Applications of Cell-Penetrating Peptides for Tumor Targeting and Future Cancer Therapies

Abstract
:1. Introduction

{kind=link}
| Peptide | Sequence | Origin | Reference |
|---|---|---|---|
| TAT (48–60) | GRKKRRQRRRQC | Protein-derived | [1] |
| Penetratin | RQIKIWFQNRRMKWKK-NH2 | Protein-derived | [2] |
| pVEC | LLIILRRRIRKQAHAHSK-NH2 | Protein-derived | [9] |
| MPG8 | AFLGWLGAWGTMGWSPKKKRK-cya | Chimeric | [10] |
| Transportan | GWTLNSAGYLLGKINLKALAALAKKIL-NH2 | Chimeric | [3] |
| Transportan10 | AGYLLGKINLKALAALAKKIL-NH2 | Chimeric, modified | [11] |
| PepFect3 | Stearyl-AGYLLGKINLKALAALAKKIL-NH2 | Chimeric, modified | [6] |
| PepFect 6 | Stearyl-AGYLLGK(εNHa)INLKALAALAKKIL-NH2 | Chimeric, modified | [12] |
| PepFect 14 | Stearyl-AGYLLGKLLOOLAAAALOOLL-NH2 | Chimeric, modified | [13] |
| Polyarginine | Rn (n = 6–12) | Designed | [14] |
| Stearyl-polyarginine | Stearyl-Rn (n = 6–12) | Designed | [5] |
| Pep-1 | Ac-KETWWETWWTEWSQPKKKRKV-cya | Designed | [15] |
| Pep-3 | KWFETWFTEWPKKRK-cya | Designed | [16] |
| CADY | Ac-GLWRALWRLLRSLWRLLWRA-cya | Designed | [17] |
| YTA2 | YTAIAWVKAFIRKLRK-NH2 | Designed | [18] |
| YTA4 | IAWVKAFIRKLRKGPLG-NH2 | Designed | [19] |
| SynB1 | RGGRLSYSRRRFSTSTGR | Protein-derived | [20,21] |
| SynB3 | RRLSYSRRRF | Protein-derived | [20] |
| Maurocalcine | GDCLPHLKLCKENKDCCSKKCKRRGTNIEKRCR | Protein-derived | [22,23] |
| PTD4 | YARAAARQARA | Protein-derived | [24] |
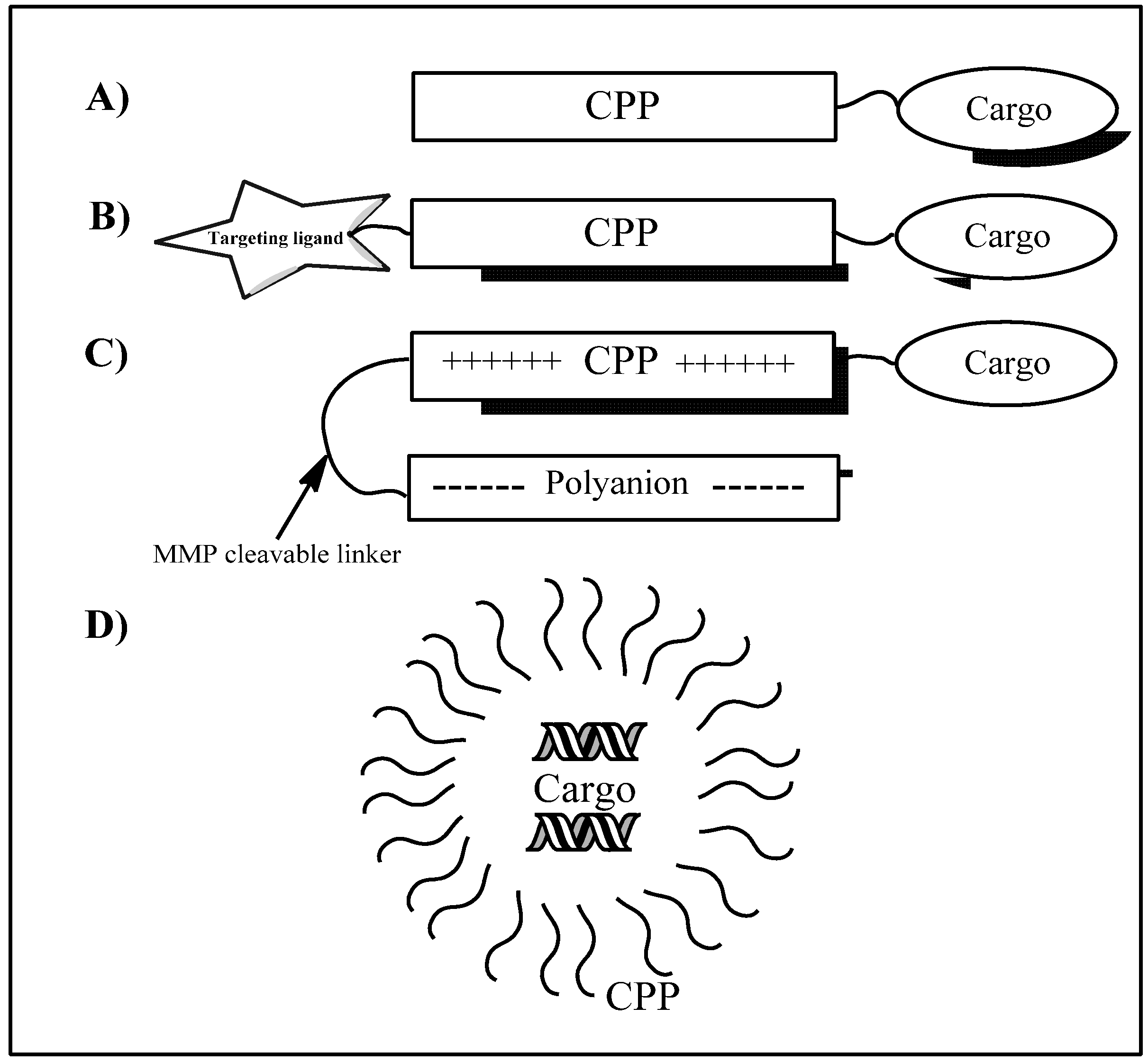
2. Uptake Mechanisms
3. The Application of CPPs in Cancer Therapies
3.1. Cell-Targeting Peptides
| Targeting peptide | Active sequence | Targets | References | |
|---|---|---|---|---|
| PEGA | CPGPEGAGC | Tumor blood vessels | [38,39] | |
| CREKA | CREKA | Tumor blood vessels and stroma | [33,36,39,40] | |
| RVG | YTIWMPENPRPGTPCDIF-TNSRGKRASNG | Acetylcholine receptor in neuronal cells | [37,41] | |
| DV3 | LGASWHRPDKG | CXC chemokine receptor 4 (CXCR4) | [33,42] | |
| DEVDG | DEVDG | Caspase 3 | [39] | |
| ACPP-MMP-2/9 | PLGLAG | Proteases in human fibrosarcoma | [45] | |
| ACPP-MMP-2 | IAGEDGDEFG | Proteases in breast cancer cells | [19] | |
3.2. Activatable CPPs
3.3. Transducible Agents of CPPs
4. Drug Loading
4.1. Small Molecules
| CPPs | Method | Cargoes | Application | References | |
|---|---|---|---|---|---|
| TAT, Penetratin, SynB1 | Covalent coupling | DOX | Breast cancer cell lines MDA-MB-231 | [20,21,22] | |
| CADY | Non-covalent complex | DOX | Increased therapeutic index and blood residence time | [55] | |
| R9PLGLAGDG-GDGGDGGDG | Covalent, activatable | DOX | Targeting ability to tumors rich MMP-2/9 | [45] | |
| YTA2 | Covalent coupling | MTX | Resistant breast cancer cells MDA-MB-231 | [57,58] | |
| YTA4 | Covalent coupling | Fluorescein and MTX | Breast cancer cells MDA-MB-231 | [19] | |
| TAT | Covalent coupling | p53 | Rabbit eyes harboring human retinoblastoma xenograft | [33,52,54] | |
| Antp | Non-covalent complex | p16 | Pancreatic cancer | [33,52,57] | |
| TAT, Antp | Non-covalent complex | Smac | Proapoptotic stimuli | [33] | |
| R8 | Non-covalent complex | SmacN7 | Reversed apoptotic resistance | [54] | |
| Antp, TAT | Covalent coupling | shepherdin | Caspase-dependent apoptosis | [33] | |
| PTD4 | Covalent coupling | Peptide D1, D3, and K4 | Antiproliferation effect on cancer cell lines | [24] | |
| MPG8, PEP3 | Non-covalent complex | siRNA, PNA | Promotes cellular uptake in cancer cell lines | [10,33] | |
| TP10 | Covalent coupling | PNA | Promotes cytosolic delivery | [57] | |
| Penetratin, Transportant | Non-covalent | siRNA | Luciferase and GFP transgenes inhibitor | [57] | |
| cholesteryl-R9 | Non-covalent | siRNA | VEGF inhibitor | [57] | |
4.2. Macromolecules
5. Future Aspects
References
- Vivès, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef]
- Derossi, D.; Joliot, H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar]
- Pooga, M.; Hällbrink, M.; Zorko, M.; Langel, Ü. Cell penetration by transportan. FASEB J. 1998, 12, 67–77. [Google Scholar]
- Kilk, K.; EL-Andaloussi, S.; Järver, P.; Meikas, A.; Valkna, A.; Bartfai, T.; Kogerman, P.; Metsis, M.; Langel, Ü. Evaluation of transportan 10 in PEI mediated plasmid delivery assay. J. Control. Release 2005, 103, 511–523. [Google Scholar] [CrossRef]
- Futaki, S.; Ohashi, W.; Suzuki, T.; Niwa, M.; Tanaka, S.; Ueda, K.; Harashima, H.; Sugiura, Y. Stearylated arginine-rich peptides: A new class of transfection systems. Bioconjugate Chem. 2001, 12, 1005–1011. [Google Scholar] [CrossRef]
- Mäe, M.; EL-Andaloussi, S.; Lundin, P.; Oskolkov, N.; Johansson, H. J.; Guterstam, P.; Langel, Ü. A stearylated CPP for delivery of splice correcting oligonucleotides using a non-covalent co-incubation strategy. J. Control. Release 2009, 134, 221–227. [Google Scholar] [CrossRef]
- Lehto, T.; Kurrikoff, K.; Langel, Ü. Cell-penetrating peptides for the delivery of nucleic acids. Expert Opin. Drug Del. 2012, 9, 823–836. [Google Scholar] [CrossRef]
- Sarko, D.; Beijer, B.; Boy, R. G.; Nothelfer, E.-M.; Leotta, K.; Eisenhut, M.; Altmann, A.; Haberkorn, U.; Walter, M. The pharmacokinetics of cell-penetrating peptides. Mol. Biopharm. 2010, 7, 2224–2231. [Google Scholar]
- Elmquist, A.; Lindgren, M.; Bartfai, T.; Langel, Ü. VE-cadherin-derived cell-penetrating peptide, pVEC, with carrier functions. Exp. Cell Res. 2001, 269, 237–244. [Google Scholar] [CrossRef]
- Crombez, L.; Morris, M.C.; Heitz, F.; Divita, G. A non-covalent peptide-based strategy for ex vivo and in vivo oligonucleotide delivery. Meth. Mol. Biol. 2011, 764, 59–73. [Google Scholar] [CrossRef]
- Lindgren, M.; Gallet, X.; Soomets, U.; Hällbrink, M.; Bråkenhielm, E.; Pooga, M.; Brasseur, R.; Langel, Ü. Translocation properties of novel cell penetrating transportan and penetratin analogues. Bioconjugate Chem. 2000, 11, 619–626. [Google Scholar] [CrossRef]
- EL-Andaloussi, S.; Lehto, T.; Mäger, I.; Rosenthal-Aizman, K.; Oprea, I.I.; Simonson, O.E.; Sork, H.; Ezzat, K.; Copolovici, D.M.; Kurrikoff, K.; et al. Design of a peptide-based vector, PepFect6, for efficient delivery of siRNA in cell culture and systemically in vivo. Nucleic Acids Res. 2011, 39, 3972–3987. [Google Scholar] [CrossRef]
- Ezzat, K.; EL-Andaloussi, S.; Zaghloul, E.M.; Lehto, T.; Lindberg, S.; Moreno, P.M.D.; Viola, J.R.; Magdy, T.; Abdo, R.; Guterstam, P.; et al. PepFect 14, a novel cell-penetrating peptide for oligonucleotide delivery in solution and as solid formulation. Nucleic Acids Res. 2011, 39, 5284–5298. [Google Scholar]
- Futaki, S.; Nakase, I.; Tadokoro, A.; Takeuchi, T.; Jones, T. Arginine-rich peptides and their internalization mechanisms. Biochem. Soc. Trans. 2007, 35, 784–787. [Google Scholar] [CrossRef]
- Heitz, F.; Morris, M.C.; Divita, G. Twenty years of cell penetrating peptides: From molecular mechanisms to therapeutics. Br. J. Pharmacol. 2009, 157, 195–206. [Google Scholar] [CrossRef]
- Morris, M.C.; Gros, E.; Aldrian-Herrada, G.; Choob, M.; Archdeacon, J.; Heitz, F.; Divita, G. A non-covalent peptide-based carrier for in vivo delivery of DNA mimics. Nucleic Acids Res. 2007, 35, e49. [Google Scholar] [CrossRef]
- Rydström, A.; Deshayes, S.; Konate, K.; Crombez, L.; Padari, K.; Boukhaddaoui, H.; Aldrian, G.; Pooga, M.; Divita, G. Direct translocation as major cellular uptake for CADY self-assembling peptide-based nanoparticles. PLoS One 2011, 6, e25924. [Google Scholar]
- Myrberg, H.; Lindgren, M.; Langel, Ü. Protein delivery by the cell-penetrating peptide YTA2. Bioconjugate Chem. 2007, 18, 170–174. [Google Scholar] [CrossRef]
- Mäe, M.; Rautsi, O.; Enbäck, J.; Hällbrink, M.; Aizman, K.R.; Lindgren, M.; Laakkonen, P.; Langel, Ü. Tumour targeting with rationally modified cell-penetrating peptides. Int. J. Pept. Res. and Ther. 2012. [Google Scholar]
- Rousselle, C.; Smirnova, M.; Clair, P.; Lefauconnier, J.M.; Chavanieu, A.; Calas, B.; Scherrmann, J.M.; Temsamani, J. Enhanced delivery of doxorubicin into the brain via a peptide-vector-mediated strategy: Saturation kinetics and specificity. J. Pharmacol. Exp. Ther. 2001, 296, 124–131. [Google Scholar]
- Rousselle, C.; Clair, P.; Lefauconnier, J.M.; Kaczorek, M.; Scherrmann, J.M.; Temsamani, J. New advances in the transport of doxorubicin through the blood-brain barrier by a peptide vector-mediated strategy. Mol. Pharmacol. 2000, 57, 679–686. [Google Scholar]
- Aroui, S.; Ram, N.; Appaix, F.; Ronjat, M.; Kenani, A.; Pirollet, F.; De Waard, M. Maurocalcine as a non toxic drug carrier overcomes doxorubicin resistance in the cancer cell line MDA-MB 231. Pharm. Res. 2009, 26, 836–845. [Google Scholar] [CrossRef]
- Fajloun, Z.; Kharrat, R.; Chen, L.; Lecomte, C.; Di Luccio, E.; Bichet, D.; El Ayeb, M.; Rochat, H.; Allen, P.D.; Pessah, I.N.; et al. Chemical synthesis and characterization of maurocalcine, a scorpion toxin that activates Ca(2+) release channel/ryanodine receptors. FEBS Lett. 2000, 469, 179–185. [Google Scholar] [CrossRef]
- Wang, H.; Chen, X.; Chen, Y.; Sun, L.; Li, G.; Zhai, M.; Zhai, W.; Kang, Q.; Gao, Y.; Qi, Y. Antitumor activity of novel chimeric peptides derived from cyclinD/CDK4 and the protein transduction domain 4. Amino Acids 2012. [Google Scholar]
- Richard, J.P.; Melikov, K.; Vives, E.; Ramos, C.; Verbeure, B.; Gait, M.J.; Chernomordik, L.V.; Lebleu, B. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J. Biol.Chem. 2003, 278, 585–590. [Google Scholar]
- Lundin, P.; Johansson, H.; Guterstam, P.; Holm, T.; Hansen, M.; Langel, Ü.; EL-Andaloussi, S. Distinct uptake routes of cell-penetrating peptide conjugates. Bioconjugate Chem. 2008, 19, 2535–2542. [Google Scholar] [CrossRef]
- Drin, G.; Cottin, S.; Blanc, E.; Rees, A.R.; Temsamani, J. Studies on the internalization mechanism of cationic cell-penetrating peptides. J. Biol.Chem. 2003, 278, 31192–201. [Google Scholar]
- Yamagata, M.; Kawano, T.; Shiba, K.; Mori, T.; Katayama, Y.; Niidome, T. Structural advantage of dendritic poly(L-lysine) for gene delivery into cells. Bioorg. Med. Chem. 2007, 15, 526–532. [Google Scholar] [CrossRef]
- Letoha, T.; Keller-Pintér, A.; Kusz, E.; Kolozsi, C.; Bozsó, Z.; Tóth, G.; Vizler, C.; Oláh, Z.; Szilák, L. Cell-penetrating peptide exploited syndecans. Biochim. Biophys. Acta 2010, 1798, 2258–2265. [Google Scholar] [CrossRef]
- Subrizi, A.; Tuominen, E.; Bunker, A.; Róg, T.; Antopolsky, M.; Urtti, A. Tat(48-60) peptide amino acid sequence is not unique in its cell penetrating properties and cell-surface glycosaminoglycans inhibit its cellular uptake. J. Control. Release 2012, 158, 277–285. [Google Scholar] [CrossRef]
- Ezzat, K.; Helmfors, H.; Tudoran, O.; Juks, C.; Lindberg, S.; Padari, K.; EL-Andaloussi, S.; Pooga, M.; Langel, Ü. Scavenger receptor-mediated uptake of cell-penetrating peptide nanocomplexes with oligonucleotides. FASEB J. 2012, 26, 1172–1180. [Google Scholar] [CrossRef]
- Tünnemann, G.; Martin, R.M.; Haupt, S.; Patsch, C.; Edenhofer, F.; Cardoso, M.C. Cargo-dependent mode of uptake and bioavailability of TAT-containing proteins and peptides in living cells. FASEB J. 2006, 20, 1175–1184. [Google Scholar]
- Bolhassani, A. Potential efficacy of cell-penetrating peptides for nucleic acid and drug delivery in cancer. Biochim. Biophys. Acta 2011, 1816, 232–246. [Google Scholar]
- Munyendo, W.L.L.; Lv, H.; Benza-Ingoula, H.; Baraza, L.D.; Zhou, J. Cell penetrating peptides in the delivery of biopharmaceuticals. Biomolecules 2012, 2, 187–202. [Google Scholar] [CrossRef]
- Vivès, E.; Schmidt, J.; Pèlegrin, A. Cell-penetrating and cell-targeting peptides in drug delivery. Biochim. Biophys. Acta 2008, 1786, 126–138. [Google Scholar]
- Mäe, M.; Myrberg, H.; EL-Andaloussi, S.; Langel, Ü. Design of a tumor homing cell-penetrating peptide for drug delivery. Int. J. Pept. Res. Ther. 2008, 15, 11–15. [Google Scholar]
- Zhang, X.X.; Eden, H.S.; Chen, X. Peptides in cancer nanomedicine: Drug carriers, targeting ligands and protease substrates. J. Control. Release 2012, 159, 2–13. [Google Scholar] [CrossRef]
- Myrberg, H.; Zhang, L.; Mäe, M.; Langel, Ü. Design of a tumor-homing cell-penetrating peptide. Bioconjugate Chem. 2008, 19, 70–75. [Google Scholar] [CrossRef]
- Kersemans, V.; Cornelissen, B. Targeting the tumour: Cell Penetrating peptides for molecular imaging and radiotherapy. Pharmaceuticals 2010, 3, 600–620. [Google Scholar] [CrossRef]
- Laakkonen, P.; Vuorinen, K. Homing peptides as targeted delivery vehicles. Integr. Biol. 2010, 2, 326–337. [Google Scholar] [CrossRef]
- Kumar, P.; Wu, H.; McBride, J.L.; Jung, K.E.; Kim, M.H.; Davidson, B.L.; Lee, S.K.; Shankar, P.; Manjunath, N. Transvascular delivery of small interfering RNA to the central nervous system. Nature 2007, 448, 39–43. [Google Scholar]
- Snyder, E.L.; Saenz, C.C.; Denicourt, C.; Meade, B.R.; Cui, X.S.; Kaplan, I.M.; Dowdy, S.F. Enhanced targeting and killing of tumor cells expressing the CXC chemokine receptor 4 by transducible anticancer peptides. Cancer Res. 2005, 65, 10646–10650. [Google Scholar] [CrossRef]
- Jiang, Q.Y.; Lai, L.H.; Shen, J.; Wang, Q.Q.; Xu, F.J.; Tang, G.P. Gene delivery to tumor cells by cationic polymeric nanovectors coupled to folic acid and the cell-penetrating peptide octaarginine. Biomaterials 2011, 32, 7253–7262. [Google Scholar] [CrossRef]
- Bauer, C.; Bauder-Wuest, U. 131I-labeled peptides as caspase substrates for apoptosis imaging. J. Nucl. Med. 2005, 46, 1066–1074. [Google Scholar]
- Shi, N.Q.; Gao, W.; Xiang, B.; Qi, X.R. Enhancing cellular uptake of activable cell-penetrating peptide-doxorubicin conjugate by enzymatic cleavage. Int. J. Nanomed. 2012, 7, 1613–1621. [Google Scholar]
- Olson, E.; Aguilera, T.; Jiang, T.; Ellies, L.G.; Nguyen, Q.T.; Wong, E.H.; Gross, L.A.; Tsien, R.Y. In vivo characterization of activatable cell penetrating peptides for targeting protease activity in cancer. Integr. Biol. 2009, 1, 382–393. [Google Scholar] [CrossRef]
- van Duijnhoven, S.M.J.; Robillard, M.S.; Nicolay, K.; Grüll, H. Tumor targeting of MMP-2/9 activatable cell-penetrating imaging probes is caused by tumor-independent activation. J. Nucl. Med. 2011, 52, 279–286. [Google Scholar] [CrossRef]
- Aguilera, T.; Olson, E.S.; Timmers, M.M.; Jiang, T.; Tsien, R.Y. Systemic in vivo distribution of activatable cell penetrating peptides is superior to that of cell penetrating peptides. Integr. Biol. 2009, 1, 371–381. [Google Scholar] [CrossRef]
- Curran, S.; Murray, G.I. Matrix metalloproteinases: Molecular aspects of their roles in tumour invasion and metastasis. Eur. J. Cancer 2000, 36, 1621–1630. [Google Scholar] [CrossRef]
- Polette, M.; Nawrocki-Raby, B.; Gilles, C.; Clavel, C.; Birembaut, P. Tumour invasion and matrix metalloproteinases. Crit. Rev. Oncol./Hematol. 2004, 49, 179–186. [Google Scholar] [CrossRef]
- Deryugina, E.I.; Quigley, J.P. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006, 25, 9–34. [Google Scholar] [CrossRef]
- Snyder, E.L.; Dowdy, S.F. Cell penetrating peptides in drug delivery. Pharm. Res. 2004, 21, 389–393. [Google Scholar] [CrossRef]
- Harada, H.; Hiraoka, M.; Kizaka-Kondoh, S. Antitumor effect of TAT-oxygen-dependent degradation-caspase-3 fusion protein specifically stabilized and activated in hypoxic tumor cells. Cancer Res. 2002, 1, 2013–2018. [Google Scholar]
- Harada, H.; Kizaka-Kondoh, S.; Hiraoka, M. Antitumor protein therapy; application of the protein transduction domain to the development of a protein drug for cancer treatment. Breast Cancer 2006, 13, 16–26. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, X.; Cao, Z.; Xu, W.; Zhang, J.; Gong, M. Self-assembled peptide (CADY-1) improved the clinical application of doxorubicin. Int. J. Pharm. 2012, 434, 209–214. [Google Scholar] [CrossRef]
- Aroui, S.; Brahim, S.; Waard, M.D.; Kenani, A. Cytotoxicity, intracellular distribution and uptake of doxorubicin and doxorubicin coupled to cell-penetrating peptides in different cell lines: A comparative study. Biochem. Biophys. Res. Commun. 2010, 391, 419–425. [Google Scholar] [CrossRef]
- Patel, L.N.; Zaro, J.L.; Shen, W.C. Cell penetrating peptides: Intracellular pathways and pharmaceutical perspectives. Pharm. Res. 2007, 24, 1977–1992. [Google Scholar] [CrossRef]
- Lindgren, M.; Rosenthal-Aizman, K.; Saar, K.; Eiríksdóttir, E.; Jiang, Y.; Sassian, M.; Ostlund, P.; Hällbrink, M.; Langel, Ü. Overcoming methotrexate resistance in breast cancer tumour cells by the use of a new cell-penetrating peptide. Biochem. Pharmacol. 2006, 71, 416–425. [Google Scholar] [CrossRef]
- Chaloin, L.; Bigey, P.; Loup, C.; Marin, M.; Galeotti, N.; Piechaczyk, M.; Heitz, F.; Meunier, B. Improvement of porphyrin cellular delivery and activity by conjugation to a carrier peptide. Bioconjugate Chem. 2001, 12, 691–700. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Regberg, J.; Srimanee, A.; Langel, Ü. Applications of Cell-Penetrating Peptides for Tumor Targeting and Future Cancer Therapies. Pharmaceuticals 2012, 5, 991-1007. https://doi.org/10.3390/ph5090991
Regberg J, Srimanee A, Langel Ü. Applications of Cell-Penetrating Peptides for Tumor Targeting and Future Cancer Therapies. Pharmaceuticals. 2012; 5(9):991-1007. https://doi.org/10.3390/ph5090991
Chicago/Turabian StyleRegberg, Jakob, Artita Srimanee, and Ülo Langel. 2012. "Applications of Cell-Penetrating Peptides for Tumor Targeting and Future Cancer Therapies" Pharmaceuticals 5, no. 9: 991-1007. https://doi.org/10.3390/ph5090991
APA StyleRegberg, J., Srimanee, A., & Langel, Ü. (2012). Applications of Cell-Penetrating Peptides for Tumor Targeting and Future Cancer Therapies. Pharmaceuticals, 5(9), 991-1007. https://doi.org/10.3390/ph5090991