Involvement of the Antioxidant Effect and Anti-inflammatory Response in Butyrate-Inhibited Vascular Smooth Muscle Cell Proliferation

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Cell Culture
2.3. GPx Activity Assay
2.3.1. Preparation of Cell Lysate
2.3.2. Measurement of GPx Activity
2.4. Western Analysis
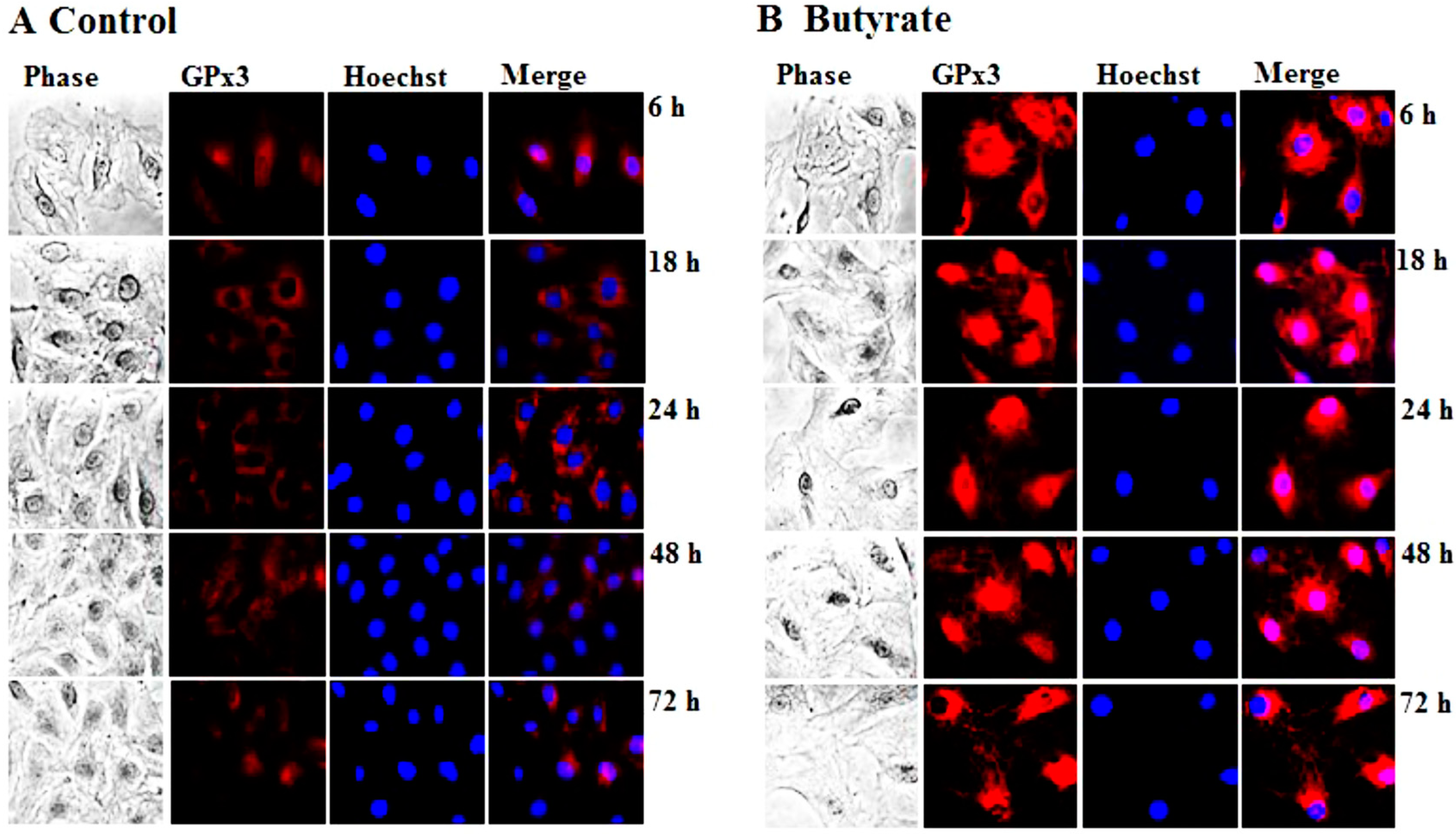
2.5. Immunofluorescence Staining
2.6. Statistical Analysis
3. Results and Discussion
3.1. Upregulation of GPXs by Butyrate in VSMC
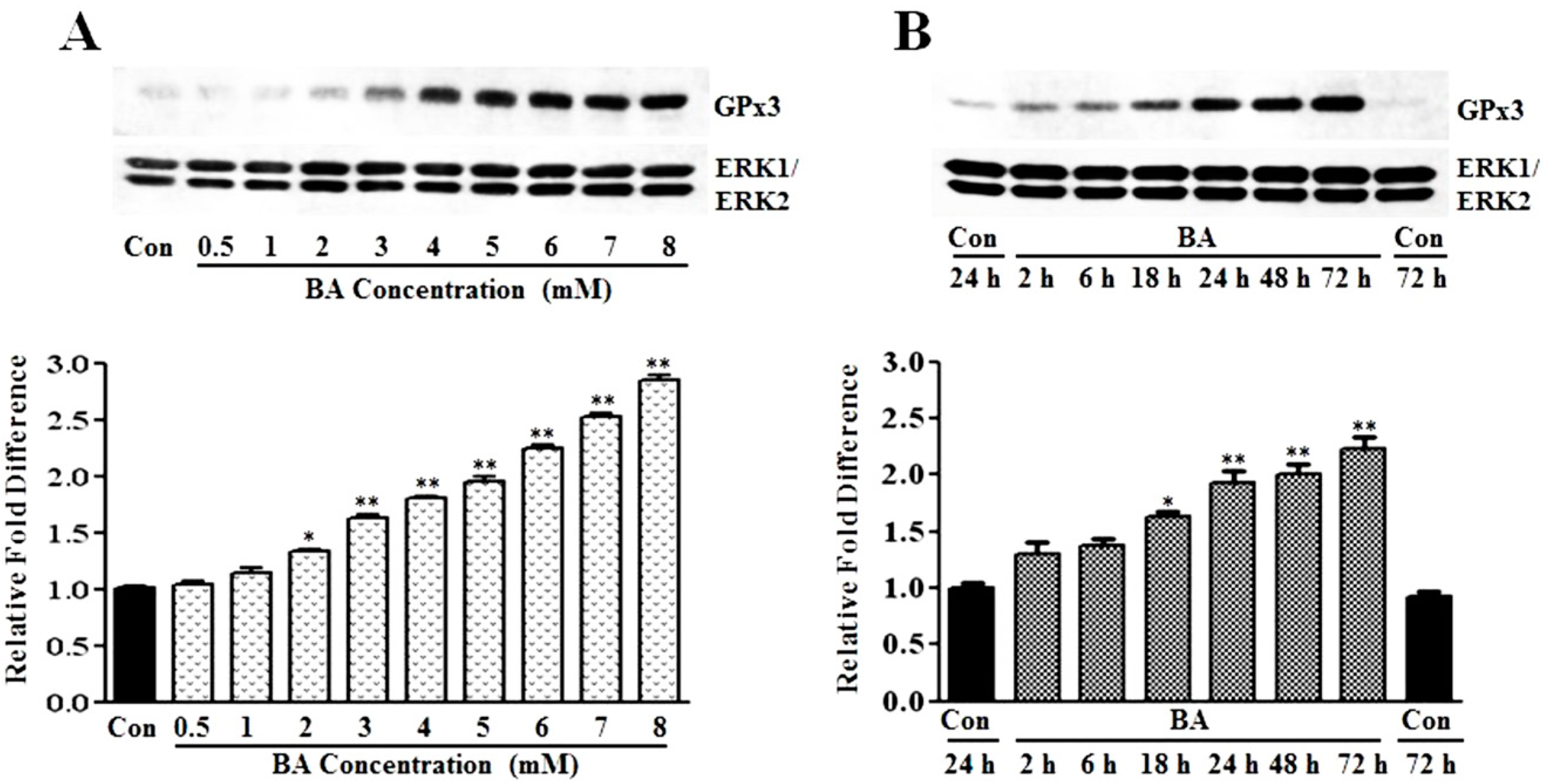
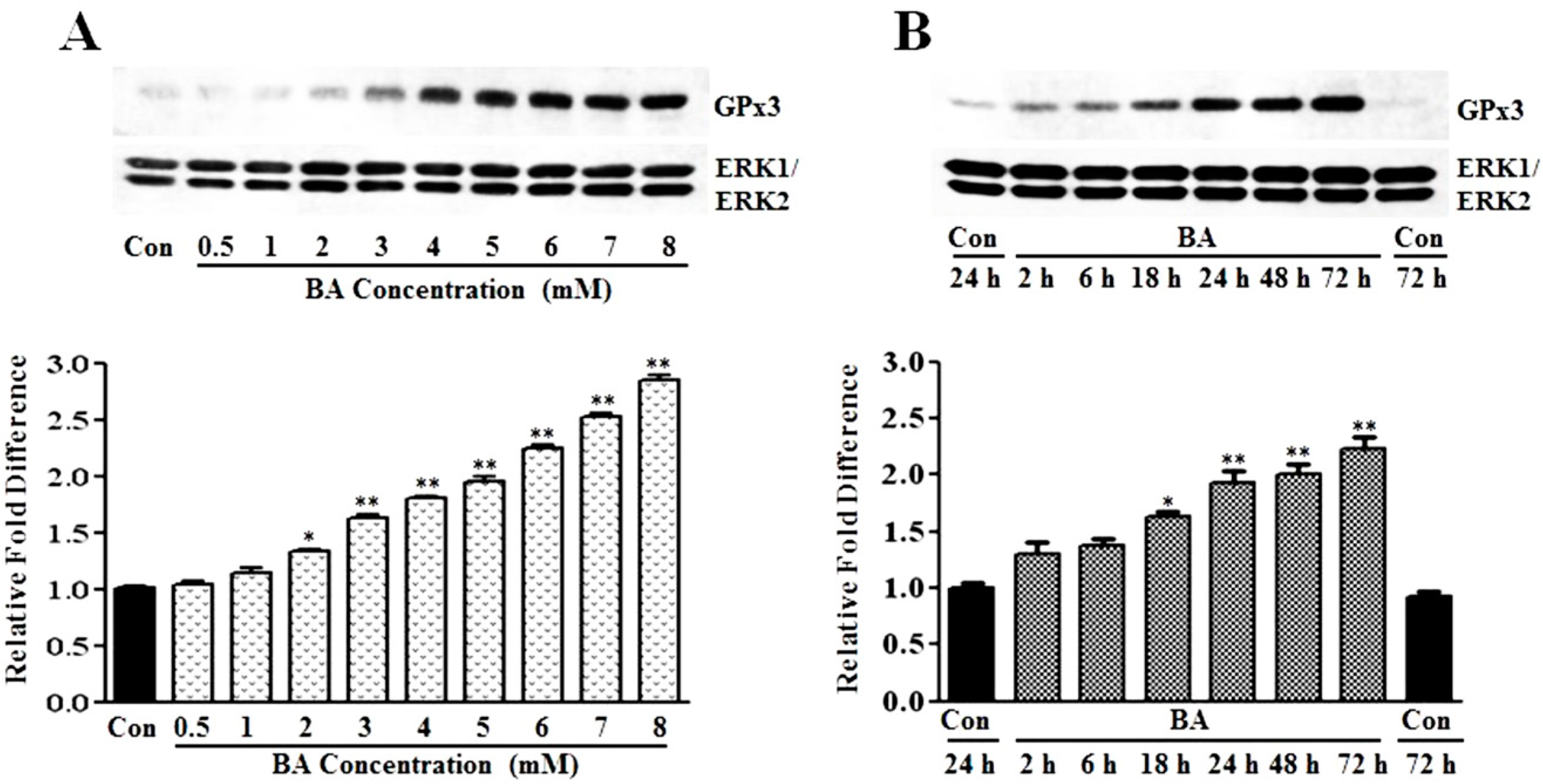
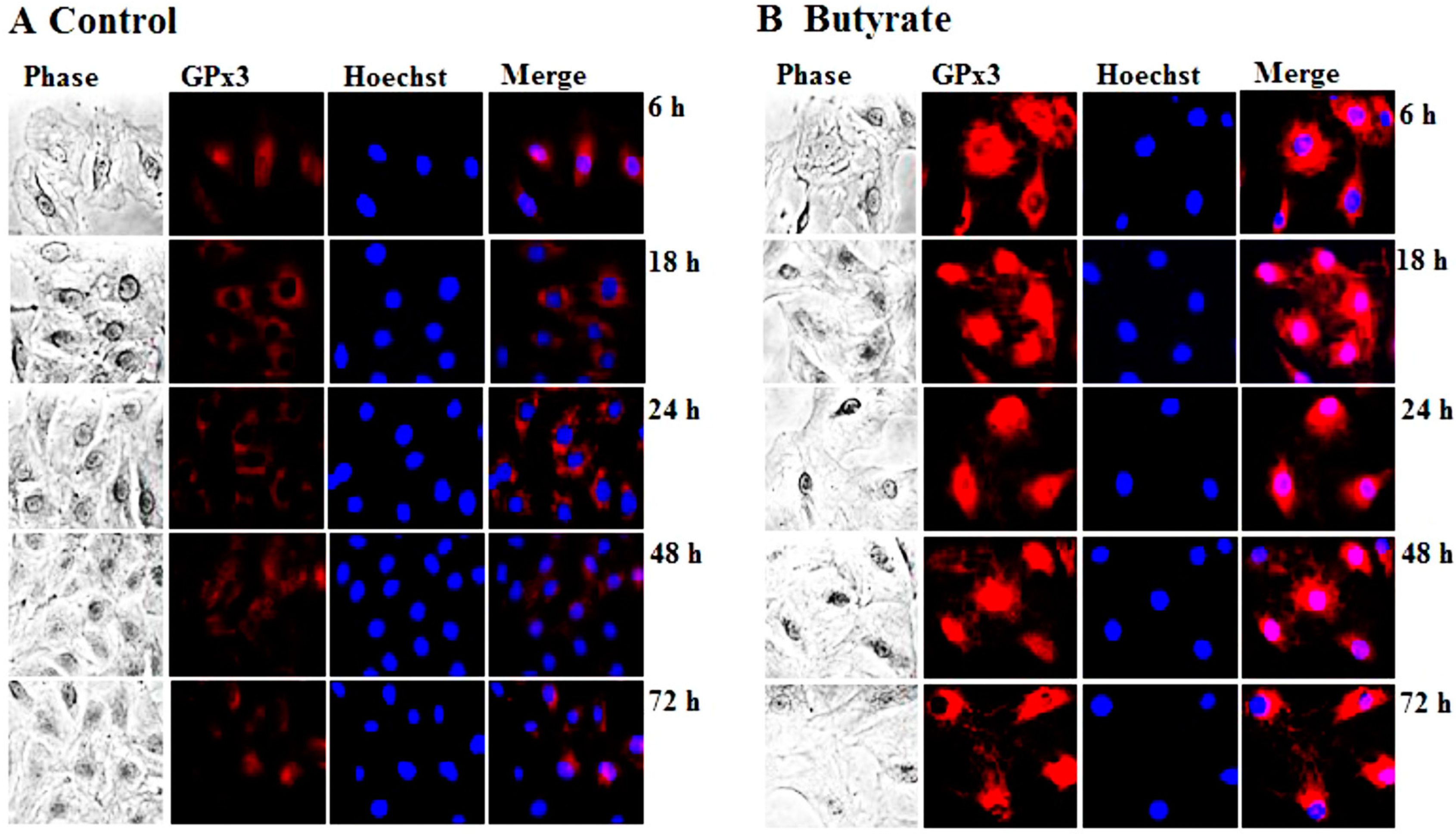
3.1.1. Induction of GPx3 Expression by Butyrate
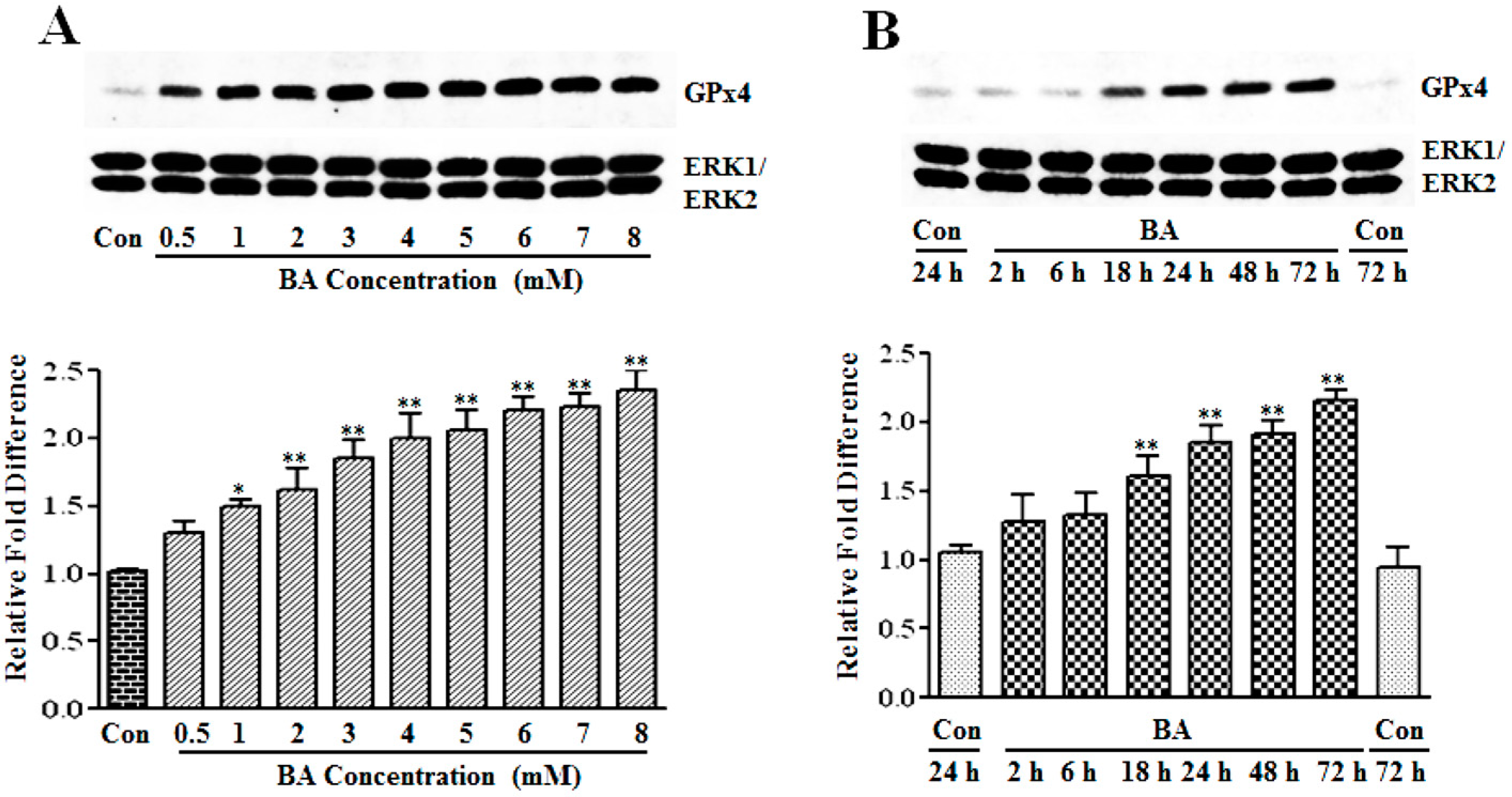
3.1.2. Upregulation of GPx4 by Butyrate
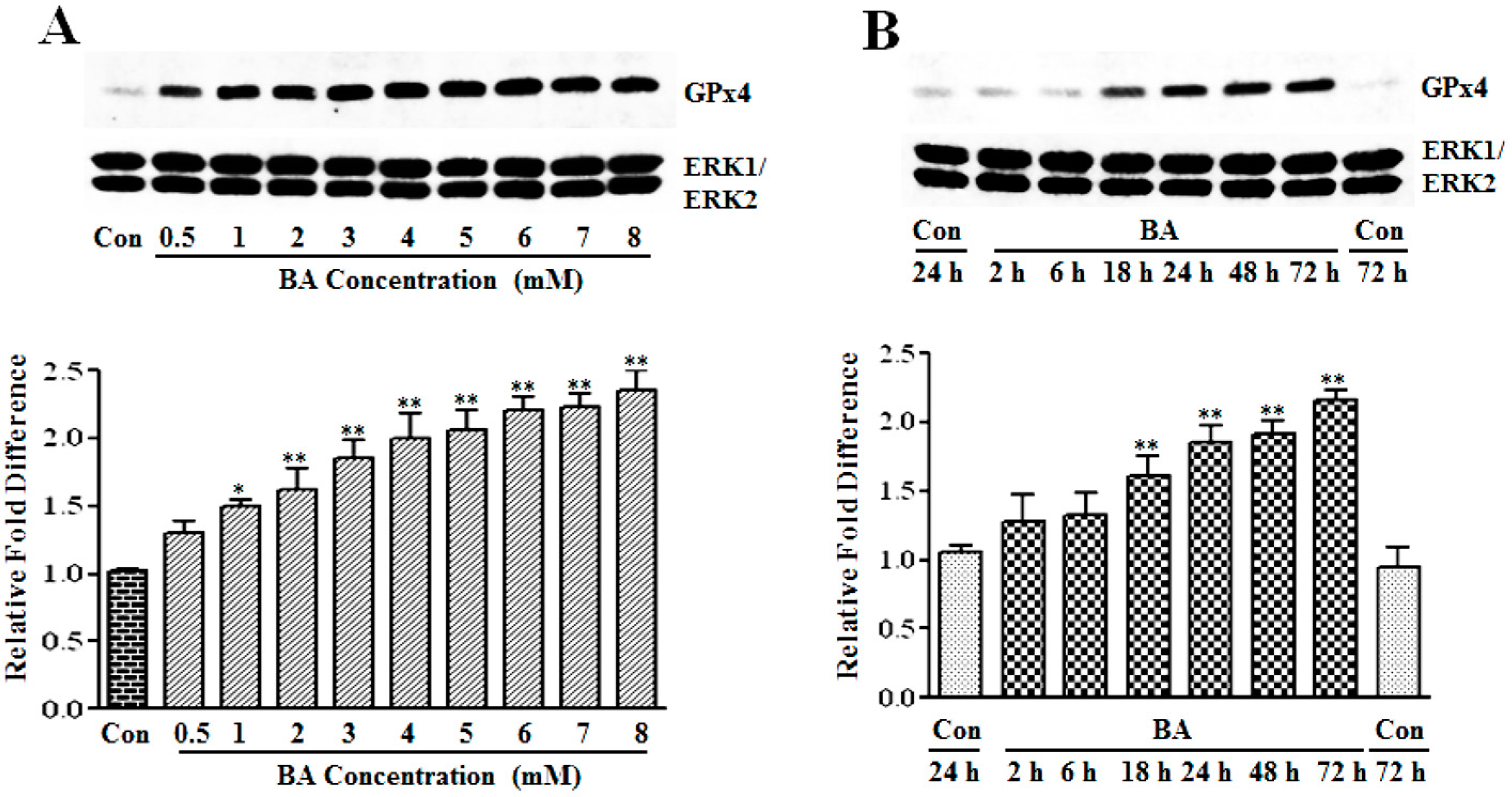
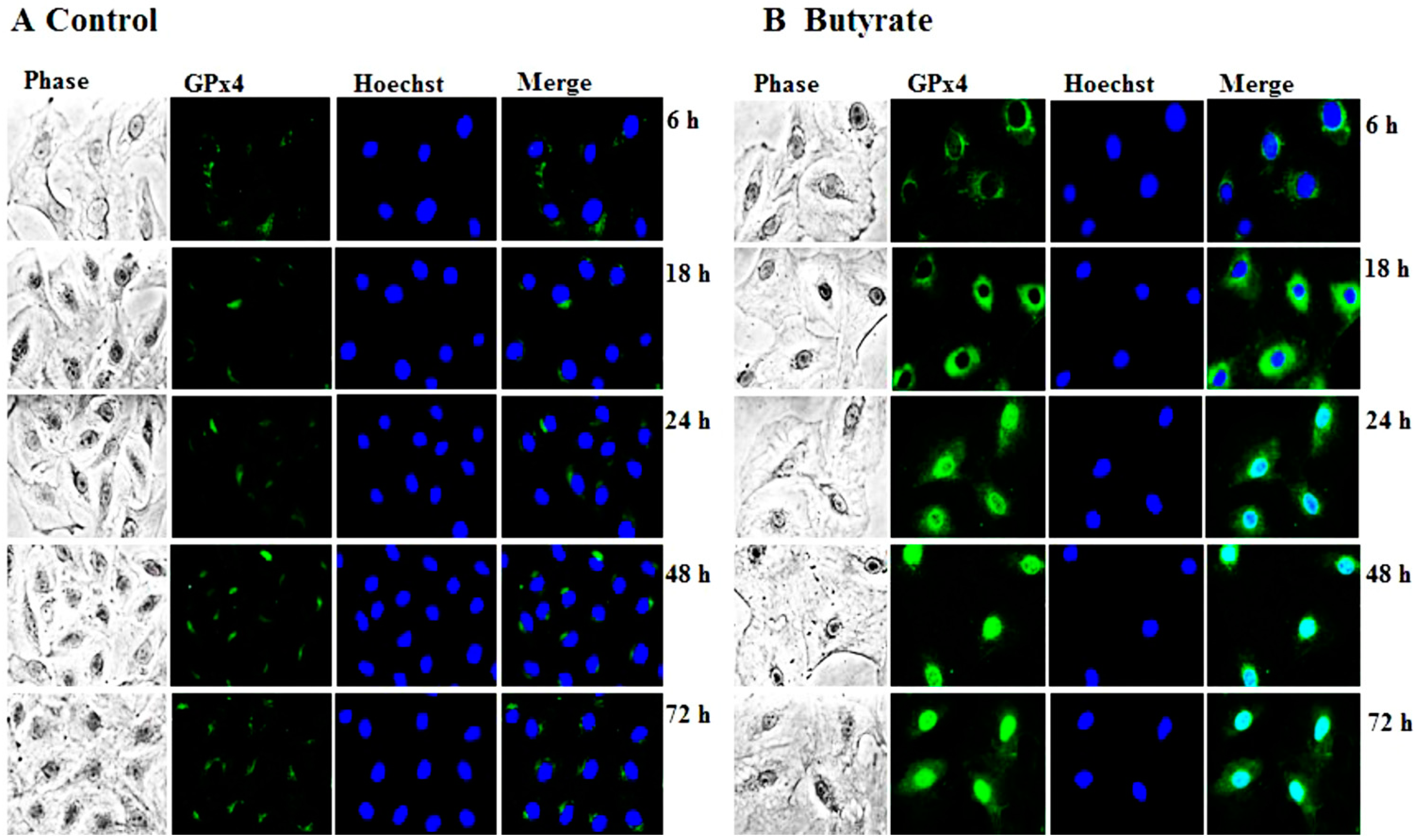
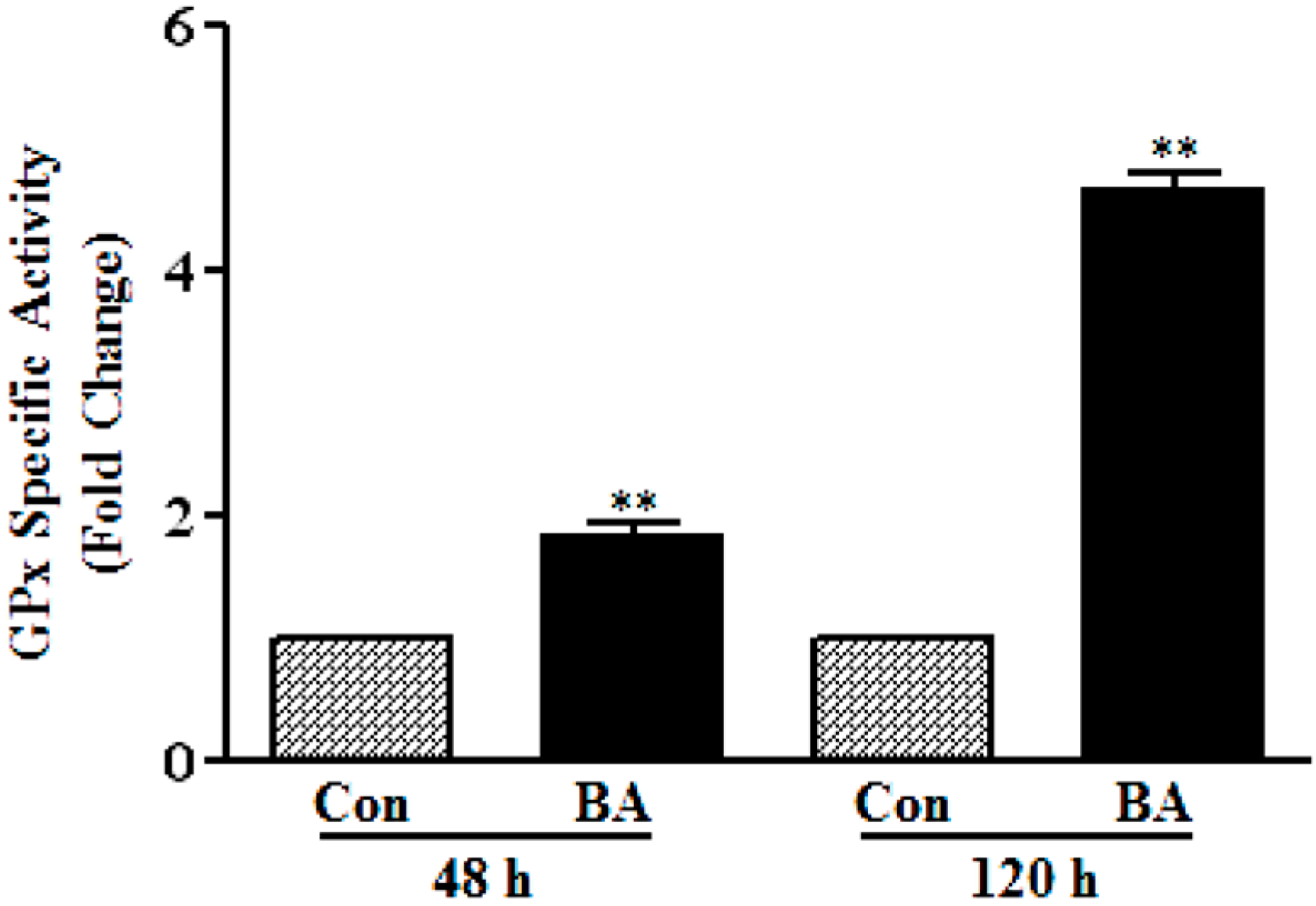
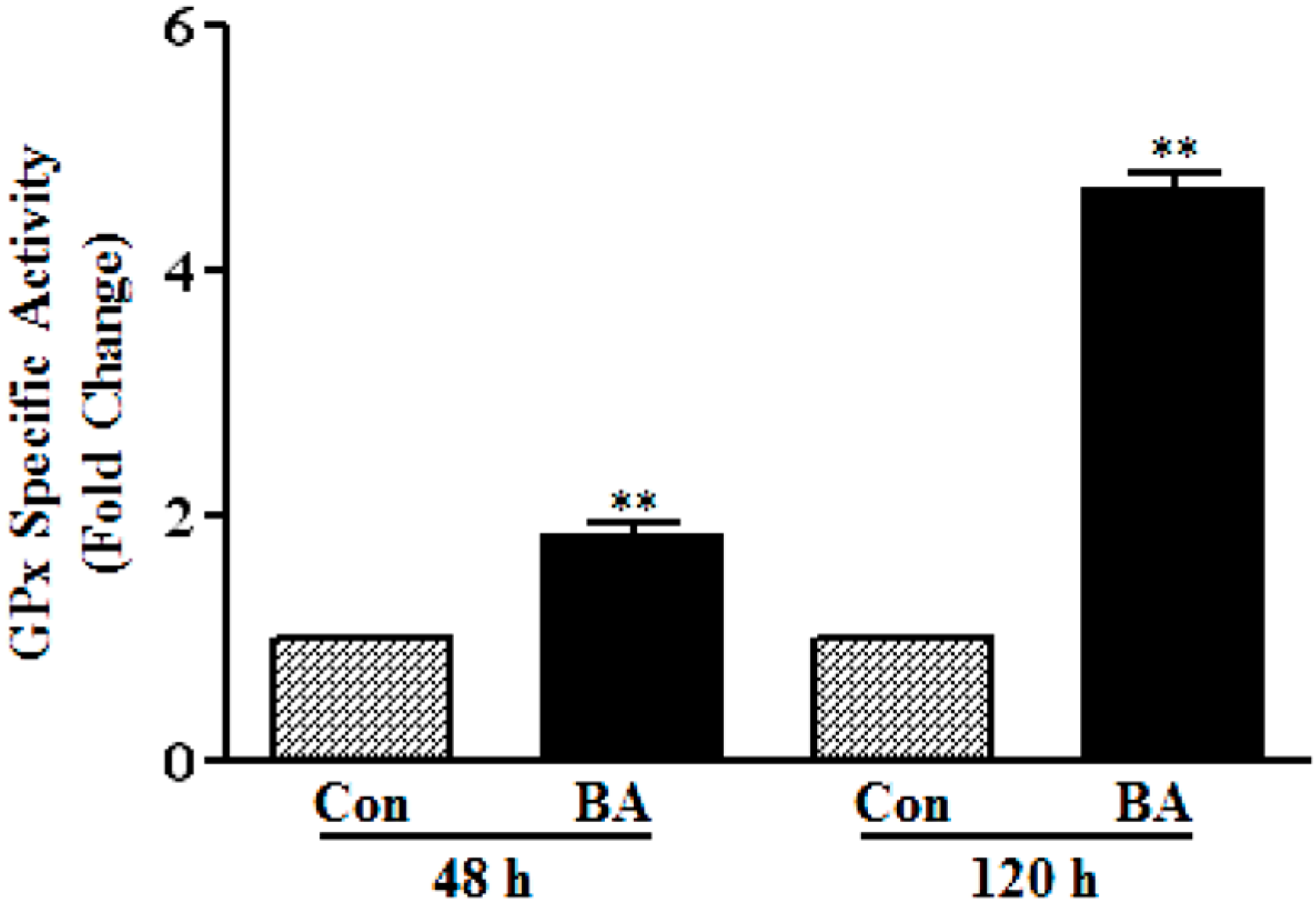
3.1.3. Induction of GPx Catalytic Activity by Butyrate
3.2. Influence of Butyrate Treatment on NF-κB Pathway and Its Targets
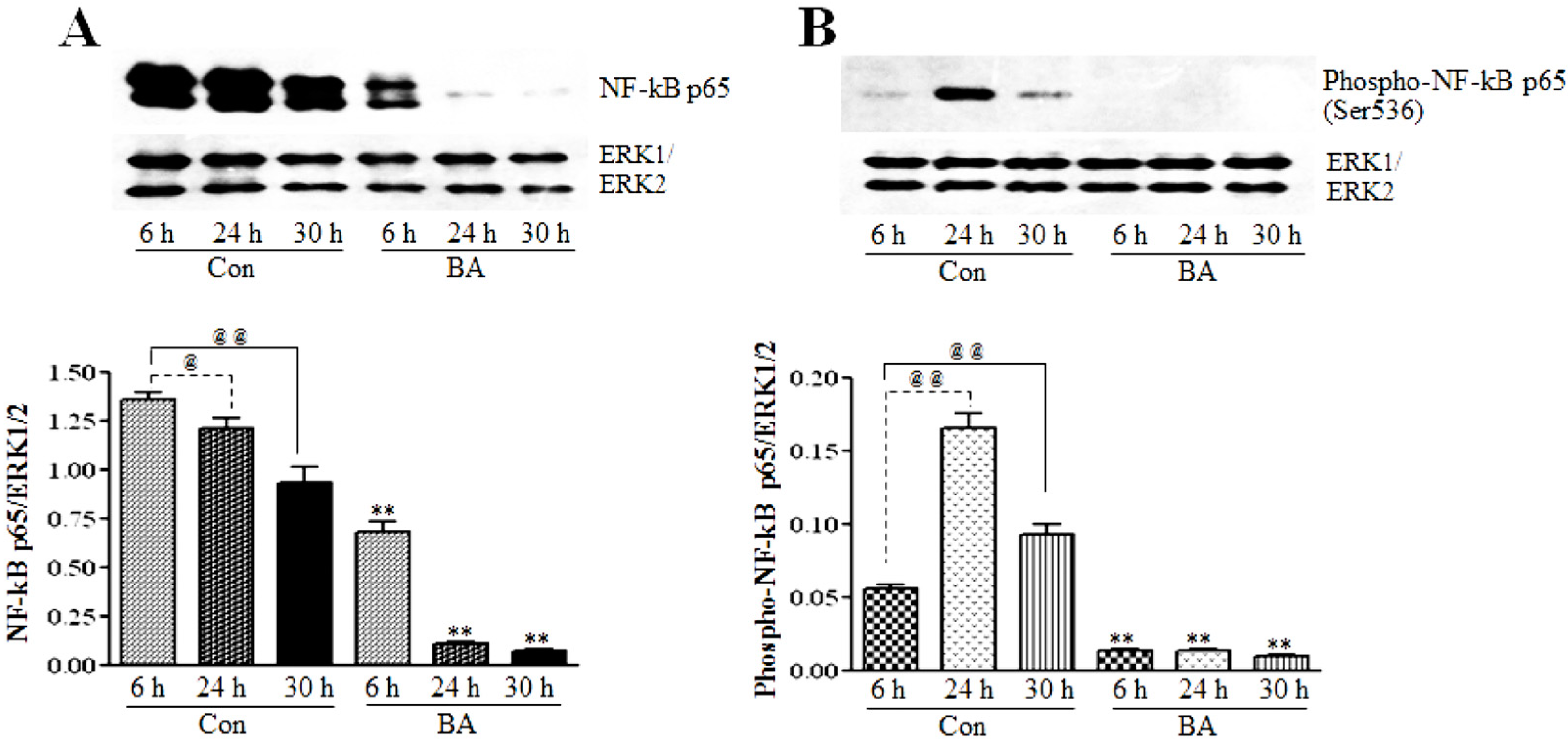
3.2.1. Butyrate Treatment Causes Inhibition of NF-κBp65 Expression and Activation
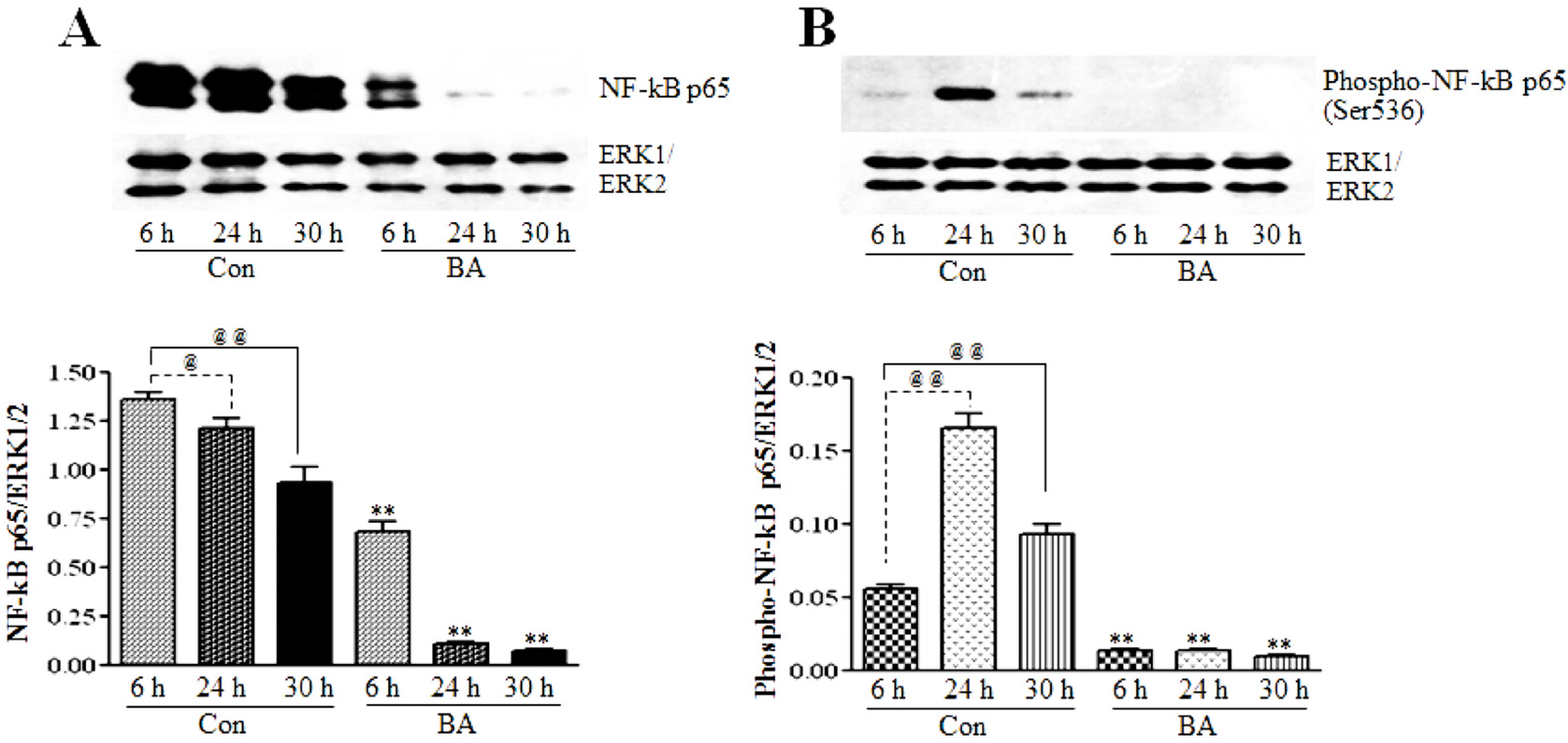
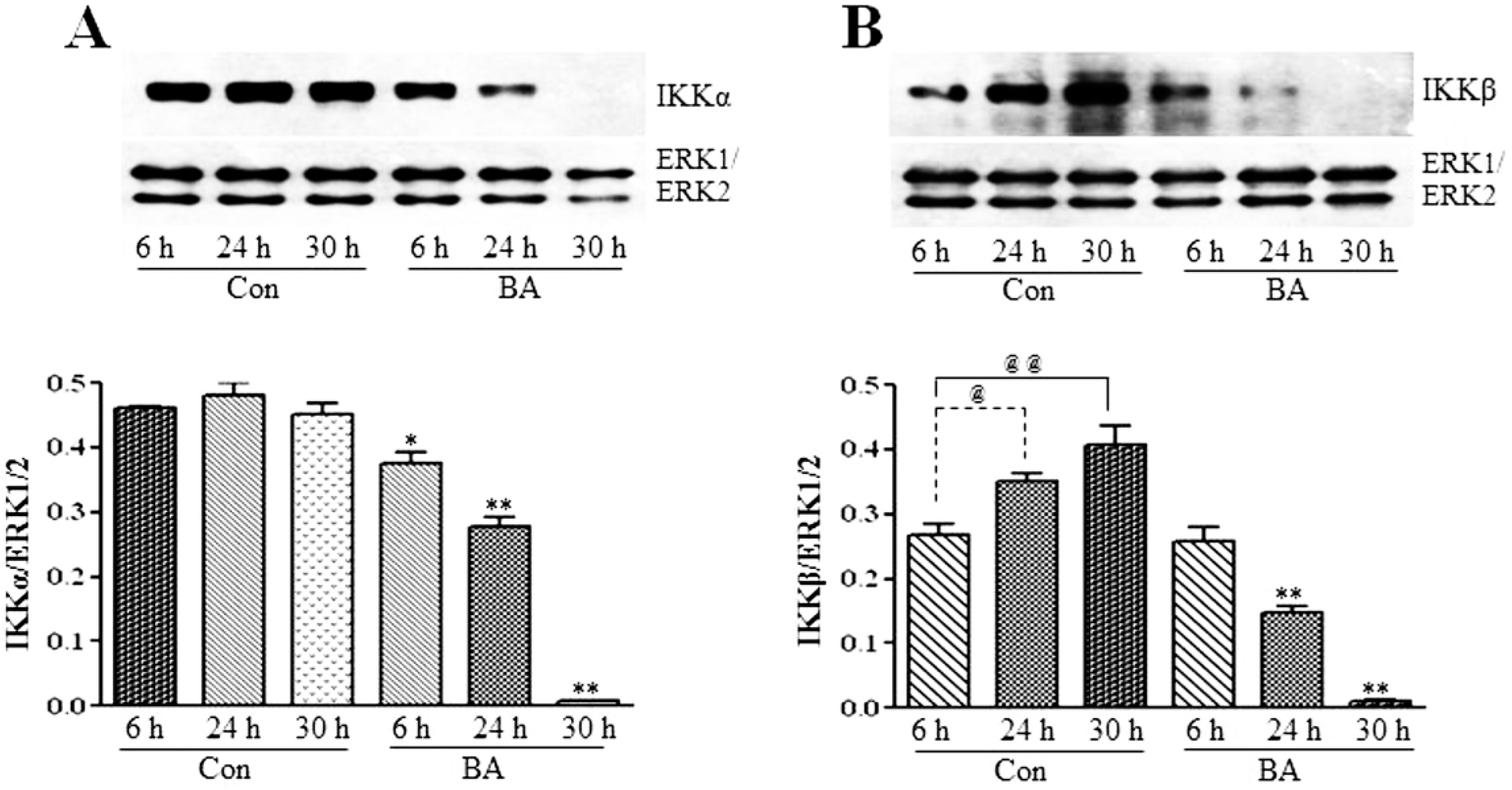
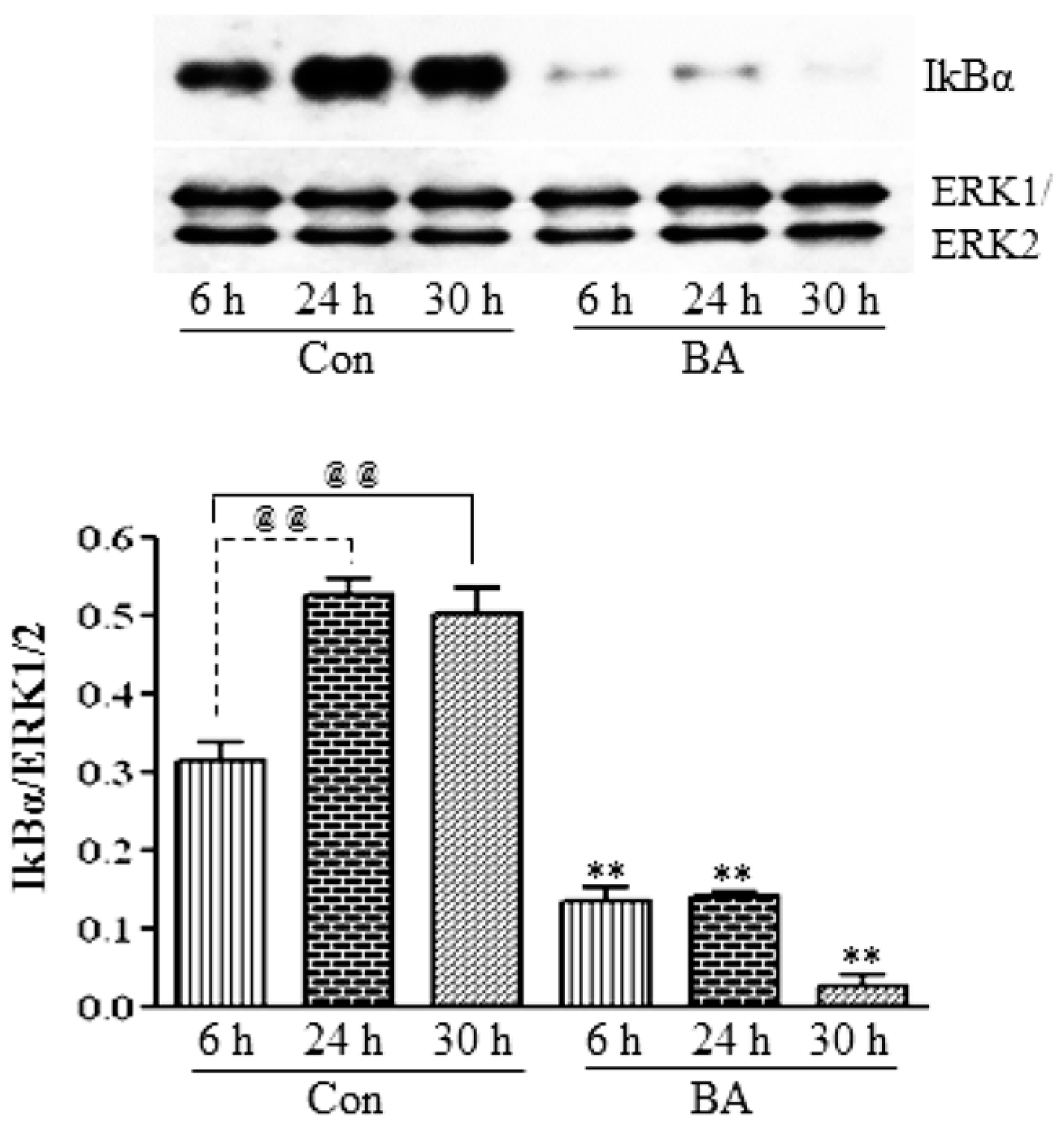
3.2.2. Butyrate Treatment Downregulates IKKα and IKKβ and Blocks IkBα Expression in VSMC
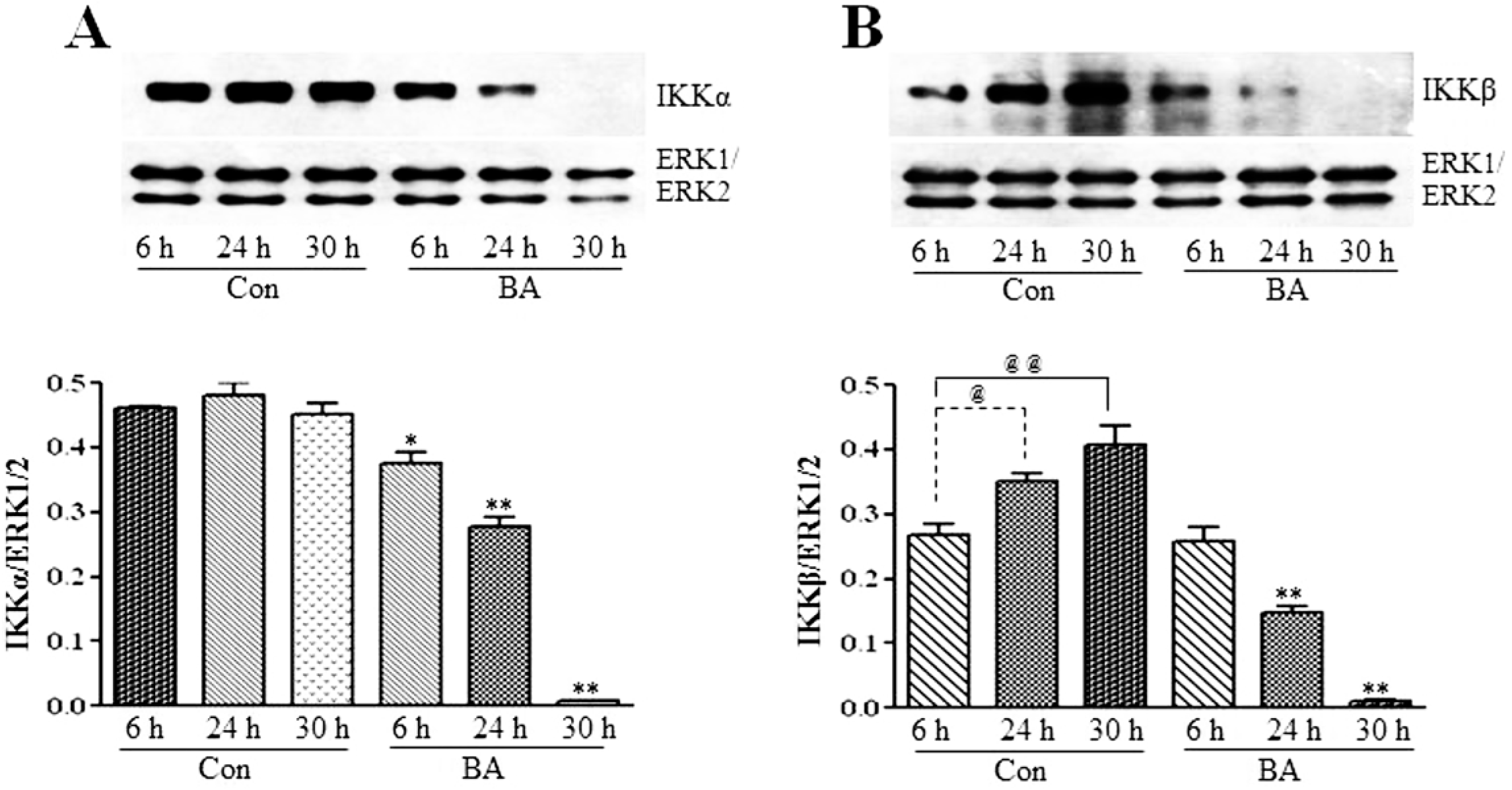
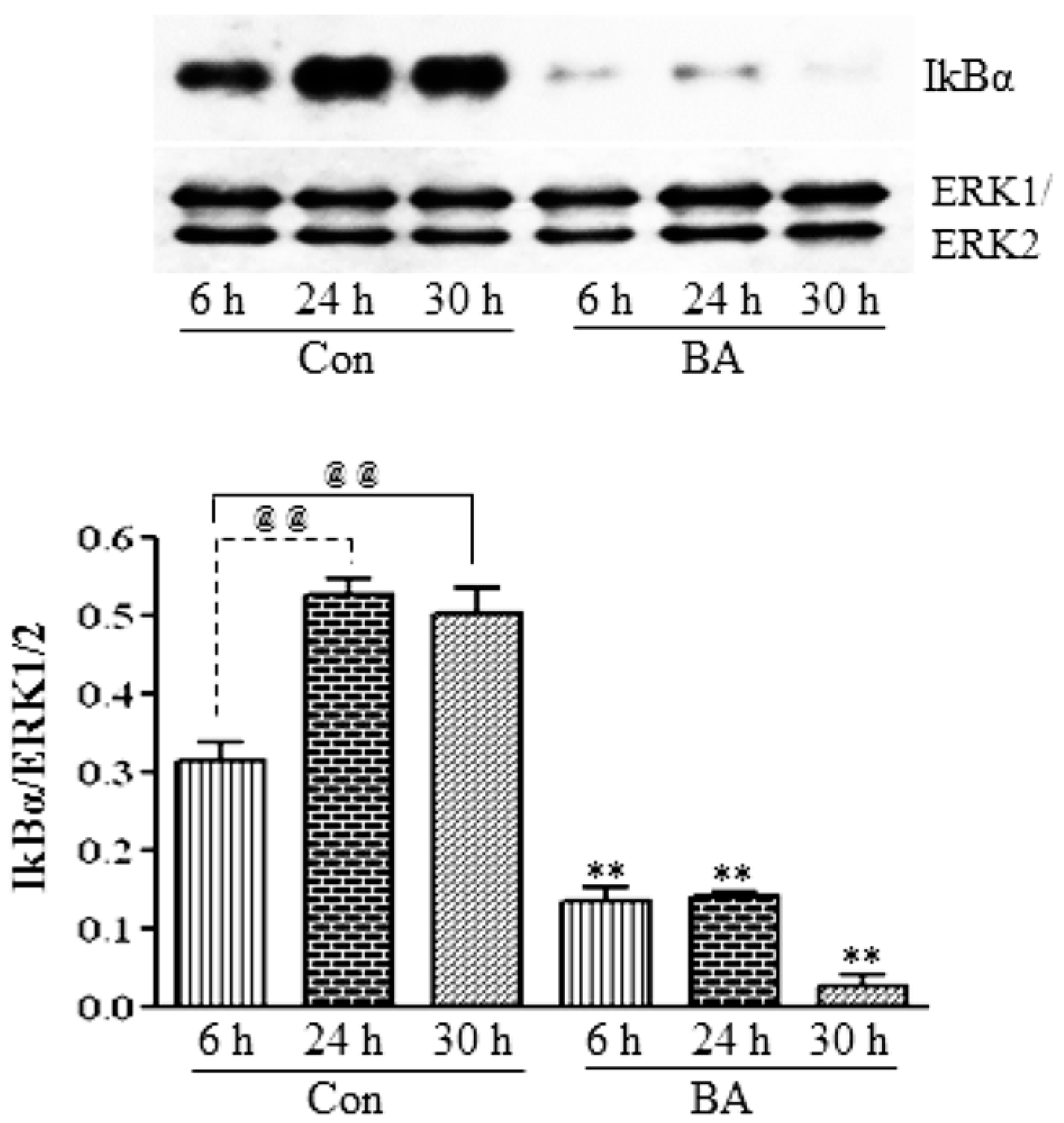
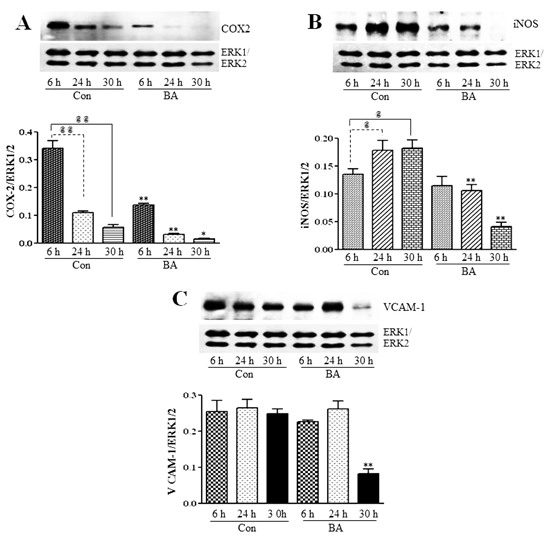
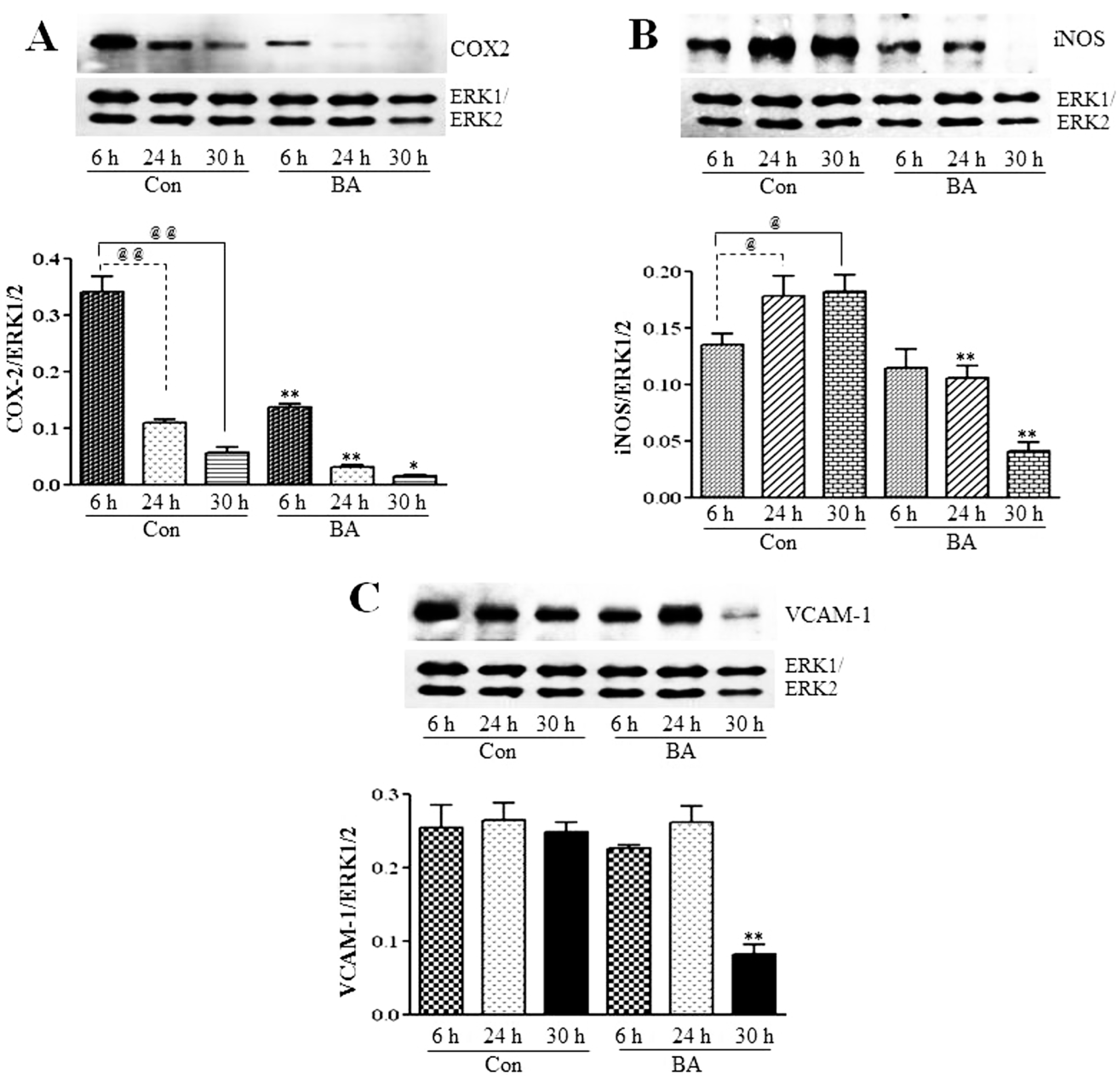
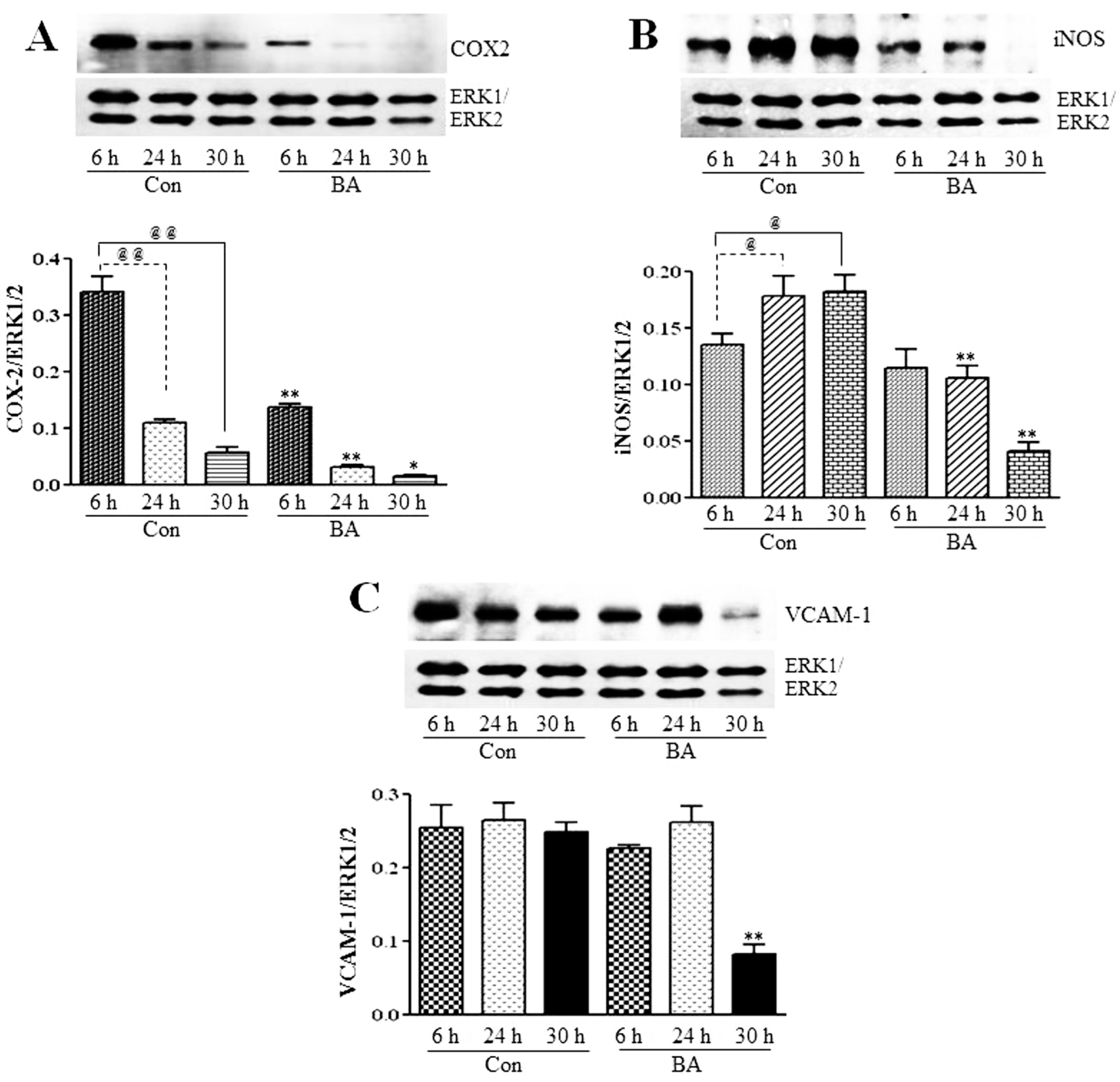
3.2.3. Attenuation of NF-κB Targets in Butyrate-Treated VSMC
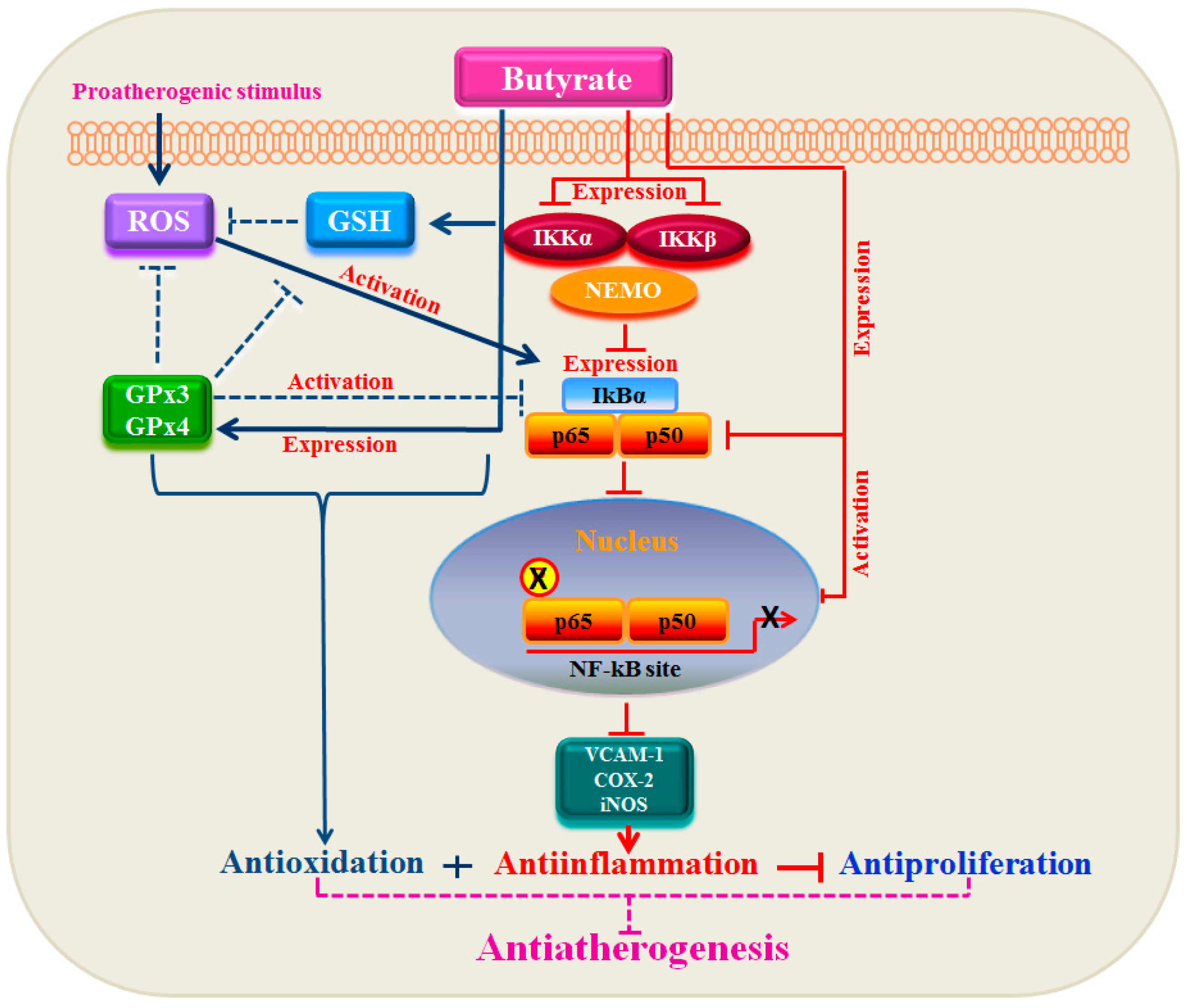
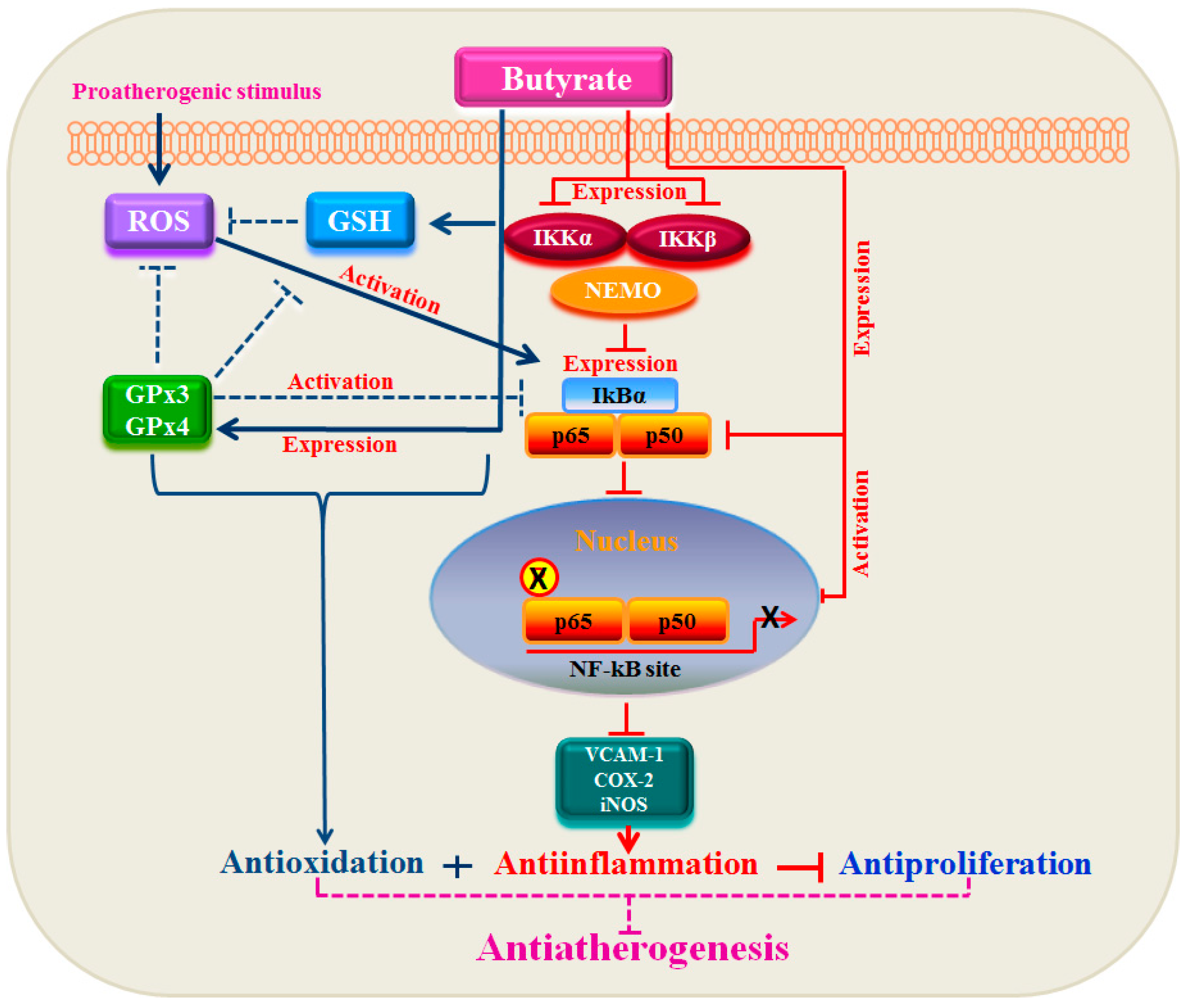
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Dzau, V.J.; Braun-Dullaeus, R.C.; Sedding, D.G. Vascular proliferation and atherosclerosis: New perspectives and therapeutic strategies. Nat. Med. 2002, 8, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Holmes, D.R., Jr. State of the art in coronary intervention. Am. J. Cardiol. 2003, 91, 50A–53A. [Google Scholar] [CrossRef] [PubMed]
- Melo, L.G.; Gnecchi, M.; Pachori, A.S.; Wang, K.; Dzau, V.J. Gene- and cell-based therapies for cardiovascular diseases: Current status and future directions. Eur. Heart. J. Suppl. 2004, 6, E24–E35. [Google Scholar] [CrossRef]
- Ranganna, K.; Yatsu, F.M.; Mathew, O.P. Insights into the pathogenesis and intervention of atherosclerosis. Vasc. Dis. Prev. 2006, 3, 375–390. [Google Scholar] [CrossRef]
- Ranganna, K.; Yatsu, F.M.; Hayes, B.E. Butyrate, a small pleiotropic molecule with multiple cellular and molecular actions: Its role as an anti-atherogenic agent. Recent Res. Dev. Mol. Cell. Biochem. 2005, 2, 123–151. [Google Scholar]
- Ekström, T.J. Epigenetic control of gene expression. Biochim. Biophys. Acta 2009, 1790, 845–846. [Google Scholar] [CrossRef] [PubMed]
- Pons, D.; de Vries, F.R.; van den Elsen, P.J.; Heijmans, B.T.; Quax, P.H.; Jukema, J.W. Epigenetic histone acetylation modifiers in vascular remodeling: New targets for therapy in cardiovascular disease. Eur. Heart J. 2009, 30, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Turunen, M.P.; Aavik, E.; Yla-Herttuala, S. Epigenetics and atherosclerosis. Biochim. Biophys. Acta 2009, 1790, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Ranganna, K.; Yatsu, F.M.; Mathew, O.P. Emerging Epigenetic Therapy for Vascular Proliferative Diseases. In Atherogenesis; Parthasarathy, S., Ed.; InTech: Rijeka, Croatia, 2011. [Google Scholar]
- Mathew, O.P.; Ranganna, K.; Yatsu, F.M. Butyrate, an HDAC inhibitor, stimulates interplay between different posttranslational modifications of histone H3 and differently alters G1- specific cell cycle proteins in vascular smooth muscle cells. Biomed. Pharmacother. 2010, 64, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Findeisen, H.M.; Gizard, F.; Zhao, Y.; Qing, H.; Heywood, E.B.; Jones, K.L.; Cohn, D.; Bruemmer, D. Epigenetic regulation of vascular smooth muscle cell proliferation and neointima formation by histone deacetylase inhibition. Arterioscler Thromb. Vasc. Biol. 2011, 31, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Ranganna, K.; Mathew, O.P.; Yatsu, F.M.; Yousefipour, Z.; Hayes, B.E.; Milton, S.G. Involvement of glutathione/glutathione S-transferase antioxidant system in butyrate-inhibited vascular smooth muscle cell proliferation. FEBS J. 2007, 274, 5962–5978. [Google Scholar] [CrossRef] [PubMed]
- Ranganna, K.; Yousefipour, Z.; Yatsu, F.M.; Milton, S.G.; Hayes, B.E. Gene expression profile of butyrate-inhibited vascular smooth muscle cell proliferation. Mol. Cell. Biochem. 2003, 254, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.F. Protection against free radical injury by selenoenzymes. Pharmacol. Ther. 1990, 45, 383–385. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.R. The glutathione peroxidases. Cell. Mol. Life. Sci. 2000, 57, 1825–1835. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R. Glutathione peroxidases and redox-regulated transcription factors. Biol. Chem. 2006, 387, 1329–1335. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohe, R.; Maiorino, M. Glutathione peroxidases. Biochim Biophys Acta. 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Rueckschloss, U.; Duerrschmidt, N.; Morawietz, H. NADPH oxidase in endothelial cells: Impact on atherosclerosis. Antioxid Redox Signal. 2003, 5, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Oxidant signals and oxidative stress. Curr. Opin. Cell Biol. 2003, 15, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, M.L.; Defranco, A.L. Prolonged production of reactive oxygen species in response to B cell receptor stimulation promotes B cell activation and proliferation. J Immunol. 2012, 189, 4405–4416. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Handy, D.E.; Loscalzo, J. Adenosine-dependent induction of glutathione peroxidase 1 in human primary endothelial cells and protection against oxidative stress. Circ. Res. 2005, 96, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Weiss, N.; Zhang, Y.Y.; Heydrick, S.; Bierl, C.; Loscalzo, J. Overexpression of cellular glutathione peroxidase rescues homocyst(e)ine-induced endothelial dysfunction. Proc. Natl. Acad. Sci. USA 2001, 98, 12503–12508. [Google Scholar] [CrossRef] [PubMed]
- Hoehn, B.; Yenari, M.A.; Sapolsky, R.M.; Steinberg, G.K. Glutathione peroxidase overexpression inhibits cytochrome C release and proapoptotic mediators to protect neurons from experimental stroke. Stroke 2003, 34, 2489–2494. [Google Scholar] [CrossRef] [PubMed]
- Shiomi, T.; Tsutsui, H.; Matsusaka, H.; Murakami, K.; Hayashidani, S.; Ikeuchi, M.; Wen, J.; Kubota, T.; Utsumi, H.; Takeshita, A. Overexpression of glutathione peroxidase prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation. 2004, 109, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Ran, Q.; Gu, M.; Van Remmen, H.; Strong, R.; Roberts, J.L.; Richardson, A. Glutathione peroxidase 4 protects cortical neurons from oxidative injury and amyloid toxicity. J. Neurosci. Res. 2006, 84, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Wenk, J.; Schüller, J.; Hinrichs, C.; Syrovets, T.; Azoitei, N.; Podda, M.; Wlaschek, M.; Brenneisen, P.; Schneider, L.A.; Sabiwalsky, A.; et al. Overexpression of phospholipid-hydroperoxide glutathione peroxidase in human dermal fibroblasts abrogates UVA irradiation-induced expression of interstitial collagenase/matrix metalloproteinase-1 by suppression of phosphatidylcholine hydroperoxide-mediated NFkappaB activation and interleukin-6 release. J. Biol. Chem. 2004, 279, 45634–45642. [Google Scholar] [CrossRef] [PubMed]
- Crack, P.J.; Taylor, J.M.; Ali, U.; Mansell, A.; Hertzog, P.J. Potential contribution of NF-kappaB in neuronal cell death in the glutathione peroxidase-1 knockout mouse in response to ischemia-reperfusion injury. Stroke 2006, 37, 1533–1538. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; West, A.P.; Ghosh, S. NF-kappaB and the immune response. Oncogene 2006, 25, 6758–6580. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef] [PubMed]
- Banning, A.; Schnurr, K.; Böl, G.F.; Kupper, D.; Müller-Schmehl, K.; Viita, H.; Ylä- Herttuala, S.; Brigelius-Flohé, R. Inhibition of basal and interleukin-1-induced VCAM-1 expression by phospholipid hydroperoxide glutathione peroxidase and 15-lipoxygenase in rabbit aortic smooth muscle cells. Free Radic. Biol. Med. 2004, 36, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Friedrichs, B.; Maurer, S.; Schultz, M.; Streicher, R. Interleukin-1-induced nuclear factor kB activation is inhibited by overexpression of phospholipid hydroperoxide glutathione peroxidase in a human endothelial cell line. Biochem. J. 1997, 328, 199–203. [Google Scholar] [PubMed]
- Lührs, H.; Gerke, T.; Müller, J.G.; Melcher, R.; Schauber, J.; Boxberge, F.; Scheppach, W.; Menzel, T. Butyrate inhibits NF-kappaB activation in lamina propria macrophages of patients with ulcerative colitis. Scand. J. Gastroenterol. 2002, 37, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Segain, J.P.; Raingeard de la Blétière, D.; Bourreille, A.; Leray, V.; Gervois, N.; Rosales, C.; Ferrier, L.; Bonnet, C.; Blottière, H.M.; Galmiche, J.P. Butyrate inhibits inflammatory responses through NFkappaB inhibition: implications for Crohn’s disease. Gut 2000, 47, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Milton, S.G.; Mathew, O.P.; Yatsu, F.M.; Ranganna, K. Differential cellular and molecular effects of butyrate and trichostatin A on vascular smooth muscle cells. Pharmaceuticals 2012, 5, 925–943. [Google Scholar] [CrossRef] [PubMed]
- Chabory, E.; Damon, C.; Lenoir, A.; Henry-Berger, J.; Vernet, P.; Cadet, R.; Saez, F.; Drevet, J.R. Mammalian glutathione peroxidases control acquisition and maintenance of spermatozoa integrity. J. Anim. Sci. 2010, 88, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Miranda, S.G.; Wang, Y.J.; Purdie, N.G.; Osborne, V.R.; Coomber, B.L.; Cant, J.P. Senenomethionine stimulates expression of glutathione peroxidase 1 and 3 and growth of bovine mammary epithelial cell in primary culture. J. Diary Sci. 2009, 92, 2670–2683. [Google Scholar] [CrossRef]
- Kang, D.H.; Kang, S.W. Targeting cellular antioxidant enzymes for targeting atherosclerotin vascular disease. Biomol. Ther. 2013, 21, 89–96. [Google Scholar] [CrossRef]
- Buijsse, B.; Lee, D-H.; Steffen, L.; Erickson, R.R.; Luepker, R.V.; Jacobs, D.R., Jr.; Holtzman, J.L. Low serum dlutathione peroxidase activity is associated with cardiovascular mortality in individuals with low HDLc’s. PloS ONE 2012, 7, e38901. [Google Scholar] [CrossRef] [PubMed]
- Voetsch, B.R.; Jin, C.; Bierl, C.; Benke, K.S.; Kenet, G.; Simioni, P.; Ottaviano, F.; Damasceno, B.P.; Annichino-Bizacchi, J.M.; Handy, D.E.; Loscalzo, J. Promoter polymorphisms in the plasma glutathione peroxidase (GPx-3) gene: a novel risk factor for arterial ischemic stroke among young adults and children. Stroke 2007, 38, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Savaskan, N.E.; Ufer, C.; Kühn, H.; Borchert, A. Molecular biology of glutathione peroxidase 4: from genomic structure to developmental expression and neural function. Biol. Chem. 2007, 388, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Ran, Q.; Roberts, L.J., 2nd.; Zhou, L.; Richardson, A.; Sharan, C.; Wu, D.; Yang, H. Suppression of atherogenesis by overexpression of glutathione peroxidase-4 in apolipoprotein E-deficient mice. Free Radic. Biol. Med. 2008, 44, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Borchert, A.; Wang, C.C.; Ufer, C.; Schiebel, H.; Savaskan, N.E.; Kuhn, H. The role of phospholipid hydroperoxide glutathione peroxidase isoforms in murine embryogenesis. J. Biol. Chem. 2006, 28, 19655–19664. [Google Scholar] [CrossRef]
- Witztum, J.L.; Steinberg, D. The oxidative modification hypothesis of atherosclerosis: Does it hold for humans? Trends Cardiovasc. Med. 2001, 11, 93–102. [Google Scholar]
- Raines, E.W.; Ross, R. Smooth muscle cells and the pathogenesis of the lesions of atherosclerosis. Br. Heart J. 1993, 69, S30–S37. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Maurer, S.; Lötzer, K.; Böl, G.; Kallionpää, H.; Lehtolainen, P.; Viita, H.; Ylä-Herttuala, S. Overexpression of PHGPx inhibits hydroperoxide-induced oxidation, NFkappaB activation and apoptosis and affects oxLDL-mediated proliferation of rabbit aortic smooth muscle cells. Atherosclerosis 2000, 152, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Ran, Q.; Liang, H.; Gu, M.; Qi, W.; Walter, C.A.; Roberts, L.J., 2nd.; Herman, B.; Richardson, A.; Van Remmen, H. Transgenic mice overexpressing glutathione peroxidase 4 are protected against oxidative stress-induced apoptosis. J. Biol. Chem. 2004, 279, 55137–55146. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Tang, E.; Guan, K.; Wang, C.Y. IKK beta plays an essential role in the phosphorylation of RelA/p65 on serine 536 induced by lipopolysaccharide. J. Immunol. 2003, 170, 5630–5635. [Google Scholar] [CrossRef] [PubMed]
- Häcker, H.; Karin, M. Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006, 2006, re13. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.F.; Williams, S.A.; Mu, Y.; Nakano, H.; Duerr, J.M.; Buckbinder, L.; Greene, W.C. NF-κB RelA phosphorylation regulates RelA acetylation. Mol. Cell Biol. 2005, 25, 7966–7975. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene. 2006, 25, 6717–6730. [Google Scholar] [CrossRef] [PubMed]
- Melnyk, J.; Tsao, R.; Marcone, M.F. How natural dietary antioxidants in fruits, vegetables and legumes promote vascular health. Food Res. Intl. 2011, 44, 14–22. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, H.J.; Um, S.H.; Sohn, E.H.; Kim, B.O.; Moon, E.Y.; Rhee, D.K.; Pyo, S. ulforaphane suppresses vascular adhesion molecule-1 expression in TNF-α-stimulated mouse vascular smooth muscle cells: involvement of the MAPK, NF-κB and AP-1 signaling pathways. Vascul. Pharmacol. 2012, 56, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Van Kuiken, M.E.; Iyer, L.H.; Harikumar, K.B.; Sung, B. Molecular targets of nutraceuticals derived from dietary spices: potential role in suppression of inflammation and tumorigenesis. Exp. Biol. Med. 2009, 234, 825–849. [Google Scholar] [CrossRef]
- Vinolo, M.A.; Rodrigues, H.G.; Nachbar, R.T.; Curi, R. Regulation of inflammation by short chain fatty acids. Nutrients 2011, 3, 858–876. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.; Luciani, A.; De Cicco, P.; Troncone, E.; Ciacci, C. Butyrate attenuates lipopolysaccharide-induced inflammation in intestinal cells and Crohn’s mucosa through modulation of antioxidant defense machinery. PLoS One 2012, 7, e32841. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathew, O.P.; Ranganna, K.; Milton, S.G. Involvement of the Antioxidant Effect and Anti-inflammatory Response in Butyrate-Inhibited Vascular Smooth Muscle Cell Proliferation. Pharmaceuticals 2014, 7, 1008-1027. https://doi.org/10.3390/ph7111008
Mathew OP, Ranganna K, Milton SG. Involvement of the Antioxidant Effect and Anti-inflammatory Response in Butyrate-Inhibited Vascular Smooth Muscle Cell Proliferation. Pharmaceuticals. 2014; 7(11):1008-1027. https://doi.org/10.3390/ph7111008
Chicago/Turabian StyleMathew, Omana P., Kasturi Ranganna, and Shirlette G. Milton. 2014. "Involvement of the Antioxidant Effect and Anti-inflammatory Response in Butyrate-Inhibited Vascular Smooth Muscle Cell Proliferation" Pharmaceuticals 7, no. 11: 1008-1027. https://doi.org/10.3390/ph7111008
APA StyleMathew, O. P., Ranganna, K., & Milton, S. G. (2014). Involvement of the Antioxidant Effect and Anti-inflammatory Response in Butyrate-Inhibited Vascular Smooth Muscle Cell Proliferation. Pharmaceuticals, 7(11), 1008-1027. https://doi.org/10.3390/ph7111008