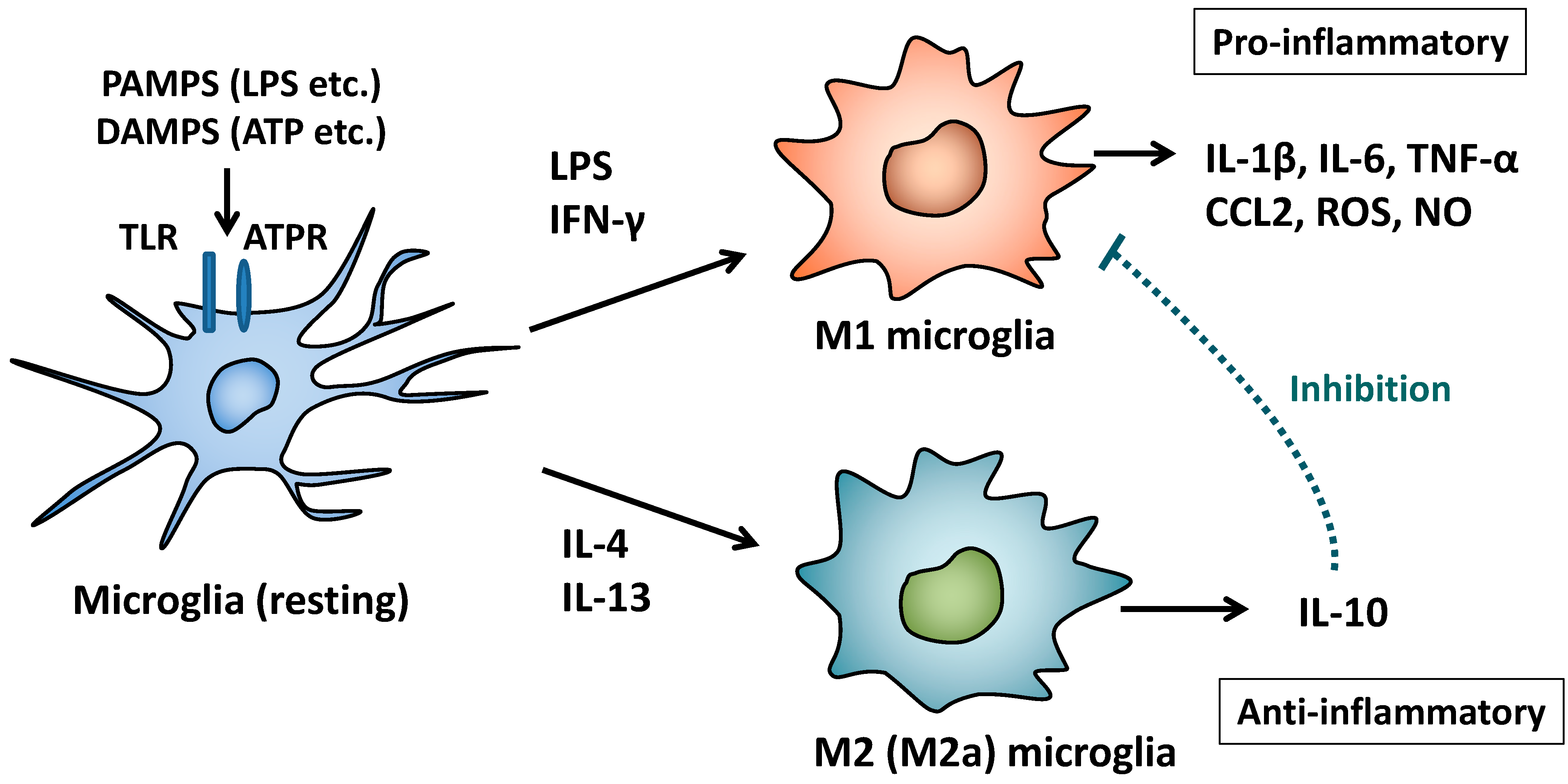
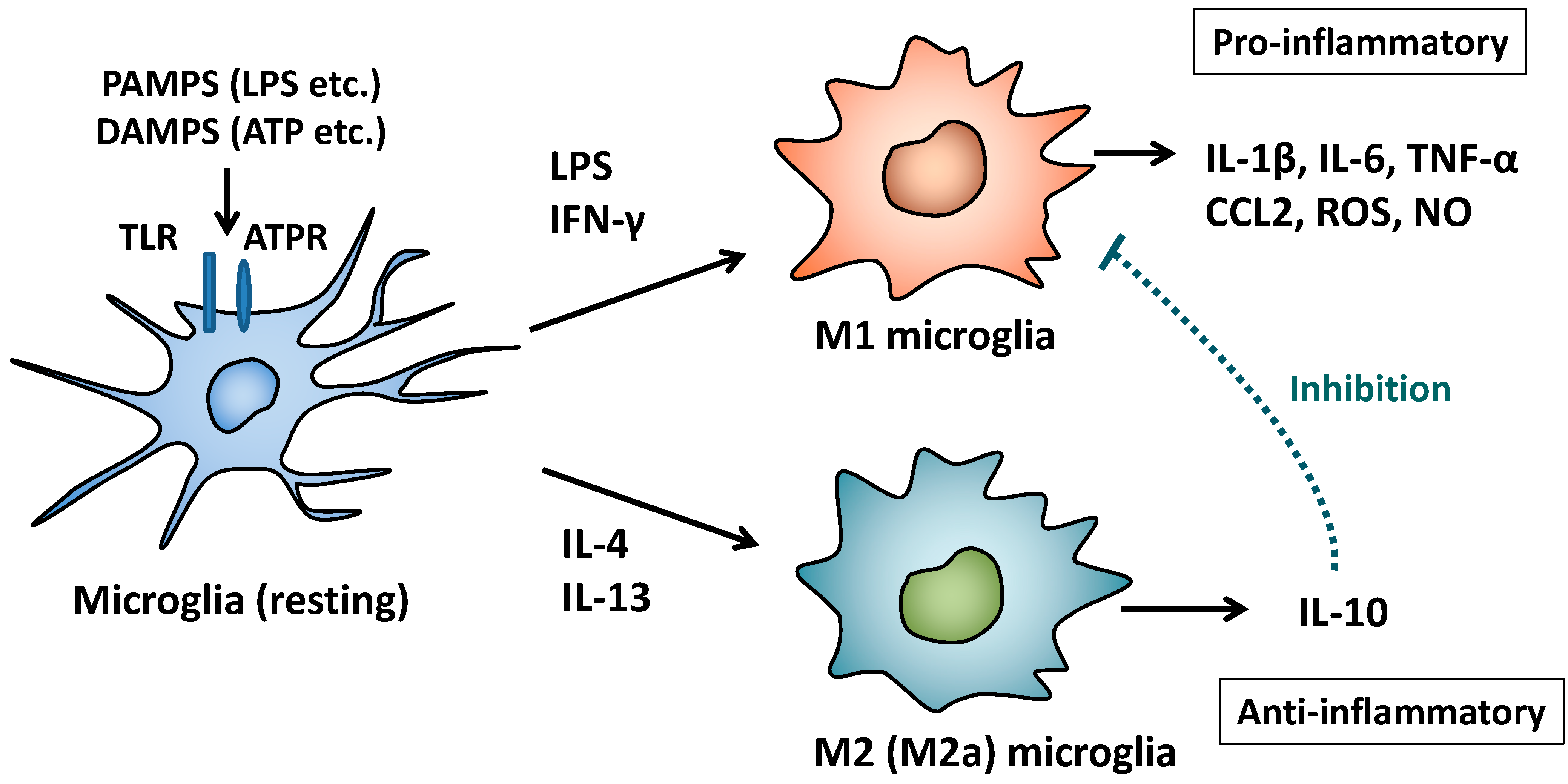
2. M1/M2 Polarization of Macrophages and Microglia
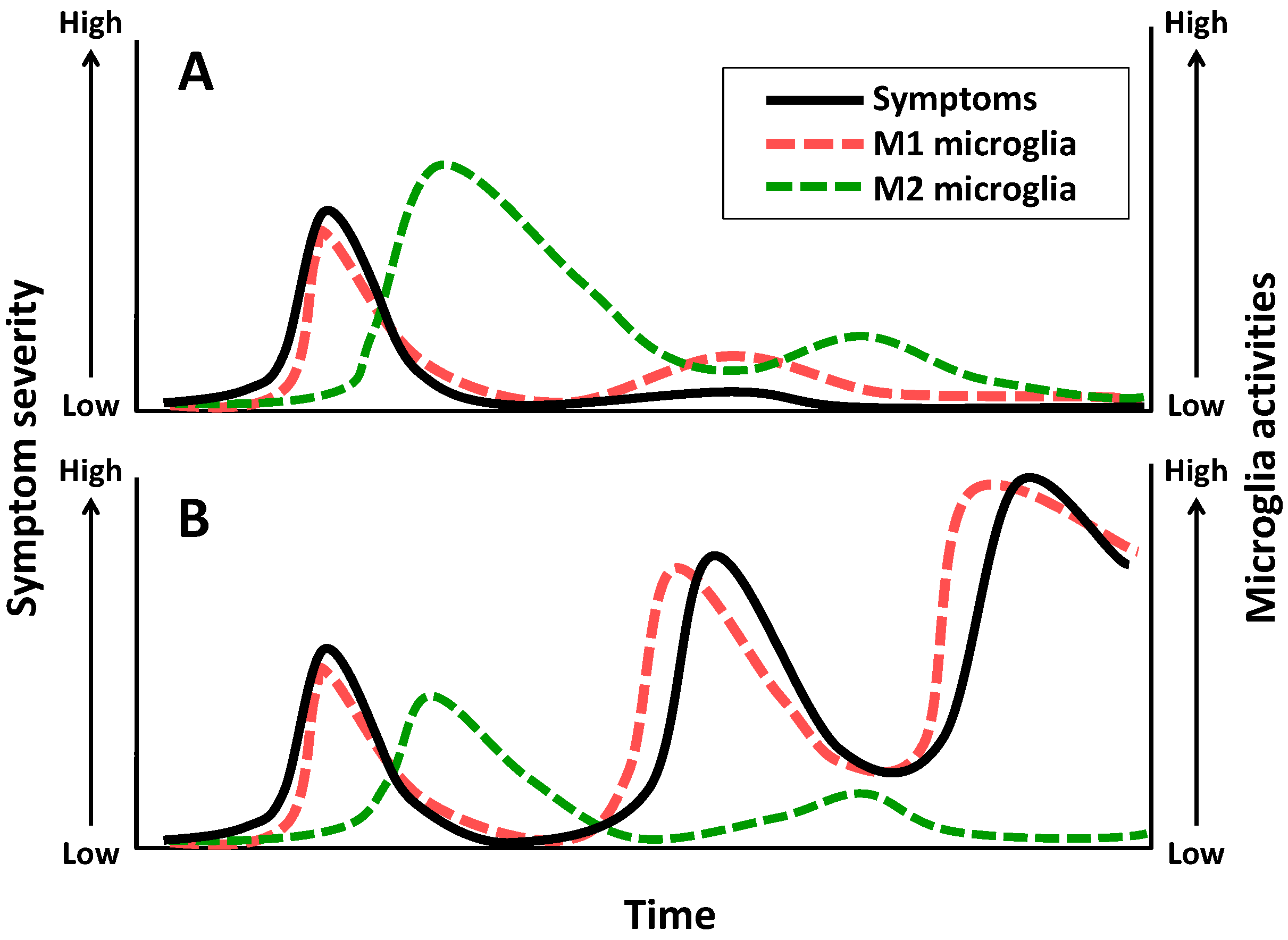
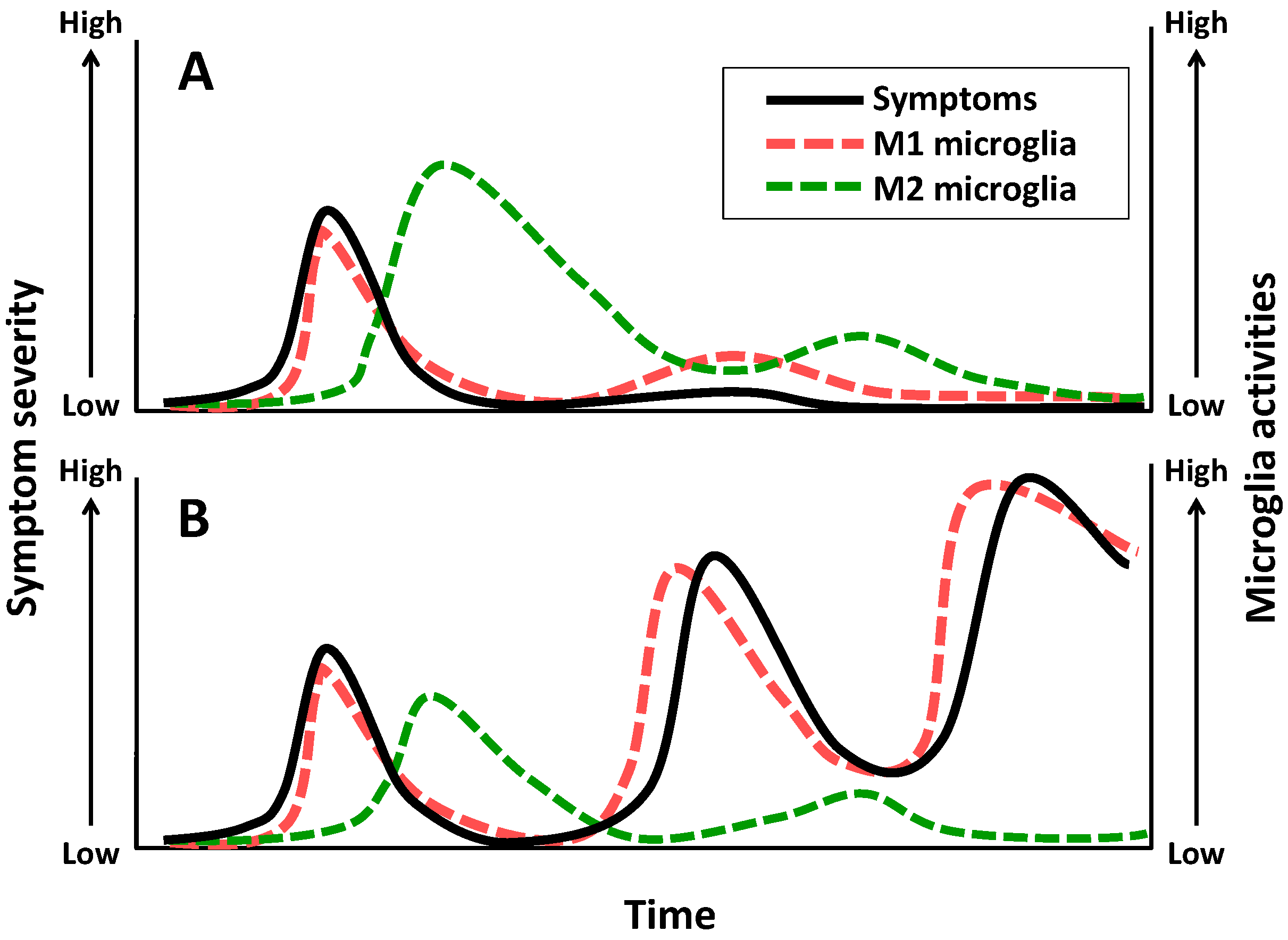
3. Schizophrenia
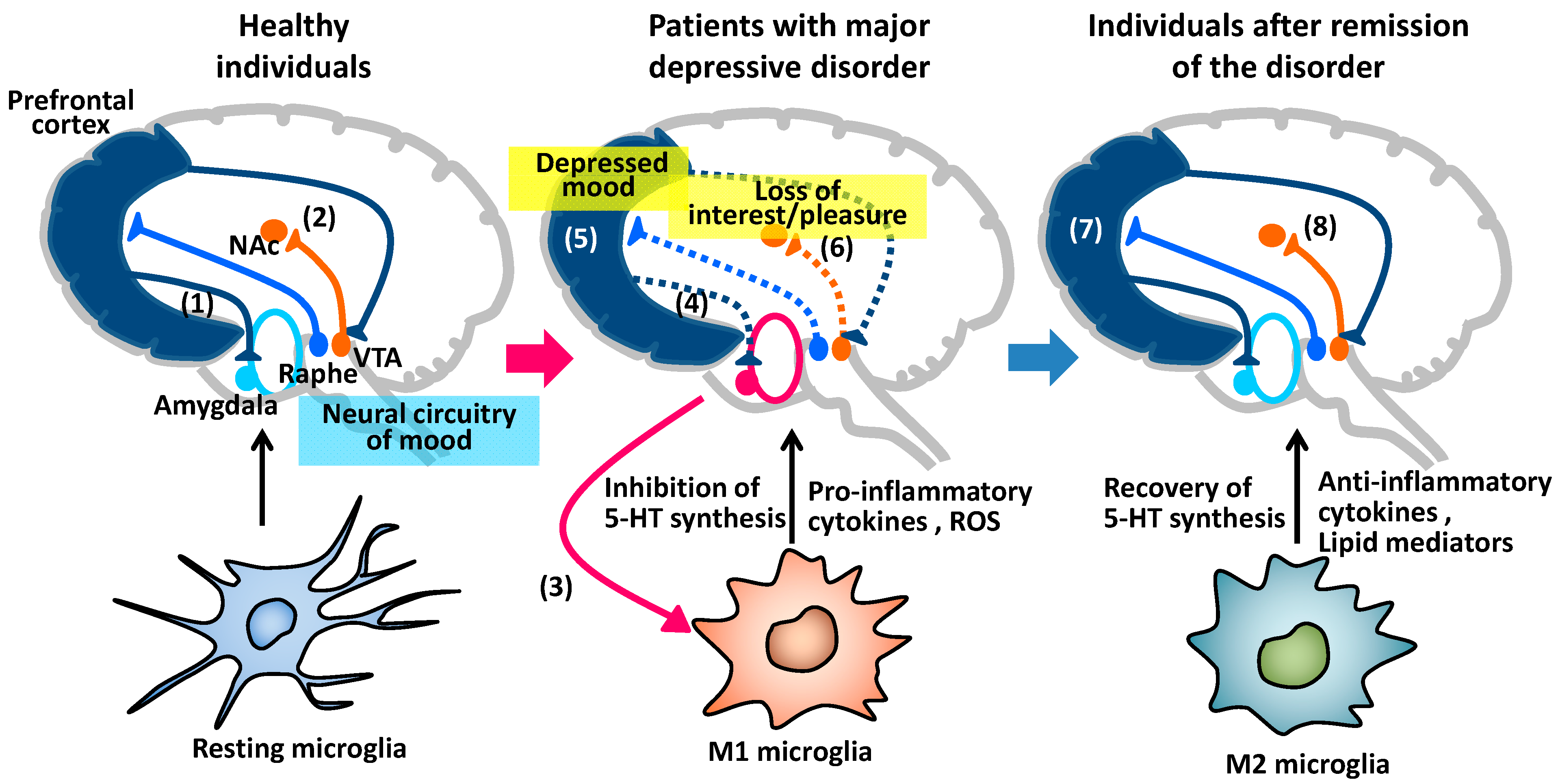
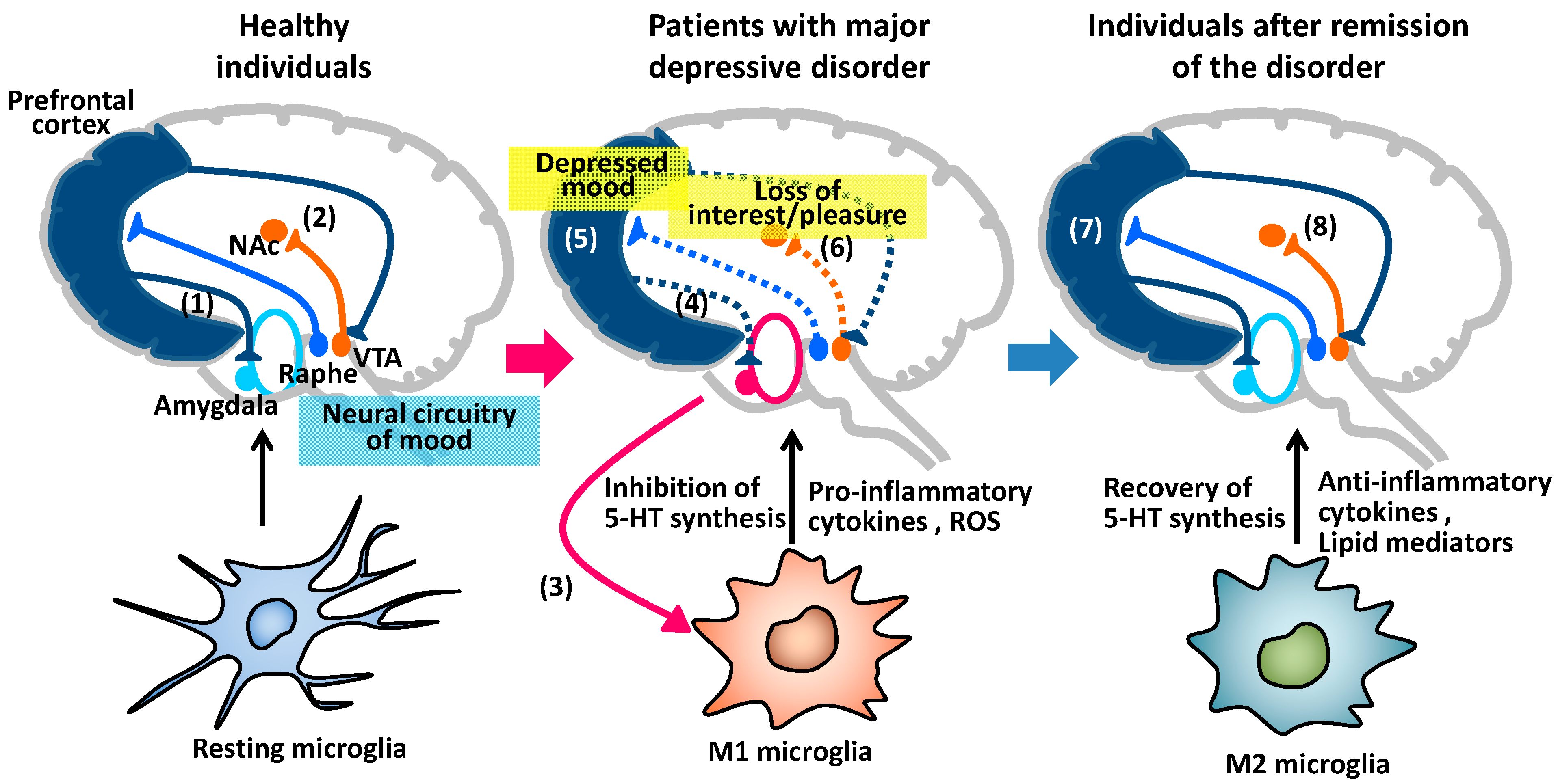
4. Major Depressive Disorder
5. Vascular Depression
6. Chronic Pain
7. Molecules to Skew M2 Polarization of Microglia
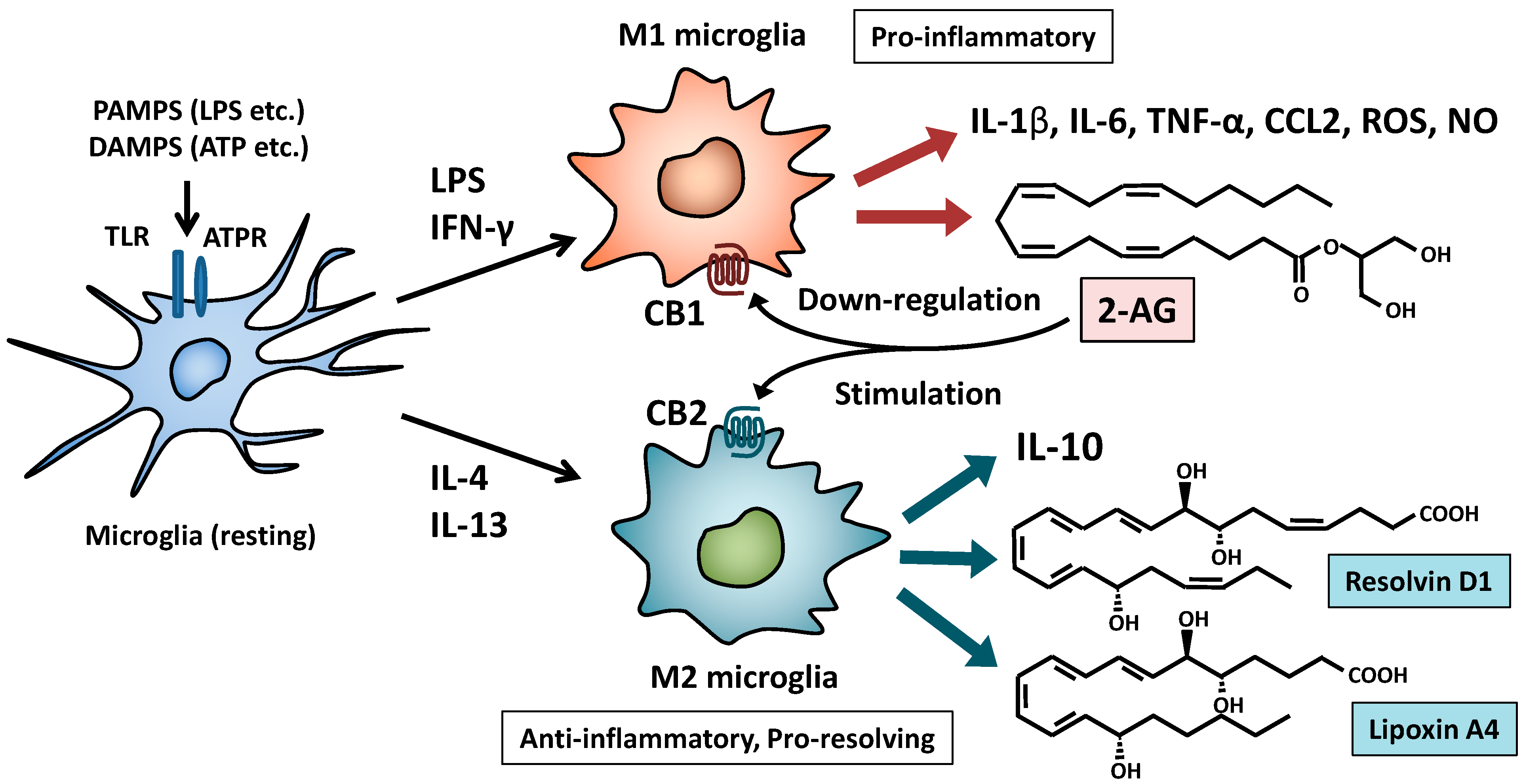
7.1. Endocannabinoids and Cannabinoid Receptors
7.2. Anti-Inflammatory and Pro-Resolving Lipid Mediators

8. Conclusions
Author contributions
Conflicts of Interest
References
- Dutta, R.; Trapp, B.D. Pathogenesis of axonal and neuronal damage in multiple sclerosis. Nerology 2007, 68, S22–S31. [Google Scholar] [CrossRef]
- Lasmann, H.; Bruck, W.; Lucchinetti, C.F. The immunopathology of multiple sclerosis: an overview. Brain Pathol. 2007, 17, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Frohman, E.M.; Racke, M.K.; Raine, C.S. Multiple sclerosis—The plaque and its pathogenesis. N. Engl. J. Med. 2006, 354, 942–955. [Google Scholar] [CrossRef] [PubMed]
- Pelfrey, C.M.; Tranquill, L.R.; Vogt, A.B.; McFarland, H.F. T cell response to two immunedominant proteolipid protein (PLP) peptides in multiple sclerosis patients and healthy controls. Mut. Scler. 1996, 1, 270–278. [Google Scholar]
- McFarland, H.F.; Martin, R. Multiple sclerosis: A complicated picture of autoimmunity. Nat. Immunol. 2007, 8, 913–919. [Google Scholar] [CrossRef]
- Steinman, L. Mixed results with modulation of TH-17cells in human autoimmune diseases. Nat. Immunol. 2010, 11, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Steinman, L. A molecular trio in relapse and remission in multiple sclerosis. Nat. Rev. Immuno. 2009, 9, 440–447. [Google Scholar] [CrossRef]
- Mikiat, J.; Doubourdieu-Cassagno, N.; Deloire, M.S.; Vekrie, A.; Biran, M.; Raffard, G.; Brochet, B.; Canron, M.H.; Franconi, J.M.; Boiziau, C.; et al. Amelioration of clinical status by M2 activated monocyte administration. Mult. Scler. 2011, 17, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Stahl, S.M. Stahl’s Essential Psychopharmacology: Neuroscientific Basis and Practical Applications, 3rd ed.; Cambridge University Press: Cambridge, USA, 2008; pp. 247–666. [Google Scholar]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verhratskym, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Perry, V.H. Microgilal physiology: Unique stimuli, specialized resonses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef]
- Meyer, U.; Schwarz, M.J.; Müller, N. Inflammatory processes in schizophrenia: A promising neuroimmunological target for the treatment of negative/cognitive symptoms and beyond. Pharmacol. Ther. 2011, 132, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Saijo, K.; Glass, C.K. Microglial cell origin and phenotypes in health and disease. Nat Rev. Immunol. 2011, 11, 775–787. [Google Scholar] [CrossRef]
- Beumer, W.; Gibney, S.M.; Drexhage, R.C.; Pont-Lezica, L.; Doorduin, J.; Klein, H.C.; Steiner, J.; Connor, T.J.; Harkin, A.; Versnel, M.A.; et al. The immune theory of psychiatric diseases: a key role for activated microglia and circulating monocytes. J. Leukoc. Biol. 2012, 92, 959–975. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.; Brown, B.D.; Merrad, M. Studying the mononuclear phagocyte system in the molecular age. Nat. Rev. Immunol. 2011, 11, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Jung, S.; Littman, D.R. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 2003, 19, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Leym, K. Development of monocytes, macrophages, and dendritic cells. Science 2010, 327, 656–661. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis, and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Roca, H.; Varsos, Z.S.; Sud, S.; Craig, M.J.; Ying, C.; Pienta, K.J. CCL2 and interleukin-6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2-type macrophage polarization. J. Biol. Chem. 2009, 284, 34342–34354. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Natoli, G. Transcriptinal regulation of macrophage polarization enabling diversity with identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, K.; Suenobu, M.; Yoshida, A.; Koga, K.; Hyodo, A.; Ohtsuka, H.; Kuniyasu, A.; Tamamaki, N.; Sugimoto, Y.; Nakayama, H. Intracerebral microinjection of interleukin-4/interleukin-13 reduces β-amyloid accumulation in the ipsilateral side and improves cognitive deficits in young amyloid precursor protein 23 mice. Neuroscience 2012, 207, 243–260. [Google Scholar] [CrossRef] [PubMed]
- Mittelbronn, M. The M1/M2 immune polarization concept in microglia: A fair transfer? Neuroimmunol. Neuroinflamm. 2014, 1, 6–7. [Google Scholar] [CrossRef]
- Tandon, R.; Keshavan, M.S.; Nasrallah, H.A. Schizophrenia, “just the facts” what we know in 2008. 2. Epidemiology and etiology. Schizophr. Res. 2008, 102, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Couture, S.M.; Penn1, D.L.; Roberts, D.L. The functional significance of social cognition in schizophrenia: a review. Schizophr. Bull. 2006, 32, S44–S63. [Google Scholar] [CrossRef] [PubMed]
- Leucht, S.; Tardy, M.; Komossa, K.; Heres, S.; Kissling, W.; Salanti, G.; Davis, J.M. Antipsychotic drugs versus placebo for relapse prevention in schizophrenia: A systematic review and meta-analysis. Lancet 2012, 379, 2063–2071. [Google Scholar] [CrossRef] [PubMed]
- Hinterkeuser, S.; Schroder, W.; Hanger, G.; Seifert, G.; Blumcke, I.; Elger, C.E.; Schramm, J.; Steinhauser, C. Astrocytes in the hippocampus of patients with temporal lobe epilepsy display changes in potassium conductances. Eur. J. Neurosci. 2000, 12, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Seifert, G.; Carmignoto, G.; Steinhäuser, C. Astrocyte dysfunction in epilepsy. Brain Res. Rev. 2010, 63, 212–221. [Google Scholar] [CrossRef]
- Wetherington, J.; Serrano, G.; Dingleline, R. Astrocytes in the epileptic brain. Neuron 2008, 58, 168–178. [Google Scholar] [CrossRef] [PubMed]
- David, S.; Kroner, A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat. Rev. Neurosci. 2011, 12, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Borgwardt, S.J.; Riecher-Rössler, A.; Dazzan, P.; Chitnis, X.; Aston, J.; Drewe, M.; Gschwandtner, U.; Haller, S.; Pflüger, M.; Rechsteiner, E.; et al. Regional gray matter volume abnormalities in the At Risk Mental State. Biol. Psychiatry. 2007, 61, 1148–1156. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.C.K.; Di, X.; McAlonan, G.M.; Gong, Q. Brain anatomical abnormalities in high-risk individuals, first-episode, and chronic schizophrenia: An activation likelihood estimation meta-analysis of illness progression. Schizophr. Bull. 2011, 37, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Van Berckel, B.N.; Bossong, M.G.; Boellaard, R.; Kloet, R.; Schuitemaker, A.; Caspers, E.; Luurtsema, G.; Windhorst, A.D.; Cahn, W.; Lammertsma, A.A.; et al. Microglia activation in recent-onset schizophrenia: a quantitative (R)-[11C]PK11195 positron emission tomography study. Biol. Psychiatry 2008, 64, 820–822. [Google Scholar] [CrossRef] [PubMed]
- Doorduin, J.; de Vries, E.F.J.; Willemsen, A.T.M.; de Groot, J.C.; Dierckx, R.A.; Klein, H.C. Neuroinflammation in schizophrenia-related psychosis: a PET study. J. Nucl. Med. 2009, 50, 1801–1807. [Google Scholar] [CrossRef]
- Barak, V.; Barak, Y.; Levine, J.; Nisman, B.; Roisman, I. Changes in interleukin-1β and soluble interleukin-2 receptor levels in CSF and serum of schizophrenic patients. J. Basic. Clin. Physiol. Pharmacol. 1995, 6, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Theodoropoulou, S.; Spanakos, G.; Baxevanis, C.N.; Economou, M.; Gritzapis, A.D.; Papamichail, M.P.; Stefanis, C.N. Cytokine serum levels, autologous mixed lymphocyte reaction and surface marker analysis in never medicated and chronically medicated schizophrenic patients. Schizophr. Res. 2001, 47, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Zhou, D.F.; Zhang, P.Y.; Wu, G.Y.; Cao, L.Y.; Shen, Y.C. Elevated interleukin-2, interleukin-6 and interleukin-8 serum levels in neuroleptic-free schizophrenia: Association with psychopathology. Schizophr. Res. 2002, 57, 247–258. [Google Scholar] [CrossRef]
- Garver, D.L.; Tamas, R.L.; Holcomb, J.A. Elevated interleukin-6 in the cerebrospinal fluid of a previously delineated schizophrenia subtype. Neuropsychopharmacology 2003, 28, 1515–1520. [Google Scholar] [CrossRef] [PubMed]
- Bezzi, P.; Domercq, M.; Brambilla, L.; Galli, R.; Schols, D.; de Clercq, E.; Vescovi, A.; Bagetta, G.; Kollias, G.; Meldolesi, J.; et al. CXCR4-activated astrocyte glutamate release via TNFα: amplification by microglia triggers neurotoxicity. Nat. Neurosci. 2001, 4, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.L.; Jones, F.; Kubota, E.S.F.C.S.; Pocock, J.M. Stimulation of microglial metabotropic glutamate receptor mGlu2 triggers tumor necrosis factor α-induced neurotoxicity in concert with microglial-derived Fas ligand. J. Neurosci. 2005, 25, 2952–2964. [Google Scholar] [CrossRef] [PubMed]
- Patneau, D.K.; Wright, P.W.; Winters, C.; Mayer, M.L.; Gallo, V. Glial cells of the oligodendrocyte lineage express both kainate- and AMPA-preferring subtypes of glutamate receptor. Neuron 1994, 12, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Káradóttir, R.; Attwell, D. Neurotransmitter receptors in the life and death of oligodendrocytes. Neuroscience 2006, 145, 1426–1438. [Google Scholar] [CrossRef] [PubMed]
- Flynn, S.W.; Lang, D.J.; Mackay, A.L.; Goghari, V.; Vavasour, I.M.; Whittall, K.P.; Smith, G.N.; Arango, V.; Mann, J.J.; Dwork, A.J.; et al. Abnormalities of myelination in schizophrenia detected in vivo with MRI, and post-mortem with analysis of oligodendrocyte proteins. Mol. Psychiatry 2003, 8, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Palaniyappan, L.; Simmonite, M.; White, T.P.; Liddle, E.B.; Liddle, P.F. Neural primacy of the salience processing system in schizophrenia. Neuron 2013, 79, 814–828. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Chiavetto, L.B.; Bignotti, S.; Tura, G.J.B.; Oioli, R.; Boin, F.; Kenis, G.; Bosmans, E.; de Jongh, R.; Altamura, C.A. Increased serum interkeukin-8 and interleukin-10 in schizophrenic patients resistant to treatment with neuroleptics and the stimulatory effects of clozapine on leukemia inhibitory factor receptor. Schizophr. Res. 2002, 54, 281–291. [Google Scholar] [CrossRef]
- Maxeiner, H.G.; Schneider, E.M.; Kurfiss, S.T.; Brettschneider, J.; Tumani, H.; Bechter, K. Cerebrospinal fluid and serum cytokine profiling to detect immune control of infectious and inflammatory neurological and psychiatric diseases. Cytokine 2014, 69, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Müller, N.; Riedel, M.; Scheppach, C.; Brandstätter, B.; Sokullu, S.; Krampe, K.; Ulmschneider, M.; Engel, R.R.; Möller, H.J.; Schwarz, M.J. Beneficial antipsychotic effects of celecoxib add-on therapy compared to risperidone alone in schizophrenia. Am. J. Psychiatry 2002, 159, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Krause, D.; Dehning, S.; Musil, R.; Schennach-Wolff, R.; Obermeier, M; Möller, H.J.; Klauss, V.; Schwarz, M.J.; Riedel, M. Celecoxib treatment in an early stage of schizophrenia: results of a randomized, double-blind, placebo-controlled trial of celecoxib augmentation of amisulpride treatment. Schizophr. Res. 2010, 121, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chun Chen, D.; Long Tan, Y.; Zhou, D.F. A doubleblind, placebo-controlled trial of celecoxib added to risperidone in first-episode and drug-naive patients with schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 2006, 256 (Suppl. 2), 50. [Google Scholar]
- Kaizaki, A.; Tien, L.T.; Pang, Y.; Cai, Z.; Tanaka, S.; Numazawa, S.; Bhatt, A.J.; Fan, L.W. Celecoxib reduces brain dopaminergic neuronaldysfunction, and improves sensorimotor behavioral performance in neonatal rats exposed to systemic lipopolysaccharide. J. Neuroinflamm. 2013, 10, 45. [Google Scholar] [CrossRef]
- Hung, Y.; Liu, J.; Wang, L.; Zhang, W.; Zhu, X. Neuroprotective effects of cyclooxygenase-2 inhibitor celecoxib against toxicity of LPS-stimulated macrophages toward motor neurons. Acta Paharmacol. Sin. 2005, 26, 952–958. [Google Scholar] [CrossRef]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar]
- Ortega-Gomez, A.; Perretti, M.; Soehnlein, O. Resolution of inflammation: An integrated view. EMBO Mol. Med. 2013, 5, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Alessandri, A.L.; Sousa, L.P.; Lucas, C.D.; Rossi, A.G.; Pinho, V.; Teixeira, M.M. Resolution of inflammation: Mechanisms and opportunity for drug development. Pharmacol. Ther. 2013, 139, 189–212. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Kato, N. Mitochondrial dysfunction in bipolar disorder. Bipolar Disord. 2000, 2, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.U.; Jeong, J.; Lee, H.; Young Mun, J.; Kim, J.H.; Seo Lee, J.; Dang Nguyen, M.; Sik Han, S.; Suh, P.G.; Ki Park, S. Disrupted-in-schizophrenia 1 (DISC1) plays essential roles in mitochondria in collaboration with Mitofilin. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 17785–17790. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; McPhie, D.L.; Lange, N.T.; Punzell, S.; Elmiligy, S.; Ye, N.Z.; Froimowitz, M.P.; Hassinger, L.C.; Menesale, E.B.; Sargent, L.W.; et al. Abnormalities in mitochondrial structure in cells from patients with bipolar disorder. Am. J. Pathol. 2010, 177, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Ferger, A.I.; Campanelli, L.; Reimer, V.; Muth, K.N.; Merdian, I.; Ludolph, A.C.; Witting, A. Effects of mitochondrial dysfunction on the immunological properties of microglia. J. Neuroinflamm. 2010, 7, 45. [Google Scholar] [CrossRef]
- Li, X.; Chauhn, A.; Shiekh, A.M.; Patil, S.; Chauhn, V.; Li, X.M.; Ji, L.; Brown, T.; Malika, M. Elevated immune response in the brain of autistic patients. J. Neuroimmunol. 2009, 207, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Sekine, Y.; Ouchi, Y.; Tsujii, M.; Yoshikawa, E.; Futatsubashi, M.; Tsuchiya, K.J.; Sugihara, G.; Iwata, Y.; Suzuki, K.; et al. Brain serotonin and dopamine transporter bindings in adults with high-functioning autism. Am. J. Psychiatry 2010, 67, 59–68. [Google Scholar]
- Zhong, M.; Wang, X.; Xiao, J.; Yi, J.; Zhu, X.; Liao, J.; Wang, W.; Yao, S. Amygdala hyperactivation and prefrontal hypoactivation in subjects with cognitive vulnerability to depression. Biol. Psychol. 2011, 88, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Berton, O.; Nestler, E.J. New approaches to antidepressant drug discovery: beyond monoamines. Nat. Rev. Neurosci. 2006, 7, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Price, J.L.; Drevets, W.C. Neurocircuitry of mood disorders. Neuropsychopharmacol. Rev. 2010, 35, 192–216. [Google Scholar] [CrossRef]
- Kang, H.J.; Voleti, B.; Hajszan, T.; Rajkowska, G.; Stockmeier, C.A.; Licznerski, P.; Lepack, A.; Majik, M.S.; Jeong, L.S.; Banasr, M.; et al. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat. Med. 2012, 18, 1413–1419. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Cai, L.; Yu, Y.; Gao, L.; Xiao, L.; He, Q.; Ren, Z.; Liu, Y. Activation of brain indoleamine 2,3-dioxygenase contributes to epilepsy-associated depressive-like behavior in rats with chronic temporal lobe epilepsy. J. Neuroinflamm. 2014, 11, 41. [Google Scholar] [CrossRef]
- Steiner, S.; Walter, M; Gos, T.; Guillemin, G.J.; Bernstein, H.G.; Sarnyai, Z.; Mawrin, C.; Brisch, R.; Bielau, H.; zu Schwabedissen, L.M.; et al. Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: evidence for an immune-modulated glutamatergic neurotransmission? J. Neuroinflamm. 2011, 8, 94. [Google Scholar] [CrossRef]
- Fattore, L.; Melis, M.; Fadda, P.; Pistis, M.; Fratta, W. The endocannabinoid system and nondrug rewarding behaviours. Exp. Neurol. 2010, 224, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Müller, N.; Schwarz, M.J.; Dehning, S.; Douhe, A.; Cerovecki, A.; Goldstein-Muller, B.; Spellmann, I.; Hetzel, G.; Maino, K.; Kleindienst, N.; et al. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol. Psychiatry 2006, 11, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, S.H.; Hosseini, F.; Modabbernia, A.; Ashrafi, M.; Akhondzadeh, S. Effect of celecoxib add-on treatment on symptoms and serum IL-6 concentrations in patients with major depressive disorder: Randomized double-blind placebo-controlled study. J. Affect. Disord. 2012, 141, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.G.; Price, T. Post-stroke depressive disorders: a follow-up study of 103 Patients. Stroke 1982, 13, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.R.R.; Hays, J.C.; Blazer, D.G. MRI-defined vascular depression. Am. J. Psychiatry 1997, 154, 497–501. [Google Scholar] [PubMed]
- Fujikawa, T.; Yamawaki, S.; Touhouda, Y. Incidence of silent cerebral infarction in patients with major depression. Stroke 1993, 24, 1631–1634. [Google Scholar] [CrossRef]
- Alexopoulos, G.S.; Bruce, M.L.; Silbersweig, D.; Kalayam, B.; Stern, E. Vascular depression: a new view of late-onset depression. Dialogues Clin. Neurosci. 1999, 1, 68–80. [Google Scholar] [PubMed]
- Taylor, W.D.; Aizenstein, H.J.; Alexopoulos, G.S. The vascular depression hypothesis: mechanisms linking vascular disease with depression. Mol. Psychiatry 2013, 18, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Naismith, S.L.; Norrie, L.M.; Mowszowski, L.; Hickie, I.B. The neurobiology of depression in later-life: Clinical, neuropsychological, neuroimaging and pathophysiological features. Prog. Neurobiol. 2012, 98, 99–143. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, M.I.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M.B. History of depression, depressive symptoms, and medial temporal lobe atrophy and the risk of Alzheimer disease. Neurology 2008, 70, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, P.B.; Lyketsos, C.G. Mild cognitive impairment: searching for the prodrome of Alzheimer’s disease. World Psychiatry 2008, 7, 72–78. [Google Scholar] [PubMed]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [PubMed]
- Jackson, S.P. Arterial thrombosis—Insidious, unpredictable and deadly. Nat. Med. 2011, 17, 1423–1436. [Google Scholar] [CrossRef] [PubMed]
- Phillipson, M.; Kubes, P. The neutrophil in vascular inflammation. Nat. Med. 2011, 17, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Abramsson, A.; Lindblom, P.; Betsholtz, C. Endothelial and nonendothelial sources of PDGF-B regulate pericyte recruitment and influence vascular pattern formation in tumors. J. Clin. Invest. 2003, 112, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Darland, D.C.; Massingham, L.J.; Smith, S.R.; Piek, E.; Saint-Geniez, M.; D’Amorea, P.A. Pericyte production of cell-associated VEGF is differentiationdependent and is associated with endothelial survival. Dev. Biol. 2003, 264, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Argaw, A.T.; Asp, L.; Zhang, J.; Navrazhina, K.; Pham, T.; Mariani, J.N.; Mahase, S.; Dutta, D.J.; Seto, J.; Kramer, E.G.; et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J. Clin. Invest. 2012, 122, 2454–2468. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, R.; Zhou, Z.; Kleinerman, E.S. SDF-1a Induces PDGF-B Expression and the differentiation of bone marrow cells into pericytes. Mol. Cancer Res. 2011, 9, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Ödemis, V.; Boosmann, K.; Heinen, A.; Küry, P.; Engele1, J. CXCR7 is an active component of SDF-1 signalling in astrocytes and Schwann cells. J. Cell Sci. 2010, 123, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Huang, Y.; Shi, H.; Yuan, S.; Ding, Y.; Song, X.; Fu, Y.; Luo, Y. Overexpression of platelet-derived growth factor-BB increases tumor pericyte content via stromal-derived factor-1A/CXCR4 axis. Cancer Res. 2009, 69, 6057–6064. [Google Scholar] [CrossRef] [PubMed]
- Bonkowski, D.; Katyshev, V.; Balabanov, R.D.; Borisov, A.; Dore-Duffy, P. The CNS microvascular pericyte: pericyte-astrocyte crosstalk in the regulation of tissue survival. Fluids Barriers CNS 2011, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat. Res. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Lavretsky, H.; Meeks, T. Late-life depression: managing mood in patients with vascular disease. Cur. Psychiatry 2009, 8, 20–38. [Google Scholar]
- Namekawa, Y.; Baba, H.; Maeshima, H.; Nakano, Y.; Satomura, E.; Takebayashi, N.; Nomoto, H.; Suzuki, T.; Arai, H. Heterogeneity of elderly depression: Increased risk of Alzheimer’s disease and Aβ protein metabolism. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 43, 203–208. [Google Scholar] [CrossRef]
- Levy, R.L.; Langer, S.L.; Whitehead, W.E. Social learning contributions to the etiology and treatment of functional abdominal pain and inflammatory bowel disease in children and adults. World J. Gastroenterol. 2007, 13, 2397–2403. [Google Scholar] [CrossRef] [PubMed]
- Wiech, K.; Ploner, M.; Tracey, I. Neurocognitive aspects of pain perception. Trends Cogn. Sci. 2008, 12, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Uceyler, N.; Rogausch, J.P.; Toyka, K.V.; Sommer, C. Differential expression of cytokines in painful and painless neuropathies. Neurology 2007, 69, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Backonja, M.M.; Coe, C.L.; Muller, D.A.; Schell, K. Altered cytokine levels in the blood and cerebrospinal fluid of chronic pain patients. J. Neuroimmunol. 2008, 195, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Tracey, I. Getting the pain you expect: mechanisms of placebo, nocebo and reappraisal effects in humans. Nat. Med. 2010, 16, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Apkarian, A.V.; Sosa, Y.; Sonty, S.; Levy, R.M.; Harden, R.N.; Parrish, T.B.; Gitelman, D.R. Chronic back pain is associated with decreased prefrontal and thalamic gray matter density. J. Neurosci. 2004, 24, 10410–10415. [Google Scholar] [CrossRef] [PubMed]
- Zubieta, J.K.; Bueller, J.A.; Jackson, L.R.; Scott, D.J.; Xu, Y.; Koeppe, R.A.; Nichols, T.E.; Stohler, C.S. Placebo effects mediated by endogenous opioid activity on mu-opioid receptors. J. Neurosci. 2005, 35, 7754–7762. [Google Scholar] [CrossRef]
- Lorenz, J.; Minoshima, S.; Casey, K.L. Keeping pain out of mind: The role of the dorsolateral prefrontal cortex in pain modulation. Brain 2003, 126, 1079–1091. [Google Scholar] [CrossRef] [PubMed]
- Maizels, M.; Aurora, S.; Heinricher, M. Beyond neurovascular: migraine as a dysfunctional neurolimbic pain network. Headache 2012, 52, 1553–1565. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, A.; Leweke, F.M.; Gerth, C.W.; Schreiber, D.; Koethe, D.; Faulhaber, J.; Klosterkotter, J.; Piomelli, D. Cerebrospinal anandamide levels are elevated in acute schizophrenia and are inversely correlated with psychotic symptoms. Neuropsychopharmacology 2004, 29, 2108–2114. [Google Scholar] [CrossRef] [PubMed]
- Leweke, F.M.; Piomelli, D.; Pahlisch, F.; Muhl, D.; Gerth, C.W.; Hoyer, C.; Klosterkotter, J.; Hellmich, M.; Koethe, D. Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl. Psychiatry 2012, 2, e94. [Google Scholar] [CrossRef] [PubMed]
- Blaas, K. Treating depression with cannabinoids. Cannabinoids 2008, 3, 8–10. [Google Scholar]
- Freund, T.F.; Katona, I.; Piomelli, D. Role of endogenous cannabinoids in synaptic signaling. Physiol. Rev. 2003, 83, 1017–1066. [Google Scholar] [PubMed]
- Varga, K.; Wagner, J.A.; Bridgen, T.; Kunosi, G. Platelet- and macrophage-derived endogenous cannabinoids are involved in endotoxin-induced hypotension. FASEB J. 1998, 12, 1035–1044. [Google Scholar] [PubMed]
- Carlisle, S.J.; Marciano-Cabral, F.; Staab, A.; Ludwick, C.; Cabral, G.A. Differential expression of the CB2 cannabinoid receptor by rodent macrophages and macrophage-like cells in relation to cell activation. Int. Immunopharmacol. 2002, 2, 69–82. [Google Scholar] [CrossRef]
- Han, K.H.; Lim, S.; Ryu, J.; Lee, C.W.; Kim, Y.; Kang, J.H.; Kang, S.S.; Ahn, Y.K.; Park, C.S.; Kim, J.J. CB1 and CB2 cannabinoid receptors differentially regulate the production of reactive oxygen species by macrophages. Cardiovasc. Res. 2009, 84, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Velasco, G.; Galve-Roperh, I.; Sanchez, C.; Blazquez, C.; Haro, A.; Guzman, M. Cannabinoids and ceramide: two lipids acting hand-by-hand. Life Sci. 2005, 77, 1723–1731. [Google Scholar] [CrossRef] [PubMed]
- Atwood, B.K.; Wager-Miller, J.; Haskins, C.; Straiker, A.; Mackie, K. Functional selectivity in CB2 cannabinoid receptor signaling and regulation: Implications for the therapeutic potential of CB2 ligands. Mol. Pharmacol. 2012, 81, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.W.; Newton, C.A.; Nakachi, N.; Friedman, H. Δ9-Tetrahydrocannabinol treatment suppresses immunity and early IFN-gamma, IL-12, and IL-12 receptor β2 responses to Legionella pneumophila infection. J. Immunol. 2000, 164, 6461–6466. [Google Scholar] [CrossRef] [PubMed]
- Newton, C.A; Chou, P.J.; Perkins, I.; Klein, T.W. CB1 and CB2 cannabinoid receptors mediate different aspects of Δ9-tetrahydrocannabinol (THC)-induced T helper cell shift following immune activation by Legionella Pneumophila infection. J. Neuroimmune. Pharmacol. 2009, 4, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Paintlia, A.S.; Paintlia, M.K.; Singh, I.; Singh, A.K. IL-4-induced peroxisome proliferator-activated receptor gamma activation inhibits NF-κB Trans activation in central nervous system (CNS) glial cells and protects oligodendrocytes progenitors under neuroinflammatory disease conditions: implication for CNS-demyelinating diseases. J. Immunol. 2006, 176, 4385–4398. [Google Scholar] [CrossRef] [PubMed]
- Galve-Roperh, I.; Sanchez, C.; Cortes, M.L.; Del Pulgar, T.G.; Izquierdo, M.; Guzman, M. Anti-tumoral action of cannabinoids: involvement of sustained ceramide accumulation and extracellular signal-regulated kinase activation. Nat. Med. 2000, 6, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Miller, R.A.; Wang, Z.V.; Sun, K.; Barth, B.M.; Bui, H.H.; Davis, K.E.; Bikman, B.T.; Halberg, N.; Rutkowski, J.M.; et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat. Med. 2011, 17, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Bensinger, S.J.; Tontonoz, P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature 2008, 454, 470–477. [Google Scholar] [PubMed]
- Rutherford, C.; Childs, S.; Ohotski, J.; McGlynn, L.; Riddick, M.; MacFarlane, S.; Tasker, D.; Pyne, S.; Pyne, N.J.; Edwards, J.; et al. Regulation of cell survival by sphingosine-1-phosphate receptor S1P1 via reciprocal ERK-dependent suppression of Bim and PI-3-kinase/protein kinase C-mediated upregulation of Mcl-1. Cell Death Dis. 2013, 4, e927. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Huang, M.C.; Goetzl, E.J. Type 1 sphingosine 1-phosphate G protein-coupled receptor (S1P1) mediation of enhanced IL-4 generation by CD4 T cells from S1P1 transgenic mice. J. Immunol. 2007, 178, 4885–4890. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.Y.; Ju, C.; Jalin, A.M.A.A.; Lee, D.A.; Prather, P.L.; Kim, W.K. Activation of cannabinoid CB2 receptor-mediated AMPK/CREB pathway reduces cerebral ischemic injury. Am. J. Pathol. 2013, 182, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.T.; Kola, B.; Korbonits, M. AMPK as a mediator of hormonal signaling. J. Mol. Endocrinol. 2010, 44, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Sun, L.; Yu, X.J.; Miao, Y.; Liu, J.J.; Wang, H.; Ren, J.; Zang, W.J. Acetylcholine mediates AMPK-dependent autophagic cytoprotection in H9c2 cells during hypoxia/reoxygenation injury. Cell. Physiol. Biochem. 2013, 32, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Rockwell, C.E.; Snider, N.T.; Thompson, J.T.; Vanden Heuvel, J.P.; Kaminski, N.E. Interleukin-2 suppression by 2-arachidonyl glycerol is mediated through peroxisome proliferator-activated receptor- gamma independently of cannabinoid receptors 1 and 2. Mol. Pharmacol. 2006, 70, 101–111. [Google Scholar] [PubMed]
- Andersson, U.; Filipsson, K.; Abbott, C.R.; Woods, A.; Smith, K.; Bloom, S.R.; Carling, D.; Small, C.J. AMP-activated protein kinase plays a role in the control of food intake. J. Biol. Chem. 2004, 279, 12005–12008. [Google Scholar] [CrossRef] [PubMed]
- Huypens, P.; Moens, K.; Heimberg, H.; Ling, Z.; Pipeleers, D.; Van de Casteele, M. Adiponectin-mediated stimulation of AMP-activated protein kinase (AMPK) in pancreatic β cells. Life Sci. 2005, 77, 1273–1282. [Google Scholar] [PubMed]
- Recchiuti, A.; Serhan, C.N. Pro-resolving lipid mediators (SPMs) and their actions in regulating miRNA in novel resolution circuits in inflammation. Front. Immunol. 2012, 3, 298. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, S.; Recchiuti, A.; Chiang, N.; Fredman, G.; Serhan, C.N. Resolvin D1 receptor stereoselectivity and regulation of inflammation and proresolving microRNAs. Am. J. Pathol. 2012, 180, 2018–2027. [Google Scholar] [CrossRef] [PubMed]
- Titos, E.; Rius, B.; Gonzalez-Periz, A.; Lopez-Vicario, C.; Moran-Salvador, E.; Martinez-Clemente, M.; Arroyo, V.; Claria, J. Resolvin D1 and its precursor docosahexaenoic acid promote resolution od adipose tissue inflammation by eliciting macrophage polarization toward an M2-like phenotype. J. Immunol. 2011, 187, 5408–5418. [Google Scholar] [CrossRef] [PubMed]
- Le, Y.; Gong, W.; Tiffany, H.L.; Tumanov, A.; Nedospasov, S.; Shen, W.; Dunlop, N.M.; Gao, J.L.; Murphy, P.M.; Oppenheim, J.J.; et al. Amyloid β42 activates a G-protein-coupled chemoattractant receptor, FPR-Like-1. J. Neurosci. 2001, 21, RC123. [Google Scholar] [PubMed]
- Amminger, G.P.; Schäfer, D.M.R.; Papageorgiou, K.; Klier, C.M.; Cotton, S.M.; Harrigan, S.M.; Mackinnon, A.; McGorry, P.D.; Berger, G.E. Long-chain ω-3 fatty acids for indicated prevention of psychotic disorders. Arch. Gen. Psychiatry 2010, 67, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Emsley, R.; Myburgh, C.; Oosthuizen, P.; van Rensburg, S.J. Randomized, placebo-controlled study of ethyl-eicosapentaenoic acid as supplemental treatment in schizophrenia. Am. J. Psychiatry 2002, 159, 1596–1598. [Google Scholar] [CrossRef] [PubMed]
- Baruch, K.; Ron-Harel, N.; Gal, H.; Deczkowska, A.; Shifrut, E.; Ndifon, W.; Mirlas-Neisberg, N.; Cardon, M.; Vaknin, I.; Cahalon, L.; et al. CNS-specific immunity at the choroid plexus shifts toward destructive Th2 inflammation in brain aging. Proc. Natl. Acad. Sci. USA. 2013, 110, 2264–2269. [Google Scholar] [CrossRef] [PubMed]
- Fenn, A.M.; Hall, J.C.E.; Gensel, J.C.; Popovich, P.G.; Godbout, J.P. IL-4 signaling drives a unique arginase+/IL-1β+ microglia phenotype and recruits macrophages to the inflammatory CNS: Consequences of age-related deficits in IL-4Rα after traumatic spinal cord injury. J. Neurosci. 2014, 34, 8904–8917. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakagawa, Y.; Chiba, K. Role of Microglial M1/M2 Polarization in Relapse and Remission of Psychiatric Disorders and Diseases. Pharmaceuticals 2014, 7, 1028-1048. https://doi.org/10.3390/ph7121028
Nakagawa Y, Chiba K. Role of Microglial M1/M2 Polarization in Relapse and Remission of Psychiatric Disorders and Diseases. Pharmaceuticals. 2014; 7(12):1028-1048. https://doi.org/10.3390/ph7121028
Chicago/Turabian StyleNakagawa, Yutaka, and Kenji Chiba. 2014. "Role of Microglial M1/M2 Polarization in Relapse and Remission of Psychiatric Disorders and Diseases" Pharmaceuticals 7, no. 12: 1028-1048. https://doi.org/10.3390/ph7121028
APA StyleNakagawa, Y., & Chiba, K. (2014). Role of Microglial M1/M2 Polarization in Relapse and Remission of Psychiatric Disorders and Diseases. Pharmaceuticals, 7(12), 1028-1048. https://doi.org/10.3390/ph7121028