MicroRNAs as Molecular Targets for Cancer Therapy: On the Modulation of MicroRNA Expression

Abstract
:1. Introduction
2. MiRNA Biogenesis and Gene Silencing Mechanisms
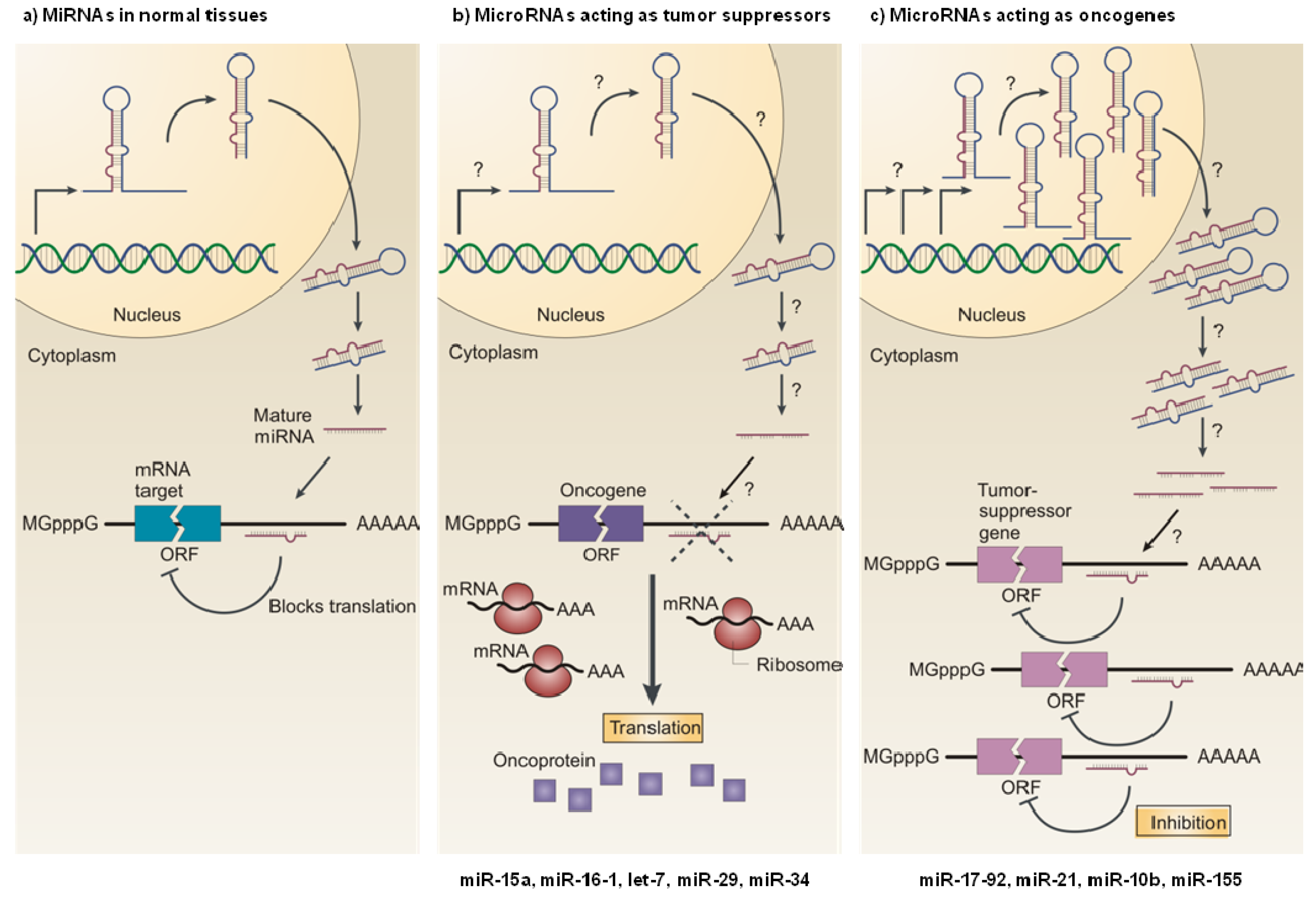
3. Role of MiRNAs in Cancer
3.1. MiRNAs as Tumor Suppressors

3.2. MiRNAs as Oncogenes
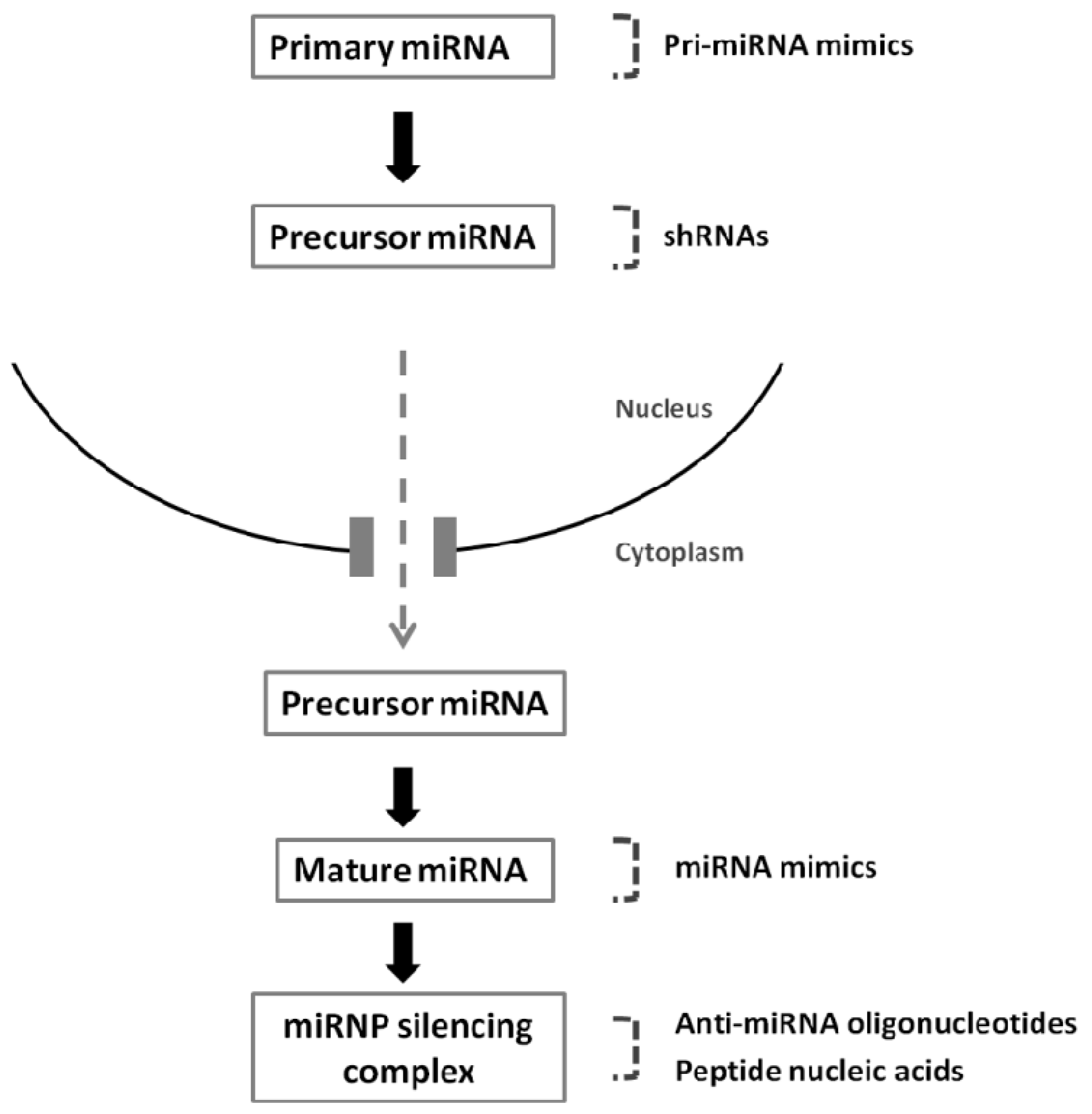
4. Therapeutic Modulation of MiRNAs
4.1. Silencing of MiRNAs: Targeting MiRNAs Overexpressed in Cancer
4.2. Overexpression of MiRNAs: Re-Expressing MiRNAs Downregulated in Cancer

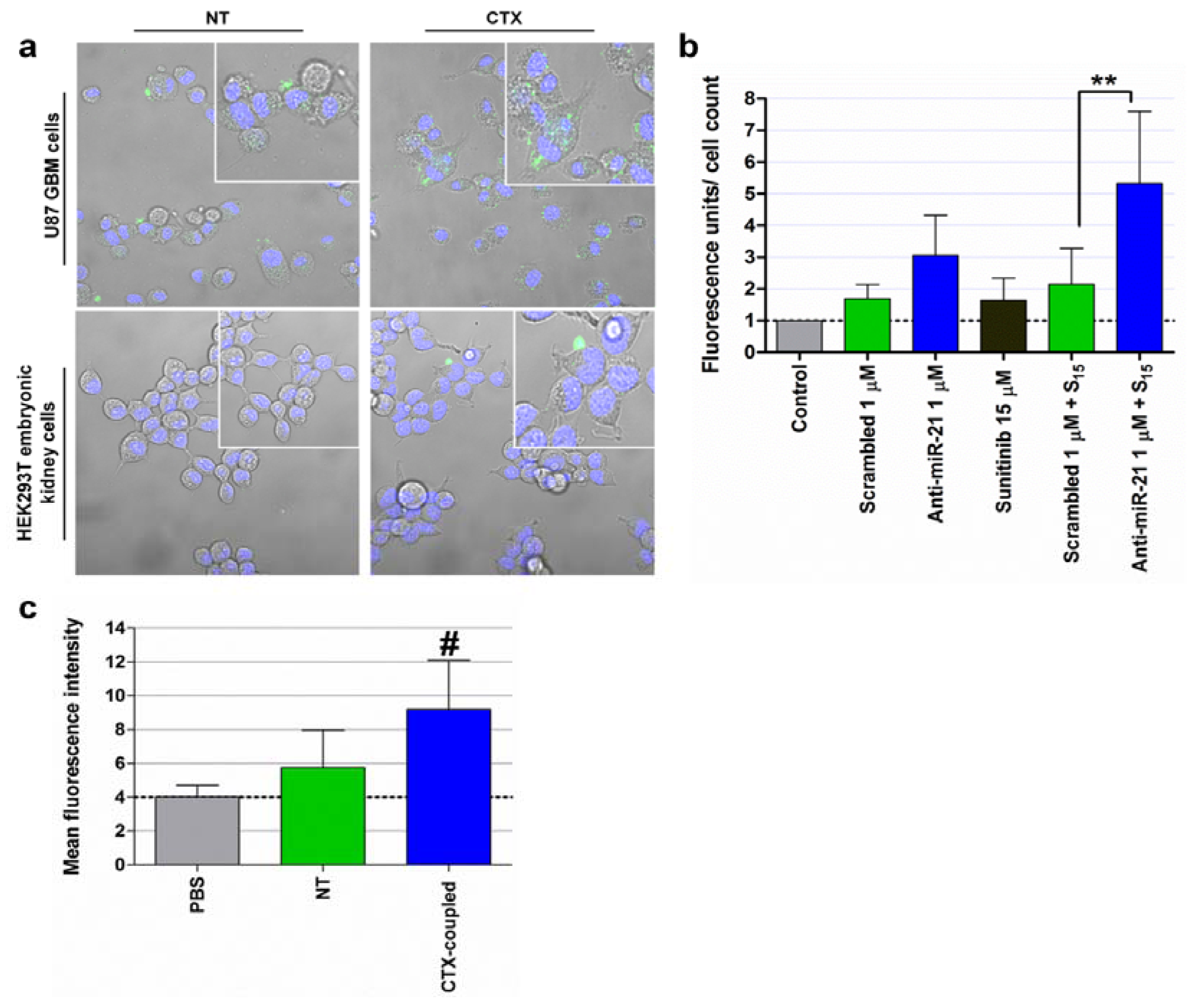
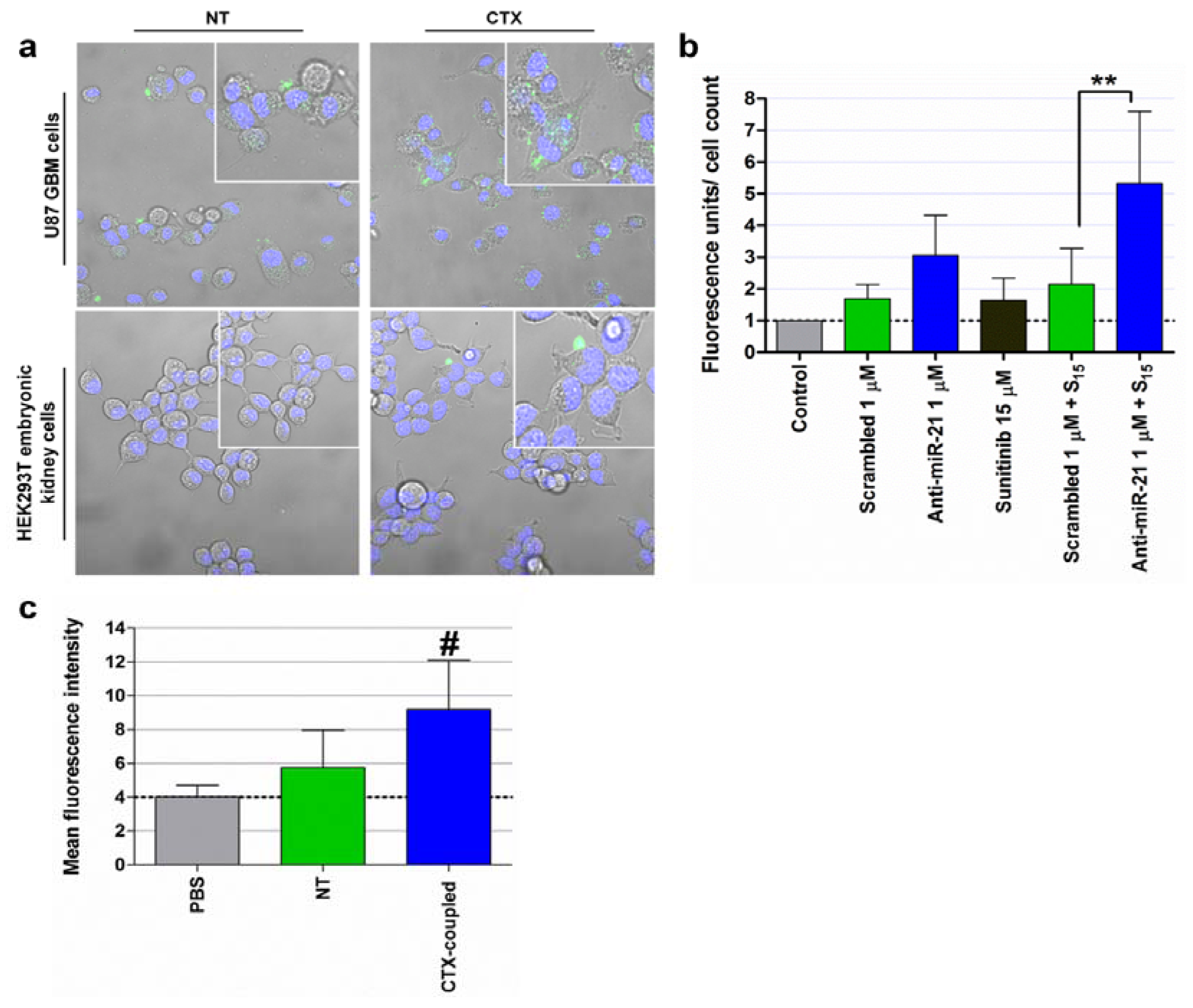
4.3. Delivery of Nucleic Acids to Modulate miRNA Function
4.3.1. Viral Vectors

{kind=link}
{kind=link}
{kind=link}
| Carrier | Disease | Target miRNA (s) and
role in cancer | References | |
|---|---|---|---|---|
| Viruses | Adeno-associated viruses(AAV8) | Hepatocellular cancer | miR-26 tumor suppressor | [140] |
| Adenoviruses | Lung cancer | let-7 tumor suppressor | [141] | |
| Adenoviruses | Glioblastoma | miR-145 tumor suppressor | [144] | |
| Adenoviruses | Glioblastoma | miR-221-222 oncogene | [145] | |
| Lentiviruses | Prostate cancer | miR-15-16 tumor suppressor | [142] | |
| Lentiviruses | Pancreatic cancer | miR-21 oncogene | [143] | |
| Lipid-based nanoparticles | Cationic liposomes | Breast cancer | miR-34a tumor suppressor | [124] |
| Cationic liposomes | Pancreatic cancer | miR-34a, miR-143-145 tumor suppressors | [146] | |
| Neutral lipid emulsion© | Lung cancer | miR-34a, let-7 tumor suppressors | [147] | |
| Stable nucleic acid lipid particles | Glioblastoma | miR-21 oncogene | [148] | |
| Polymer-based nanoparticles | Polyurethane | Glioblastoma | miR-145 tumor suppressor | [149] |
| Poly(lactic-co-glycolic acid) | Lymphoma | miR-155 oncogene | [96] | |
| Polyamidoamine | Glioblastoma | miR-21 oncogene | [150] | |
4.3.2. Non-Viral Vectors
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Alvarez-Garcia, I.; Miska, E.A. Microrna functions in animal development and human disease. Development 2005, 132, 4653–4662. [Google Scholar] [CrossRef]
- Ambros, V. The functions of animal micrornas. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Croce, C.M. Causes and consequences of microrna dysregulation in cancer. Nat. Rev. Genet. 2009, 10, 704–714. [Google Scholar] [CrossRef]
- Stark, A.; Brennecke, J.; Bushati, N.; Russell, R.B.; Cohen, S.M. Animal micrornas confer robustness to gene expression and have a significant impact on 3'utr evolution. Cell 2005, 123, 1133–1146. [Google Scholar] [CrossRef]
- Liu, N.; Olson, E.N. Microrna regulatory networks in cardiovascular development. Dev. Cell 2010, 18, 510–525. [Google Scholar] [CrossRef]
- Gangaraju, V.K.; Lin, H. Micrornas: Key regulators of stem cells. Nat. Rev. Mol. Cell Biol. 2009, 10, 116–125. [Google Scholar] [CrossRef]
- O'Connell, R.M.; Rao, D.S.; Chaudhuri, A.A.; Baltimore, D. Physiological and pathological roles for micrornas in the immune system. Nat. Rev. Immunol. 2010, 10, 111–122. [Google Scholar] [CrossRef]
- Poy, M.N.; Eliasson, L.; Krutzfeldt, J.; Kuwajima, S.; Ma, X.; Macdonald, P.E.; Pfeffer, S.; Tuschl, T.; Rajewsky, N.; Rorsman, P.; et al. A pancreatic islet-specific microrna regulates insulin secretion. Nature 2004, 432, 226–230. [Google Scholar] [CrossRef]
- Lecellier, C.H.; Dunoyer, P.; Arar, K.; Lehmann-Che, J.; Eyquem, S.; Himber, C.; Saib, A.; Voinnet, O. A cellular microrna mediates antiviral defense in human cells. Science 2005, 308, 557–560. [Google Scholar] [CrossRef]
- Nelson, P.T.; Wang, W.X.; Rajeev, B.W. Micrornas (mirnas) in neurodegenerative diseases. Brain Pathol. 2008, 18, 130–138. [Google Scholar] [CrossRef]
- Small, E.M.; Olson, E.N. Pervasive roles of micrornas in cardiovascular biology. Nature 2011, 469, 336–342. [Google Scholar] [CrossRef]
- Pandey, A.K.; Agarwal, P.; Kaur, K.; Datta, M. Micrornas in diabetes: Tiny players in big disease. Cell. Physiol. Biochem. 2009, 23, 221–232. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. Microrna signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef]
- Ruby, J.G.; Jan, C.H.; Bartel, D.P. Intronic microrna precursors that bypass drosha processing. Nature 2007, 448, 83–86. [Google Scholar] [CrossRef]
- Meister, G.; Landthaler, M.; Patkaniowska, A.; Dorsett, Y.; Teng, G.; Tuschl, T. Human argonaute2 mediates rna cleavage targeted by mirnas and sirnas. Mol. Cell 2004, 15, 185–197. [Google Scholar] [CrossRef]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by micrornas: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar]
- Perron, M.P.; Provost, P. Protein interactions and complexes in human microrna biogenesis and function. Front. Biosci. 2008, 13, 2537–2547. [Google Scholar] [CrossRef]
- Bartel, D.P. Micrornas: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Brennecke, J.; Stark, A.; Russell, R.B.; Cohen, S.M. Principles of microrna-target recognition. PLoS Biol. 2005, 3, e85. [Google Scholar] [CrossRef]
- Liu, X.; Fortin, K.; Mourelatos, Z. Micrornas: Biogenesis and molecular functions. Brain Pathol. 2008, 18, 113–121. [Google Scholar] [CrossRef]
- Kiriakidou, M.; Nelson, P.T.; Kouranov, A.; Fitziev, P.; Bouyioukos, C.; Mourelatos, Z.; Hatzigeorgiou, A. A combined computational-experimental approach predicts human microrna targets. Genes Dev. 2004, 18, 1165–1178. [Google Scholar] [CrossRef]
- Fabian, M.R.; Sonenberg, N. The mechanics of mirna-mediated gene silencing: A look under the hood of mirisc. Nat. Struct. Mol. Biol. 2012, 19, 586–593. [Google Scholar] [CrossRef]
- Chendrimada, T.P.; Finn, K.J.; Ji, X.; Baillat, D.; Gregory, R.I.; Liebhaber, S.A.; Pasquinelli, A.E.; Shiekhattar, R. Microrna silencing through risc recruitment of eif6. Nature 2007, 447, 823–828. [Google Scholar] [CrossRef]
- Fabian, M.R.; Mathonnet, G.; Sundermeier, T.; Mathys, H.; Zipprich, J.T.; Svitkin, Y.V.; Rivas, F.; Jinek, M.; Wohlschlegel, J.; Doudna, J.A.; et al. Mammalian mirna risc recruits caf1 and pabp to affect pabp-dependent deadenylation. Mol. Cell 2009, 35, 868–880. [Google Scholar] [CrossRef]
- Baek, D.; Villen, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The impact of micrornas on protein output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian micrornas predominantly act to decrease target mrna levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef]
- Selbach, M.; Schwanhausser, B.; Thierfelder, N.; Fang, Z.; Khanin, R.; Rajewsky, N. Widespread changes in protein synthesis induced by micrornas. Nature 2008, 455, 58–63. [Google Scholar] [CrossRef]
- Behm-Ansmant, I.; Rehwinkel, J.; Doerks, T.; Stark, A.; Bork, P.; Izaurralde, E. Mrna degradation by mirnas and gw182 requires both ccr4:Not deadenylase and dcp1:Dcp2 decapping complexes. Genes Dev. 2006, 20, 1885–1898. [Google Scholar] [CrossRef]
- Giraldez, A.J.; Mishima, Y.; Rihel, J.; Grocock, R.J.; Van Dongen, S.; Inoue, K.; Enright, A.J.; Schier, A.F. Zebrafish mir-430 promotes deadenylation and clearance of maternal mrnas. Science 2006, 312, 75–79. [Google Scholar] [CrossRef]
- Wu, L.; Fan, J.; Belasco, J.G. Micrornas direct rapid deadenylation of mrna. Proc. Natl. Acad. Sci. USA 2006, 103, 4034–4039. [Google Scholar] [CrossRef]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro-rna genes mir15 and mir16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microrna genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef]
- Kumar, M.S.; Lu, J.; Mercer, K.L.; Golub, T.R.; Jacks, T. Impaired microrna processing enhances cellular transformation and tumorigenesis. Nat. Genet. 2007, 39, 673–677. [Google Scholar] [CrossRef]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. Microrna expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microrna expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef]
- Calin, G.A.; Ferracin, M.; Cimmino, A.; Di Leva, G.; Shimizu, M.; Wojcik, S.E.; Iorio, M.V.; Visone, R.; Sever, N.I.; Fabbri, M.; et al. A microrna signature associated with prognosis and progression in chronic lymphocytic leukemia. N. Engl. J. Med. 2005, 353, 1793–1801. [Google Scholar] [CrossRef]
- Yanaihara, N.; Caplen, N.; Bowman, E.; Seike, M.; Kumamoto, K.; Yi, M.; Stephens, R.M.; Okamoto, A.; Yokota, J.; Tanaka, T.; et al. Unique microrna molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 2006, 9, 189–198. [Google Scholar] [CrossRef]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—Micrornas with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. Mir-15 and mir-16 induce apoptosis by targeting bcl2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef]
- Ambs, S.; Prueitt, R.L.; Yi, M.; Hudson, R.S.; Howe, T.M.; Petrocca, F.; Wallace, T.A.; Liu, C.G.; Volinia, S.; Calin, G.A.; et al. Genomic profiling of microrna and messenger rna reveals deregulated microrna expression in prostate cancer. Cancer Res. 2008, 68, 6162–6170. [Google Scholar] [CrossRef]
- Roccaro, A.M.; Sacco, A.; Thompson, B.; Leleu, X.; Azab, A.K.; Azab, F.; Runnels, J.; Jia, X.; Ngo, H.T.; Melhem, M.R.; et al. Micrornas 15a and 16 regulate tumor proliferation in multiple myeloma. Blood 2009, 113, 6669–6680. [Google Scholar] [CrossRef]
- Akao, Y.; Nakagawa, Y.; Naoe, T. Let-7 microrna functions as a potential growth suppressor in human colon cancer cells. Biol. Pharm. Bull. 2006, 29, 903–906. [Google Scholar] [CrossRef]
- Iorio, M.V.; Ferracin, M.; Liu, C.G.; Veronese, A.; Spizzo, R.; Sabbioni, S.; Magri, E.; Pedriali, M.; Fabbri, M.; Campiglio, M.; et al. Microrna gene expression deregulation in human breast cancer. Cancer Res. 2005, 65, 7065–7070. [Google Scholar] [CrossRef]
- Esquela-Kerscher, A.; Trang, P.; Wiggins, J.F.; Patrawala, L.; Cheng, A.; Ford, L.; Weidhaas, J.B.; Brown, D.; Bader, A.G.; Slack, F.J. The let-7 microrna reduces tumor growth in mouse models of lung cancer. Cell Cycle 2008, 7, 759–764. [Google Scholar] [CrossRef]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. Ras is regulated by the let-7 microrna family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef]
- Lee, Y.S.; Dutta, A. The tumor suppressor microrna let-7 represses the hmga2 oncogene. Genes Dev. 2007, 21, 1025–1030. [Google Scholar] [CrossRef]
- Sampson, V.B.; Rong, N.H.; Han, J.; Yang, Q.; Aris, V.; Soteropoulos, P.; Petrelli, N.J.; Dunn, S.P.; Krueger, L.J. Microrna let-7a down-regulates myc and reverts myc-induced growth in burkitt lymphoma cells. Cancer Res. 2007, 67, 9762–9770. [Google Scholar] [CrossRef]
- Xiong, Y.; Fang, J.H.; Yun, J.P.; Yang, J.; Zhang, Y.; Jia, W.H.; Zhuang, S.M. Effects of microrna-29 on apoptosis, tumorigenicity, and prognosis of hepatocellular carcinoma. Hepatology 2010, 51, 836–845. [Google Scholar]
- Fabbri, M.; Garzon, R.; Cimmino, A.; Liu, Z.; Zanesi, N.; Callegari, E.; Liu, S.; Alder, H.; Costinean, S.; Fernandez-Cymering, C.; et al. Microrna-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3a and 3b. Proc. Natl. Acad. Sci. USA 2007, 104, 15805–15810. [Google Scholar] [CrossRef]
- Corney, D.C.; Hwang, C.I.; Matoso, A.; Vogt, M.; Flesken-Nikitin, A.; Godwin, A.K.; Kamat, A.A.; Sood, A.K.; Ellenson, L.H.; Hermeking, H.; et al. Frequent downregulation of mir-34 family in human ovarian cancers. Clin. Cancer Res. 2010, 16, 1119–1128. [Google Scholar] [CrossRef]
- Fabbri, M.; Bottoni, A.; Shimizu, M.; Spizzo, R.; Nicoloso, M.S.; Rossi, S.; Barbarotto, E.; Cimmino, A.; Adair, B.; Wojcik, S.E.; et al. Association of a microrna/tp53 feedback circuitry with pathogenesis and outcome of b-cell chronic lymphocytic leukemia. JAMA 2011, 305, 59–67. [Google Scholar] [CrossRef]
- Tanaka, N.; Toyooka, S.; Soh, J.; Kubo, T.; Yamamoto, H.; Maki, Y.; Muraoka, T.; Shien, K.; Furukawa, M.; Ueno, T.; et al. Frequent methylation and oncogenic role of microrna-34b/c in small-cell lung cancer. Lung Cancer 2011, 76, 32–38. [Google Scholar]
- Vogt, M.; Munding, J.; Gruner, M.; Liffers, S.T.; Verdoodt, B.; Hauk, J.; Steinstraesser, L.; Tannapfel, A.; Hermeking, H. Frequent concomitant inactivation of mir-34a and mir-34b/c by cpg methylation in colorectal, pancreatic, mammary, ovarian, urothelial, and renal cell carcinomas and soft tissue sarcomas. Virchows Arch. 2011, 458, 313–322. [Google Scholar] [CrossRef]
- Li, Y.; Guessous, F.; Zhang, Y.; Dipierro, C.; Kefas, B.; Johnson, E.; Marcinkiewicz, L.; Jiang, J.; Yang, Y.; Schmittgen, T.D.; et al. Microrna-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res. 2009, 69, 7569–7576. [Google Scholar] [CrossRef]
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A microrna component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar] [CrossRef]
- Rokhlin, O.W.; Scheinker, V.S.; Taghiyev, A.F.; Bumcrot, D.; Glover, R.A.; Cohen, M.B. Microrna-34 mediates ar-dependent p53-induced apoptosis in prostate cancer. Cancer Biol. Ther. 2008, 7, 1288–1296. [Google Scholar] [CrossRef]
- Garzon, R.; Calin, G.A.; Croce, C.M. Micrornas in cancer. Annu. Rev. Med. 2009, 60, 167–179. [Google Scholar] [CrossRef]
- He, L.; Thomson, J.M.; Hemann, M.T.; Hernando-Monge, E.; Mu, D.; Goodson, S.; Powers, S.; Cordon-Cardo, C.; Lowe, S.W.; Hannon, G.J.; et al. A microrna polycistron as a potential human oncogene. Nature 2005, 435, 828–833. [Google Scholar] [CrossRef]
- Ota, A.; Tagawa, H.; Karnan, S.; Tsuzuki, S.; Karpas, A.; Kira, S.; Yoshida, Y.; Seto, M. Identification and characterization of a novel gene, c13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res. 2004, 64, 3087–3095. [Google Scholar] [CrossRef]
- Mendell, J.T. Miriad roles for the mir-17-92 cluster in development and disease. Cell 2008, 133, 217–222. [Google Scholar] [CrossRef]
- Dews, M.; Homayouni, A.; Yu, D.; Murphy, D.; Sevignani, C.; Wentzel, E.; Furth, E.E.; Lee, W.M.; Enders, G.H.; Mendell, J.T.; et al. Augmentation of tumor angiogenesis by a myc-activated microrna cluster. Nat. Genet. 2006, 38, 1060–1065. [Google Scholar] [CrossRef]
- O'Donnell, K.A.; Wentzel, E.A.; Zeller, K.I.; Dang, C.V.; Mendell, J.T. C-myc-regulated micrornas modulate e2f1 expression. Nature 2005, 435, 839–843. [Google Scholar] [CrossRef]
- Ivanovska, I.; Ball, A.S.; Diaz, R.L.; Magnus, J.F.; Kibukawa, M.; Schelter, J.M.; Kobayashi, S.V.; Lim, L.; Burchard, J.; Jackson, A.L.; et al. Micrornas in the mir-106b family regulate p21/cdkn1a and promote cell cycle progression. Mol. Cell. Biol. 2008, 28, 2167–2174. [Google Scholar] [CrossRef]
- Bloomston, M.; Frankel, W.L.; Petrocca, F.; Volinia, S.; Alder, H.; Hagan, J.P.; Liu, C.G.; Bhatt, D.; Taccioli, C.; Croce, C.M. Microrna expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. JAMA 2007, 297, 1901–1908. [Google Scholar] [CrossRef]
- Meng, F.; Henson, R.; Wehbe-Janek, H.; Ghoshal, K.; Jacob, S.T.; Patel, T. Microrna-21 regulates expression of the pten tumor suppressor gene in human hepatocellular cancer. Gastroenterology 2007, 133, 647–658. [Google Scholar] [CrossRef]
- Selcuklu, S.D.; Donoghue, M.T.; Spillane, C. Mir-21 as a key regulator of oncogenic processes. Biochem. Soc. Trans. 2009, 37, 918–925. [Google Scholar] [CrossRef]
- Novakova, J.; Slaby, O.; Vyzula, R.; Michalek, J. Microrna involvement in glioblastoma pathogenesis. Biochem. Biophys. Res. Commun. 2009, 386, 1–5. [Google Scholar] [CrossRef]
- Sana, J.; Hajduch, M.; Michalek, J.; Vyzula, R.; Slaby, O. Micrornas and glioblastoma: Roles in core signalling pathways and potential clinical implications. J. Cell. Mol. Med. 2011, 15, 1636–1644. [Google Scholar] [CrossRef]
- Zhang, Y.; Dutta, A.; Abounader, R. The role of micrornas in glioma initiation and progression. Front. Biosci. 2012, 17, 700–712. [Google Scholar] [CrossRef]
- Goke, R.; Barth, P.; Schmidt, A.; Samans, B.; Lankat-Buttgereit, B. Programmed cell death protein 4 suppresses cdk1/cdc2 via induction of p21(waf1/cip1). Am. J. Physiol. Cell. Physiol. 2004, 287, C1541–C1546. [Google Scholar] [CrossRef]
- Papagiannakopoulos, T.; Shapiro, A.; Kosik, K.S. Microrna-21 targets a network of key tumor-suppressive pathways in glioblastoma cells. Cancer Res. 2008, 68, 8164–8172. [Google Scholar] [CrossRef]
- Gabriely, G.; Wurdinger, T.; Kesari, S.; Esau, C.C.; Burchard, J.; Linsley, P.S.; Krichevsky, A.M. Microrna 21 promotes glioma invasion by targeting matrix metalloproteinase regulators. Mol. Cell. Biol. 2008, 28, 5369–5380. [Google Scholar] [CrossRef]
- Gabriely, G.; Yi, M.; Narayan, R.S.; Niers, J.M.; Wurdinger, T.; Imitola, J.; Ligon, K.L.; Kesari, S.; Esau, C.; Stephens, R.M.; et al. Human glioma growth is controlled by microrna-10b. Cancer Res. 2011, 71, 3563–3572. [Google Scholar] [CrossRef]
- Ma, L.; Teruya-Feldstein, J.; Weinberg, R.A. Tumour invasion and metastasis initiated by microrna-10b in breast cancer. Nature 2007, 449, 682–688. [Google Scholar] [CrossRef]
- Baffa, R.; Fassan, M.; Volinia, S.; O'Hara, B.; Liu, C.G.; Palazzo, J.P.; Gardiman, M.; Rugge, M.; Gomella, L.G.; Croce, C.M.; et al. Microrna expression profiling of human metastatic cancers identifies cancer gene targets. J. Pathol. 2009, 219, 214–221. [Google Scholar] [CrossRef]
- Tan, H.X.; Wang, Q.; Chen, L.Z.; Huang, X.H.; Chen, J.S.; Fu, X.H.; Cao, L.Q.; Chen, X.L.; Li, W.; Zhang, L.J. Microrna-9 reduces cell invasion and e-cadherin secretion in sk-hep-1 cell. Med. Oncol. 2010, 27, 654–660. [Google Scholar] [CrossRef]
- Ciafre, S.A.; Galardi, S.; Mangiola, A.; Ferracin, M.; Liu, C.G.; Sabatino, G.; Negrini, M.; Maira, G.; Croce, C.M.; Farace, M.G. Extensive modulation of a set of micrornas in primary glioblastoma. Biochem. Biophys. Res. Commun. 2005, 334, 1351–1358. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, J.; Zhang, P.Y.; Zhang, Y.; Sun, S.Y.; Yu, S.Y.; Xi, Q.S. Microrna-10b targets e-cadherin and modulates breast cancer metastasis. Med. Sci. Monit. 2012, 18, BR299–BR308. [Google Scholar] [CrossRef]
- Hossain, A.; Kuo, M.T.; Saunders, G.F. Mir-17-5p regulates breast cancer cell proliferation by inhibiting translation of aib1 mrna. Mol. Cell. Biol. 2006, 26, 8191–8201. [Google Scholar] [CrossRef]
- Cheng, A.M.; Byrom, M.W.; Shelton, J.; Ford, L.P. Antisense inhibition of human mirnas and indications for an involvement of mirna in cell growth and apoptosis. Nucleic Acids Res. 2005, 33, 1290–1297. [Google Scholar] [CrossRef]
- Esau, C.C. Inhibition of microrna with antisense oligonucleotides. Methods 2008, 44, 55–60. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, H.K.; Chung, S.; Kim, K.S.; Dutta, A. Depletion of human micro-rna mir-125b reveals that it is critical for the proliferation of differentiated cells but not for the down-regulation of putative targets during differentiation. J. Biol. Chem. 2005, 280, 16635–16641. [Google Scholar] [CrossRef]
- Kloosterman, W.P.; Lagendijk, A.K.; Ketting, R.F.; Moulton, J.D.; Plasterk, R.H. Targeted inhibition of mirna maturation with morpholinos reveals a role for mir-375 in pancreatic islet development. PLoS Biol. 2007, 5, e203. [Google Scholar] [CrossRef]
- Gantier, M.P.; McCoy, C.E.; Rusinova, I.; Saulep, D.; Wang, D.; Xu, D.; Irving, A.T.; Behlke, M.A.; Hertzog, P.J.; Mackay, F.; et al. Analysis of microrna turnover in mammalian cells following dicer1 ablation. Nucleic Acids Res. 2011, 39, 5692–5703. [Google Scholar] [CrossRef]
- Lee, Y.; Hur, I.; Park, S.Y.; Kim, Y.K.; Suh, M.R.; Kim, V.N. The role of pact in the rna silencing pathway. EMBO J. 2006, 25, 522–532. [Google Scholar] [CrossRef]
- Zhang, B.; Farwell, M.A. Micrornas: A new emerging class of players for disease diagnostics and gene therapy. J. Cell. Mol. Med. 2008, 12, 3–21. [Google Scholar] [CrossRef]
- Hutvagner, G.; Simard, M.J.; Mello, C.C.; Zamore, P.D. Sequence-specific inhibition of small rna function. PLoS Biol. 2004, 2, E98. [Google Scholar] [CrossRef]
- Krutzfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of micrornas in vivo with “antagomirs”. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. Mir-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef]
- Davis, S.; Lollo, B.; Freier, S.; Esau, C. Improved targeting of mirna with antisense oligonucleotides. Nucleic Acids Res. 2006, 34, 2294–2304. [Google Scholar] [CrossRef]
- Orom, U.A.; Kauppinen, S.; Lund, A.H. Lna-modified oligonucleotides mediate specific inhibition of microrna function. Gene 2006, 372, 137–141. [Google Scholar] [CrossRef]
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1500. [Google Scholar]
- Fabbri, E.; Brognara, E.; Borgatti, M.; Lampronti, I.; Finotti, A.; Bianchi, N.; Sforza, S.; Tedeschi, T.; Manicardi, A.; Marchelli, R.; et al. Mirna therapeutics: Delivery and biological activity of peptide nucleic acids targeting mirnas. Epigenomics 2011, 3, 733–745. [Google Scholar] [CrossRef]
- Torres, A.G.; Fabani, M.M.; Vigorito, E.; Williams, D.; Al-Obaidi, N.; Wojciechowski, F.; Hudson, R.H.; Seitz, O.; Gait, M.J. Chemical structure requirements and cellular targeting of microrna-122 by peptide nucleic acids anti-mirs. Nucleic Acids Res. 2011, 40, 2152–2167. [Google Scholar]
- Shiraishi, T.; Nielsen, P.E. Cellular Bioavailability of Peptide Nucleic Acids (Pnas) Conjugated to Cell Penetrating Peptides. In Delivery Technologies for Biopharmaceuticals: Peptides, Proteins, Nucleic Acids and Vaccines; Jorgensen, L., Nielsen, H.M., Eds.; John Wiley and Sons Ltd: Hoboken, NJ, USA, 2009. [Google Scholar]
- Babar, I.A.; Cheng, C.J.; Booth, C.J.; Liang, X.; Weidhaas, J.B.; Saltzman, W.M.; Slack, F.J. Nanoparticle-based therapy in an in vivo microrna-155 (mir-155)-dependent mouse model of lymphoma. Proc. Natl. Acad. Sci. USA 2012, 109, E1695–E1704. [Google Scholar]
- Ebert, M.S.; Neilson, J.R.; Sharp, P.A. Microrna sponges: Competitive inhibitors of small rnas in mammalian cells. Nat. Methods 2007, 4, 721–726. [Google Scholar] [CrossRef]
- Kluiver, J.; Gibcus, J.H.; Hettinga, C.; Adema, A.; Richter, M.K.; Halsema, N.; Slezak-Prochazka, I.; Ding, Y.; Kroesen, B.J.; van den Berg, A. Rapid generation of microrna sponges for microrna inhibition. PLoS One 2012, 7, e29275. [Google Scholar] [CrossRef]
- Li, T.; Li, D.; Sha, J.; Sun, P.; Huang, Y. Microrna-21 directly targets marcks and promotes apoptosis resistance and invasion in prostate cancer cells. Biochem. Biophys. Res. Commun. 2009, 383, 280–285. [Google Scholar] [CrossRef]
- Si, M.L.; Zhu, S.; Wu, H.; Lu, Z.; Wu, F.; Mo, Y.Y. Mir-21-mediated tumor growth. Oncogene 2007, 26, 2799–2803. [Google Scholar] [CrossRef]
- Seike, M.; Goto, A.; Okano, T.; Bowman, E.D.; Schetter, A.J.; Horikawa, I.; Mathe, E.A.; Jen, J.; Yang, P.; Sugimura, H.; et al. Mir-21 is an egfr-regulated anti-apoptotic factor in lung cancer in never-smokers. Proc. Natl. Acad. Sci. USA 2009, 106, 12085–12090. [Google Scholar] [CrossRef]
- Hatley, M.E.; Patrick, D.M.; Garcia, M.R.; Richardson, J.A.; Bassel-Duby, R.; van Rooij, E.; Olson, E.N. Modulation of k-ras-dependent lung tumorigenesis by microrna-21. Cancer Cell 2010, 18, 282–293. [Google Scholar] [CrossRef]
- Park, J.K.; Lee, E.J.; Esau, C.; Schmittgen, T.D. Antisense inhibition of microrna-21 or -221 arrests cell cycle, induces apoptosis, and sensitizes the effects of gemcitabine in pancreatic adenocarcinoma. Pancreas 2009, 38, e190–e199. [Google Scholar] [CrossRef]
- Basu, A.; Alder, H.; Khiyami, A.; Leahy, P.; Croce, C.M.; Haldar, S. Microrna-375 and microrna-221: Potential noncoding rnas associated with antiproliferative activity of benzyl isothiocyanate in pancreatic cancer. Genes Cancer 2011, 2, 108–119. [Google Scholar] [CrossRef]
- Park, J.K.; Kogure, T.; Nuovo, G.J.; Jiang, J.; He, L.; Kim, J.H.; Phelps, M.A.; Papenfuss, T.L.; Croce, C.M.; Patel, T.; et al. Mir-221 silencing blocks hepatocellular carcinoma and promotes survival. Cancer Res. 2011, 71, 7608–7616. [Google Scholar] [CrossRef]
- Ma, L.; Reinhardt, F.; Pan, E.; Soutschek, J.; Bhat, B.; Marcusson, E.G.; Teruya-Feldstein, J.; Bell, G.W.; Weinberg, R.A. Therapeutic silencing of mir-10b inhibits metastasis in a mouse mammary tumor model. Nat. Biotechnol. 2010, 28, 341–347. [Google Scholar]
- Henry, J.C.; Azevedo-Pouly, A.C.; Schmittgen, T.D. Microrna replacement therapy for cancer. Pharm. Res. 2011, 28, 3030–3042. [Google Scholar] [CrossRef]
- Behlke, M.A. Chemical modification of sirnas for in vivo use. Oligonucleotides 2008, 18, 305–319. [Google Scholar] [CrossRef]
- Choung, S.; Kim, Y.J.; Kim, S.; Park, H.O.; Choi, Y.C. Chemical modification of sirnas to improve serum stability without loss of efficacy. Biochem. Biophys. Res. Commun. 2006, 342, 919–927. [Google Scholar] [CrossRef]
- Prakash, T.P.; Allerson, C.R.; Dande, P.; Vickers, T.A.; Sioufi, N.; Jarres, R.; Baker, B.F.; Swayze, E.E.; Griffey, R.H.; Bhat, B. Positional effect of chemical modifications on short interference rna activity in mammalian cells. J. Med. Chem. 2005, 48, 4247–4253. [Google Scholar] [CrossRef]
- Kitade, Y.; Akao, Y. Micrornas and their therapeutic potential for human diseases: Micrornas, mir-143 and -145, function as anti-oncomirs and the application of chemically modified mir-143 as an anti-cancer drug. J. Pharmacol. Sci. 2010, 114, 276–280. [Google Scholar] [CrossRef]
- Hamm, S.; Latz, E.; Hangel, D.; Muller, T.; Yu, P.; Golenbock, D.; Sparwasser, T.; Wagner, H.; Bauer, S. Alternating 2'-o-ribose methylation is a universal approach for generating non-stimulatory sirna by acting as tlr7 antagonist. Immunobiology 2010, 215, 559–569. [Google Scholar] [CrossRef]
- Chorn, G.; Klein-McDowell, M.; Zhao, L.; Saunders, M.A.; Flanagan, W.M.; Willingham, A.T.; Lim, L.P. Single-stranded microrna mimics. RNA 2012, 18, 1796–1804. [Google Scholar] [CrossRef]
- Esau, C.C.; Monia, B.P. Therapeutic potential for micrornas. Adv. Drug Deliv. Rev. 2007, 59, 101–114. [Google Scholar] [CrossRef]
- Liu, Y.P.; Berkhout, B. Mirna cassettes in viral vectors: Problems and solutions. Biochim. Biophys. Acta 2011, 1809, 732–745. [Google Scholar]
- McManus, M.T.; Petersen, C.P.; Haines, B.B.; Chen, J.; Sharp, P.A. Gene silencing using micro-rna designed hairpins. RNA 2002, 8, 842–850. [Google Scholar] [CrossRef]
- Sibley, C.R.; Seow, Y.; Wood, M.J. Novel rna-based strategies for therapeutic gene silencing. Mol. Ther. 2010, 18, 466–476. [Google Scholar] [CrossRef]
- Grimm, D.; Kay, M.A. Rnai and gene therapy: A mutual attraction. Hematology Am. Soc. Hematol. Educ. Program. 2007, 473–481. [Google Scholar] [CrossRef]
- Boudreau, R.L.; Martins, I.; Davidson, B.L. Artificial micrornas as sirna shuttles: Improved safety as compared to shrnas in vitro and in vivo. Mol. Ther. 2009, 17, 169–175. [Google Scholar] [CrossRef]
- McBride, J.L.; Boudreau, R.L.; Harper, S.Q.; Staber, P.D.; Monteys, A.M.; Martins, I.; Gilmore, B.L.; Burstein, H.; Peluso, R.W.; Polisky, B.; et al. Artificial mirnas mitigate shrna-mediated toxicity in the brain: Implications for the therapeutic development of rnai. Proc. Natl. Acad. Sci. USA 2008, 105, 5868–5873. [Google Scholar] [CrossRef]
- Jackson, A.L.; Burchard, J.; Schelter, J.; Chau, B.N.; Cleary, M.; Lim, L.; Linsley, P.S. Widespread sirna “Off-target” Transcript silencing mediated by seed region sequence complementarity. RNA 2006, 12, 1179–1187. [Google Scholar] [CrossRef]
- Liu, C.; Kelnar, K.; Liu, B.; Chen, X.; Calhoun-Davis, T.; Li, H.; Patrawala, L.; Yan, H.; Jeter, C.; Honorio, S.; et al. The microrna mir-34a inhibits prostate cancer stem cells and metastasis by directly repressing cd44. Nat. Med. 2011, 17, 211–215. [Google Scholar] [CrossRef]
- Tazawa, H.; Tsuchiya, N.; Izumiya, M.; Nakagama, H. Tumor-suppressive mir-34a induces senescence-like growth arrest through modulation of the e2f pathway in human colon cancer cells. Proc. Natl. Acad. Sci. USA 2007, 104, 15472–15477. [Google Scholar] [CrossRef]
- Li, L.; Xie, X.; Luo, J.; Liu, M.; Xi, S.; Guo, J.; Kong, Y.; Wu, M.; Gao, J.; Xie, Z.; et al. Targeted expression of mir-34a using the t-visa system suppresses breast cancer cell growth and invasion. Mol. Ther. 2012, 20, 2326–2334. [Google Scholar] [CrossRef]
- Takamizawa, J.; Konishi, H.; Yanagisawa, K.; Tomida, S.; Osada, H.; Endoh, H.; Harano, T.; Yatabe, Y.; Nagino, M.; Nimura, Y.; et al. Reduced expression of the let-7 micrornas in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004, 64, 3753–3756. [Google Scholar] [CrossRef]
- Johnson, C.D.; Esquela-Kerscher, A.; Stefani, G.; Byrom, M.; Kelnar, K.; Ovcharenko, D.; Wilson, M.; Wang, X.; Shelton, J.; Shingara, J.; et al. The let-7 microrna represses cell proliferation pathways in human cells. Cancer Res. 2007, 67, 7713–7722. [Google Scholar] [CrossRef]
- Marquez, R.T.; McCaffrey, A.P. Advances in micrornas: Implications for gene therapists. Hum. Gene Ther. 2008, 19, 27–38. [Google Scholar] [CrossRef]
- Kefas, B.; Godlewski, J.; Comeau, L.; Li, Y.; Abounader, R.; Hawkinson, M.; Lee, J.; Fine, H.; Chiocca, E.A.; Lawler, S.; et al. Microrna-7 inhibits the epidermal growth factor receptor and the akt pathway and is down-regulated in glioblastoma. Cancer Res. 2008, 68, 3566–3572. [Google Scholar] [CrossRef]
- Zhang, Y.; Chao, T.; Li, R.; Liu, W.; Chen, Y.; Yan, X.; Gong, Y.; Yin, B.; Qiang, B.; Zhao, J.; et al. Microrna-128 inhibits glioma cells proliferation by targeting transcription factor e2f3a. J. Mol. Med. 2009, 87, 43–51. [Google Scholar] [CrossRef]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis c virus rna abundance by a liver-specific microrna. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef]
- Lanford, R.E.; Hildebrandt-Eriksen, E.S.; Petri, A.; Persson, R.; Lindow, M.; Munk, M.E.; Kauppinen, S.; Orum, H. Therapeutic silencing of microrna-122 in primates with chronic hepatitis c virus infection. Science 2010, 327, 198–201. [Google Scholar] [CrossRef]
- Lindow, M.; Kauppinen, S. Discovering the first microrna-targeted drug. J. Cell Biol. 2012, 199, 407–412. [Google Scholar] [CrossRef]
- Bader, A.G. Mir-34—A microrna replacement therapy is headed to the clinic. Front. Genet. 2012, 3, 120. [Google Scholar] [CrossRef]
- Banks, W.A.; Farr, S.A.; Butt, W.; Kumar, V.B.; Franko, M.W.; Morley, J.E. Delivery across the blood-brain barrier of antisense directed against amyloid beta: Reversal of learning and memory deficits in mice overexpressing amyloid precursor protein. J. Pharmacol. Exp. Ther. 2001, 297, 1113–1121. [Google Scholar]
- Catuogno, S.; Esposito, C.L.; Quintavalle, C.; Condorelli, G.; de Franciscis, V.; Cerchia, L. Nucleic acids in human glioma treatment: Innovative approaches and recent results. J. Signal. Transduct. 2012, 2012, 735135. [Google Scholar]
- Du, L.; Kayali, R.; Bertoni, C.; Fike, F.; Hu, H.; Iversen, P.L.; Gatti, R.A. Arginine-rich cell-penetrating peptide dramatically enhances amo-mediated atm aberrant splicing correction and enables delivery to brain and cerebellum. Hum. Mol. Genet. 2011, 20, 3151–3160. [Google Scholar] [CrossRef]
- Xia, H.; Mao, Q.; Paulson, H.L.; Davidson, B.L. Sirna-mediated gene silencing in vitro and in vivo. Nat. Biotechnol. 2002, 20, 1006–1010. [Google Scholar] [CrossRef]
- Lu, P.Y.; Xie, F.; Woodle, M.C. In vivo application of rna interference: From functional genomics to therapeutics. Adv. Genet. 2005, 54, 117–142. [Google Scholar]
- Tong, A.W. Small rnas and non-small cell lung cancer. Curr. Mol. Med. 2006, 6, 339–349. [Google Scholar] [CrossRef]
- Kota, J.; Chivukula, R.R.; O’Donnell, K.A.; Wentzel, E.A.; Montgomery, C.L.; Hwang, H.W.; Chang, T.C.; Vivekanandan, P.; Torbenson, M.; Clark, K.R.; et al. Therapeutic microrna delivery suppresses tumorigenesis in a murine liver cancer model. Cell 2009, 137, 1005–1017. [Google Scholar] [CrossRef]
- Trang, P.; Medina, P.P.; Wiggins, J.F.; Ruffino, L.; Kelnar, K.; Omotola, M.; Homer, R.; Brown, D.; Bader, A.G.; Weidhaas, J.B.; et al. Regression of murine lung tumors by the let-7 microrna. Oncogene 2010, 29, 1580–1587. [Google Scholar] [CrossRef]
- Bonci, D.; Coppola, V.; Musumeci, M.; Addario, A.; Giuffrida, R.; Memeo, L.; D’Urso, L.; Pagliuca, A.; Biffoni, M.; Labbaye, C.; et al. The mir-15a-mir-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat. Med. 2008, 14, 1271–1277. [Google Scholar] [CrossRef]
- Sicard, F.; Gayral, M.; Lulka, H.; Buscail, L.; Cordelier, P. Targeting mir-21 for the therapy of pancreatic cancer. Mol. Ther. 2013, 21, 986–994. [Google Scholar] [CrossRef]
- Lee, S.J.; Kim, S.J.; Seo, H.H.; Shin, S.P.; Kim, D.; Park, C.S.; Kim, K.T.; Kim, Y.H.; Jeong, J.S.; Kim, I.H. Over-expression of mir-145 enhances the effectiveness of hsvtk gene therapy for malignant glioma. Cancer Lett. 2012, 320, 72–80. [Google Scholar] [CrossRef]
- Wang, X.; Han, L.; Zhang, A.; Wang, G.; Jia, Z.; Yang, Y.; Yue, X.; Pu, P.; Shen, C.; Kang, C. Adenovirus-mediated shrnas for co-repression of mir-221 and mir-222 expression and function in glioblastoma cells. Oncol. Rep. 2011, 25, 97–105. [Google Scholar]
- Pramanik, D.; Campbell, N.R.; Karikari, C.; Chivukula, R.; Kent, O.A.; Mendell, J.T.; Maitra, A. Restitution of tumor suppressor micrornas using a systemic nanovector inhibits pancreatic cancer growth in mice. Mol. Cancer Ther. 2011, 10, 1470–1480. [Google Scholar] [CrossRef]
- Trang, P.; Wiggins, J.F.; Daige, C.L.; Cho, C.; Omotola, M.; Brown, D.; Weidhaas, J.B.; Bader, A.G.; Slack, F.J. Systemic delivery of tumor suppressor microrna mimics using a neutral lipid emulsion inhibits lung tumors in mice. Mol. Ther. 2011, 19, 1116–1122. [Google Scholar] [CrossRef]
- Costa, P.M.; Cardoso, A.L.; Mendonca, L.S.; Serani, A.; Custodia, C.; Conceicao, M.; Simoes, S.; Moreira, J.N.; Pereira de Almeida, L.; Pedroso de Lima, M.C. Tumor-targeted chlorotoxin-coupled nanoparticles for nucleic acid delivery to glioblastoma cells: A promising system for glioblastoma treatment. Mol. Ther. Nucleic Acids 2013, 2, e100. [Google Scholar] [CrossRef]
- Yang, Y.P.; Chien, Y.; Chiou, G.Y.; Cherng, J.Y.; Wang, M.L.; Lo, W.L.; Chang, Y.L.; Huang, P.I.; Chen, Y.W.; Shih, Y.H.; et al. Inhibition of cancer stem cell-like properties and reduced chemoradioresistance of glioblastoma using microrna145 with cationic polyurethane-short branch pei. Biomaterials 2012, 33, 1462–1476. [Google Scholar] [CrossRef]
- Ren, Y.; Kang, C.S.; Yuan, X.B.; Zhou, X.; Xu, P.; Han, L.; Wang, G.X.; Jia, Z.; Zhong, Y.; Yu, S.; et al. Co-delivery of as-mir-21 and 5-fu by poly(amidoamine) dendrimer attenuates human glioma cell growth in vitro. J. Biomater. Sci. Polym. Ed. 2010, 21, 303–314. [Google Scholar] [CrossRef]
- A Multicenter Phase I Study of MRX34, MicroRNA miR-RX34 Liposome Injectable Suspension. Available online: http://clinicaltrials.gov/ct2/show/NCT01829971/ (accessed on 23 September 2013).
- Guan, J.; Fujimoto, K.L.; Sacks, M.S.; Wagner, W.R. Preparation and characterization of highly porous, biodegradable polyurethane scaffolds for soft tissue applications. Biomaterials 2005, 26, 3961–3971. [Google Scholar] [CrossRef]
- Liu, X.Y.; Ho, W.Y.; Hung, W.J.; Shau, M.D. The characteristics and transfection efficiency of cationic poly (ester-co-urethane)—Short chain pei conjugates self-assembled with DNA. Biomaterials 2009, 30, 6665–6673. [Google Scholar] [CrossRef]
- Bala, I.; Hariharan, S.; Kumar, M.N. Plga nanoparticles in drug delivery: The state of the art. Crit. Rev. Ther. Drug Carrier Syst. 2004, 21, 387–422. [Google Scholar] [CrossRef]
- Pettit, M.W.; Griffiths, P.; Ferruti, P.; Richardson, S.C. Poly(amidoamine) polymers: Soluble linear amphiphilic drug-delivery systems for genes, proteins and oligonucleotides. Ther. Deliv. 2011, 2, 907–917. [Google Scholar] [CrossRef]
- D’Emanuele, A.; Attwood, D. Dendrimer-drug interactions. Adv. Drug Deliv. Rev. 2005, 57, 2147–2162. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Costa, P.M.; Pedroso de Lima, M.C. MicroRNAs as Molecular Targets for Cancer Therapy: On the Modulation of MicroRNA Expression. Pharmaceuticals 2013, 6, 1195-1220. https://doi.org/10.3390/ph6101195
Costa PM, Pedroso de Lima MC. MicroRNAs as Molecular Targets for Cancer Therapy: On the Modulation of MicroRNA Expression. Pharmaceuticals. 2013; 6(10):1195-1220. https://doi.org/10.3390/ph6101195
Chicago/Turabian StyleCosta, Pedro M., and Maria C. Pedroso de Lima. 2013. "MicroRNAs as Molecular Targets for Cancer Therapy: On the Modulation of MicroRNA Expression" Pharmaceuticals 6, no. 10: 1195-1220. https://doi.org/10.3390/ph6101195
APA StyleCosta, P. M., & Pedroso de Lima, M. C. (2013). MicroRNAs as Molecular Targets for Cancer Therapy: On the Modulation of MicroRNA Expression. Pharmaceuticals, 6(10), 1195-1220. https://doi.org/10.3390/ph6101195