Chemotherapeutic Interventions Against Tuberculosis

Abstract
:1. Introduction
2. Current Anti-Tuberculosis Drugs
2.1. First Line Drugs
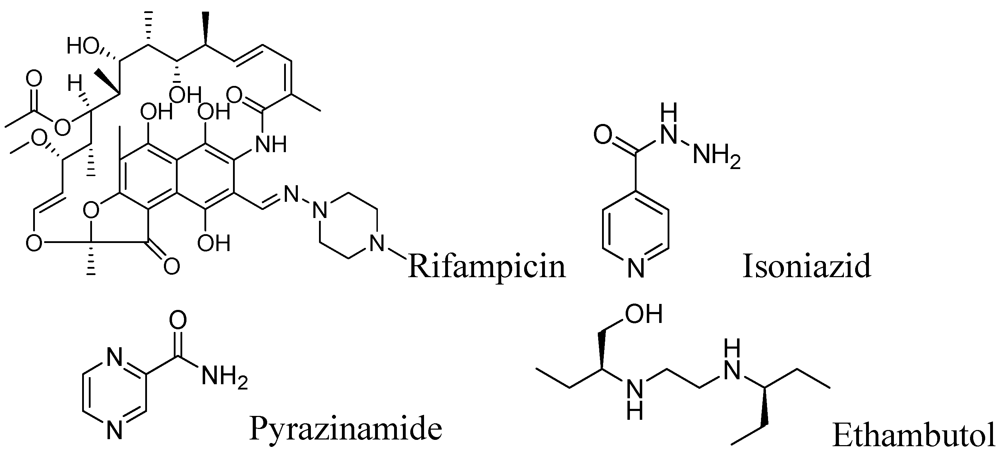
2.1.1. Rifampicin
2.1.2. Isoniazid
2.1.3. Pyrazinamide
2.1.4. Ethambutol
2.2. Second Line Drugs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug (Discovery) MIC values * | Structure | Daily dose (Max. dose) Route | Adverse effects | Mode of action |
|---|---|---|---|---|
| Capreomycin (1963) MIC 1.25–2.5 μg/mL [ 36,37] |  | 15–30 mg/kg (1 g) IM or IV | Auditory, vestibular, and renal toxicity | Inhibits protein synthesis (binds to ribosomal subunit 16S and 23S rRNA) [38] |
| Amikacin (1972) MIC 4–8 μg/mL [39] |  | 15–30 mg/kg (1 g) IM or IV | Same as capreomycin | Inhibits protein synthesis (binds to the bacterial 30S ribosome) |
| Kanamycin (1957) MIC 1–8 μg/mL |  | 15–30 mg/kg (1 g) IM or IV | Same as Capreomycin | Inhibits protein synthesis via S12 ribosomal protein & 16 S RNA. |
| Streptomycin (1944) MIC 2–8 μg/mL |  | 15–40 mg/kg (1 g) IM | Renal, ophthalmic and respiratory toxicity | Same as kanamycin |
| Cycloserine (1952) MIC 5–20 μg/mL |  | 15–20 mg/kg (1 g) Oral | Psychosis, Rashes, Convulsions Depression | Inhibition of peptidoglycan synthesis (D-alanine racemase) |
| Ethionamide (1956) MIC 0.6–2.5 μg/mL |  | 15–20 mg/kg (1 g) Oral | GI upset Hepatotoxicity Hypersensitivity | Inhibition of mycolic acid synthesis |
| Clofazimine (1954) MIC 0.12–0.24 μg/mL [40] |  | 100–300 mg/day Oral | Eosinophilic enteritis, GI irritation, discoloration of the skin (upon sun exposure) | Inhibits bacterial proliferation by binding to the guanine bases of bacterial DNA |
| Levofloxacin (1992) MIC 0.50 to 0.75 μg/mL [41] |  | 500 mg/day Oral | GI upset Dizziness Headache Hypersensitivity Restlessness | Inhibition of DNA replication and transcription by inhibiting DNA gyrase |
| Ofloxacin (1980) Oral, MIC 0.12–2 μg/mL [42] |  | 600–800 mg/day | Same as for levofloxacin | Same as for levofloxacin |
| Ciprofloxacin (1960s) MIC 0.4 to 6.2 μg/mL [43] |  | 750–1,500 mg/day Oral | Same as for levofloxacin | Same as for levofloxacin |
| PAS (1946) MIC 1–8 μg/mL |  | 150 mg/kg (16 g) Oral | Same as for ethionamide, Sodium load | Inhibition of folic acid and iron metabolism (unknown target) |
3. Drug Discovery Program
3.1. Early Stage Drug Discovery
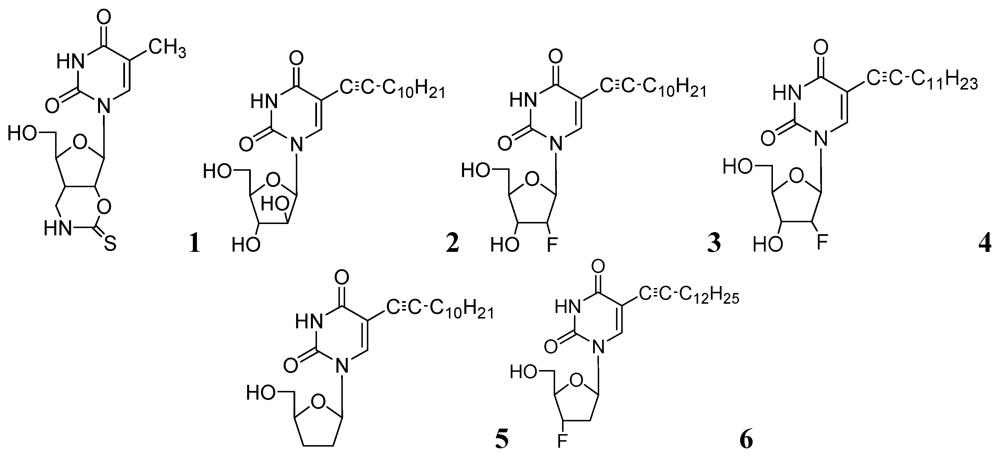
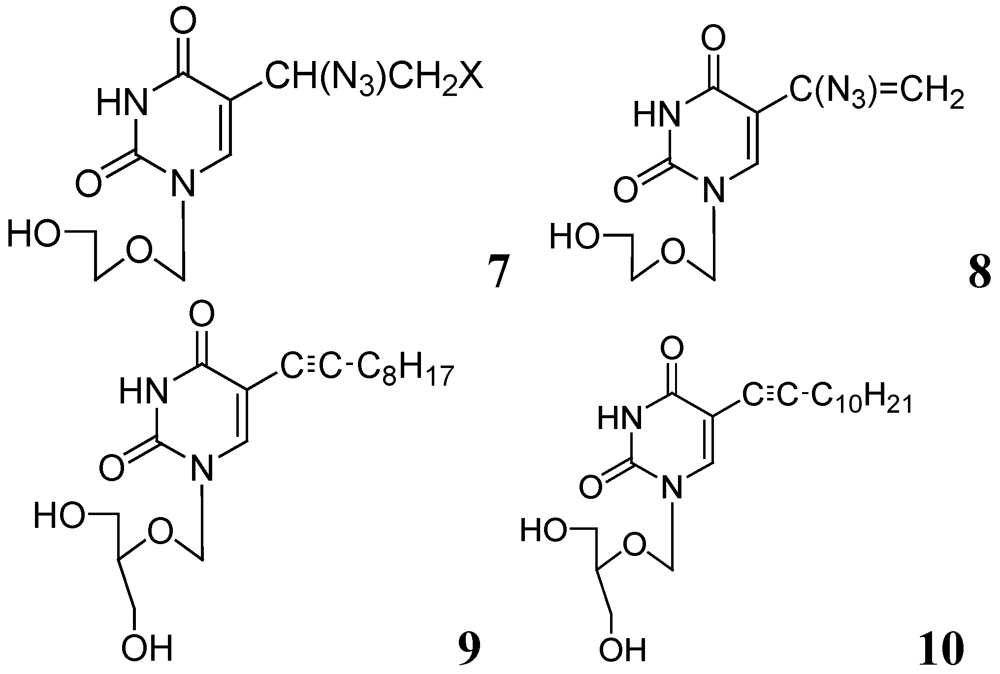
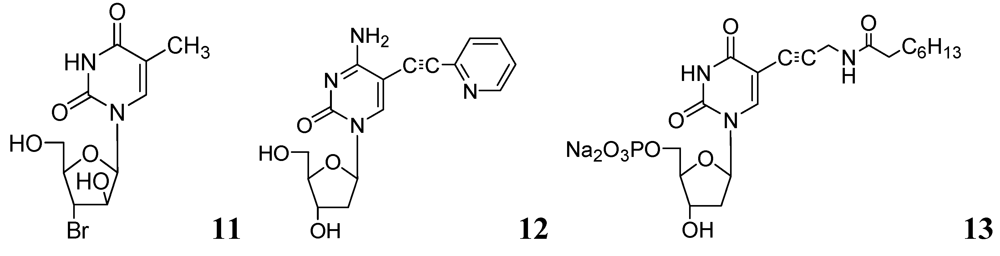
3.1.1. Nucleosides
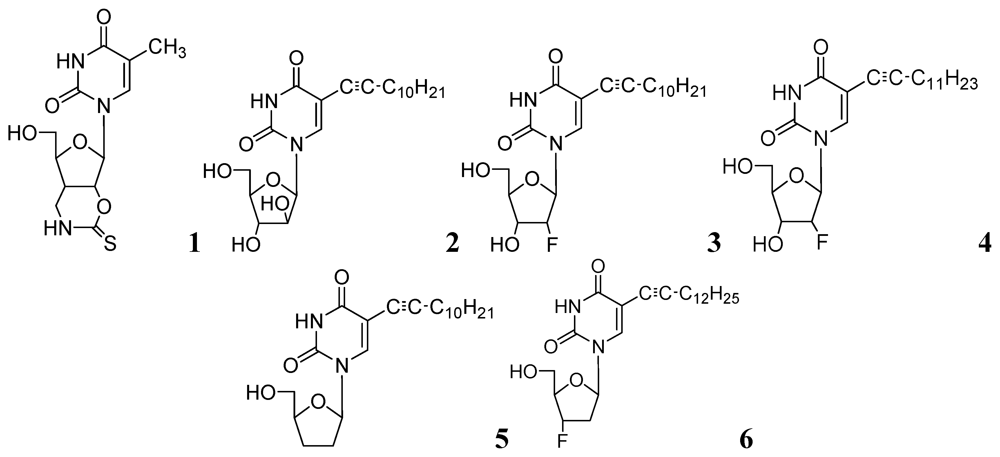
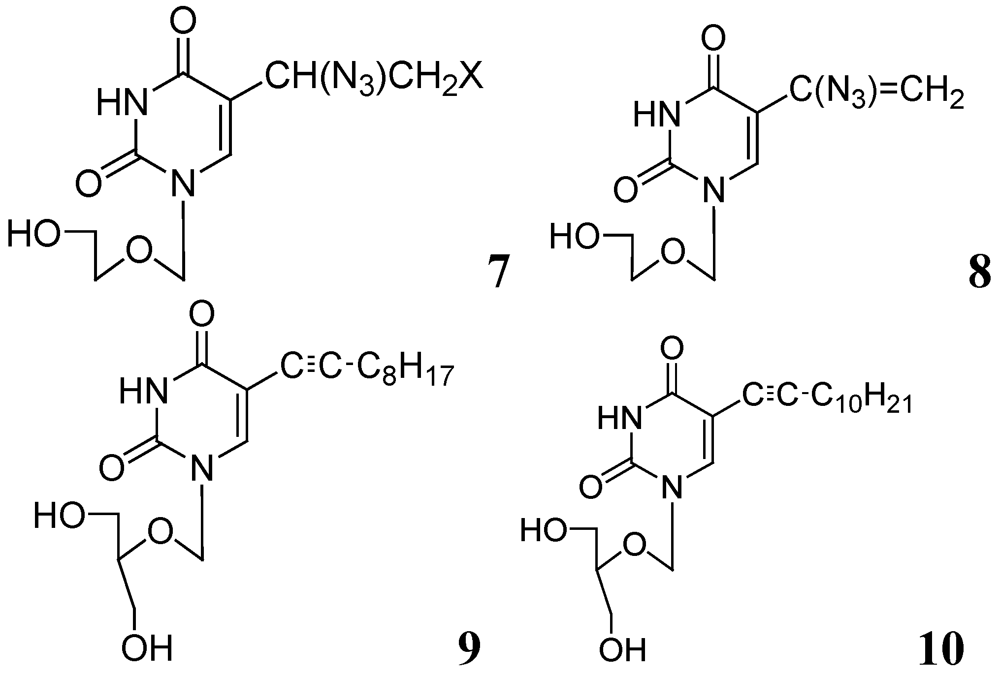
3.1.1.1. Pyrimidine Nucleosides
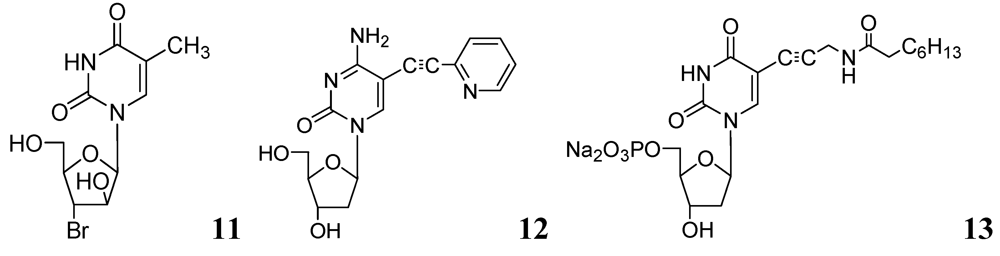
3.1.1.2. Purine Nucleosides
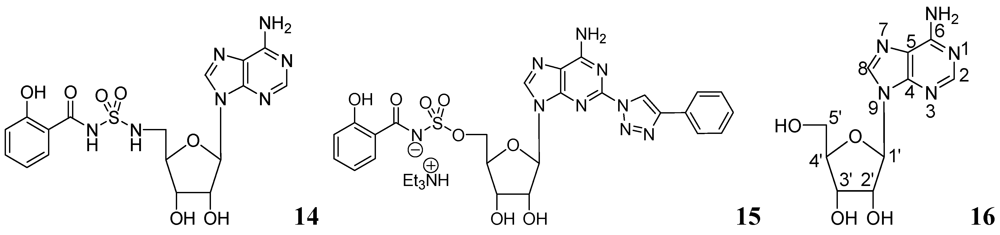
3.1.2. Carbohydrates
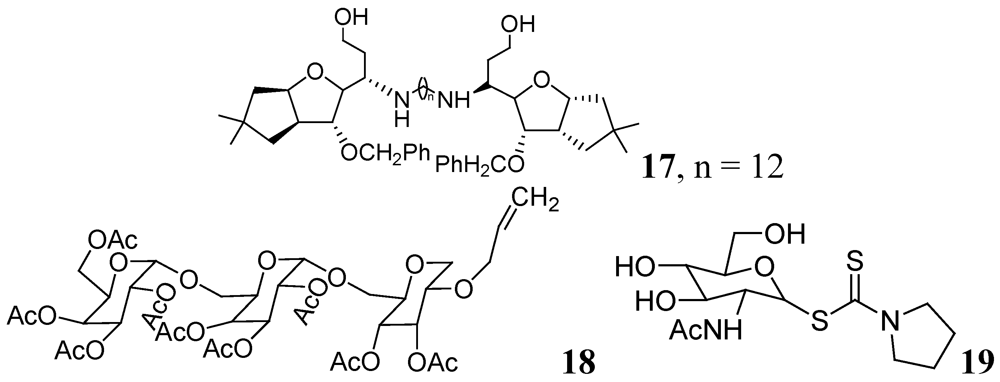
3.1.3. Heterocyclic Compounds
3.1.3.1. Quinolines and Quinoxalines
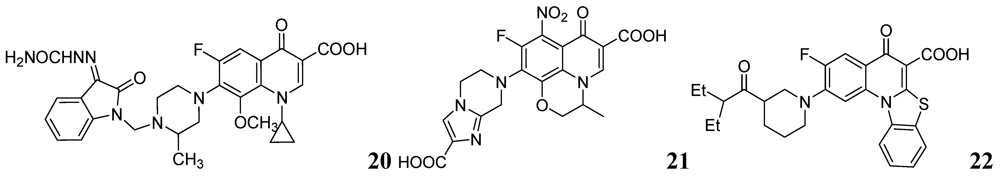
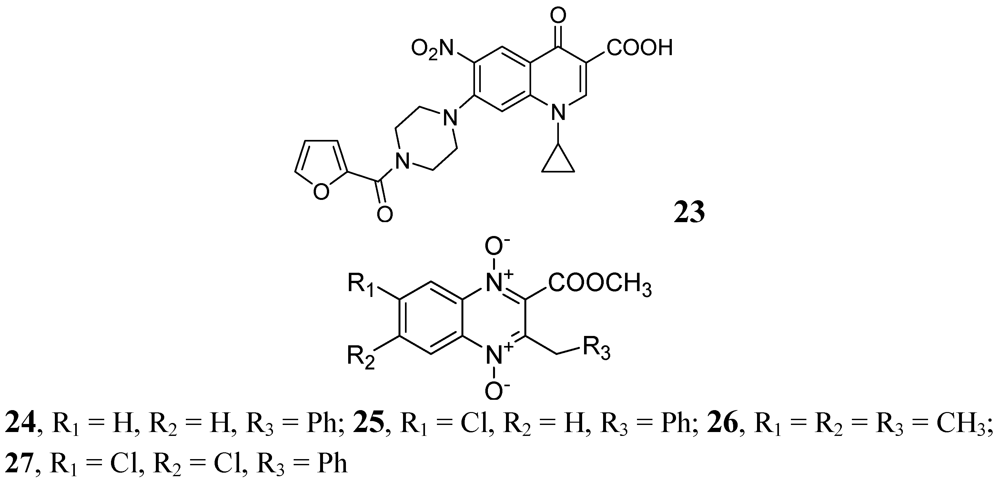
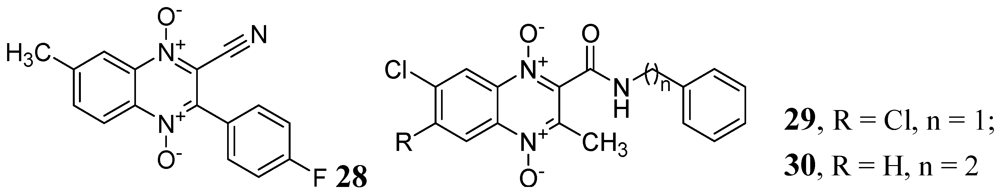
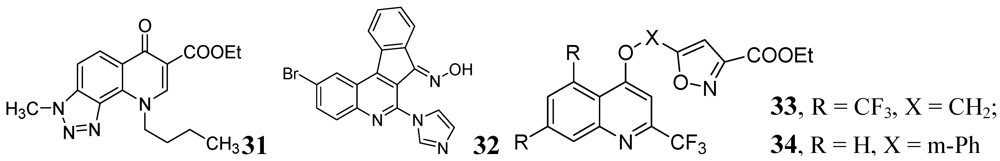
3.1.3.2. Pyrimidine and Purines
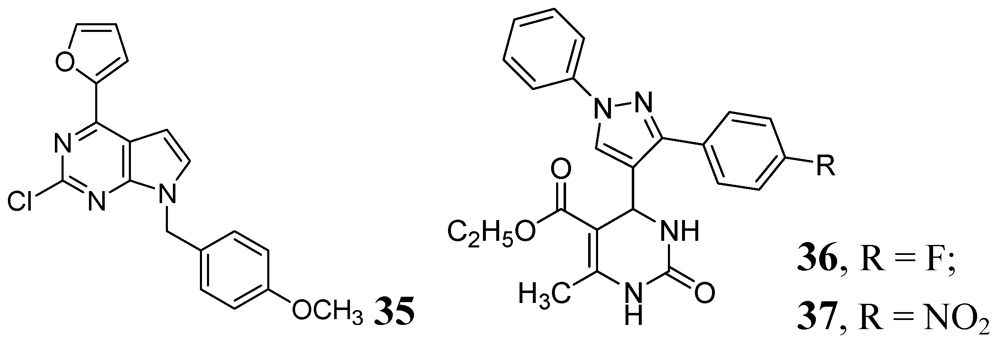
3.1.3.3. Azoles
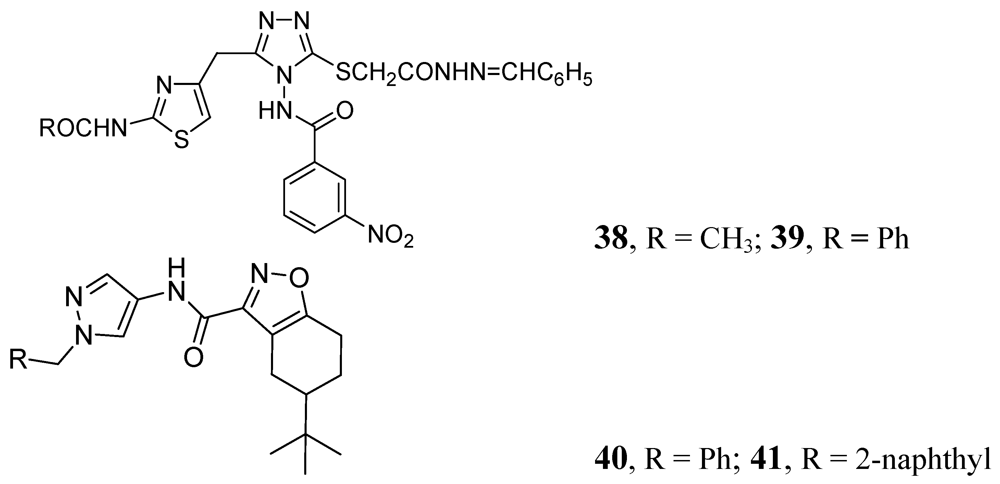
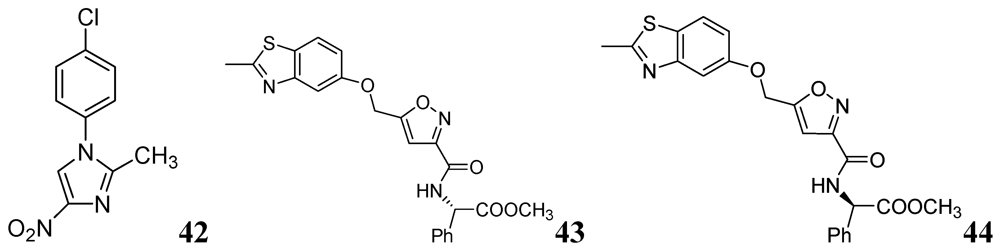
3.1.3.4. Azines
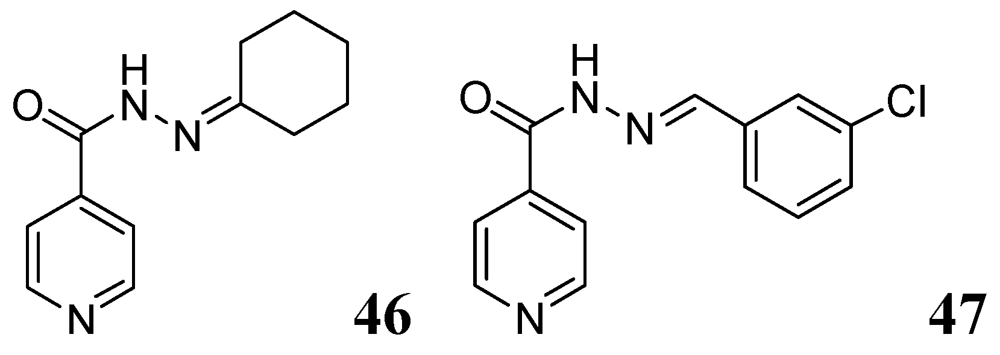
3.1.3.5. Pyridine hydrazides (INH analogs)

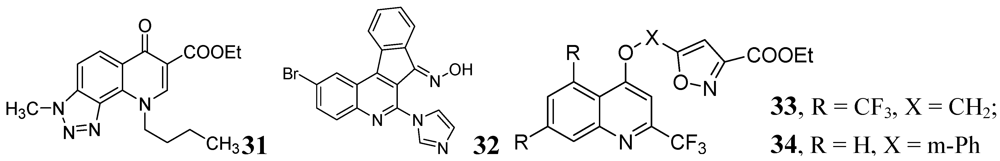
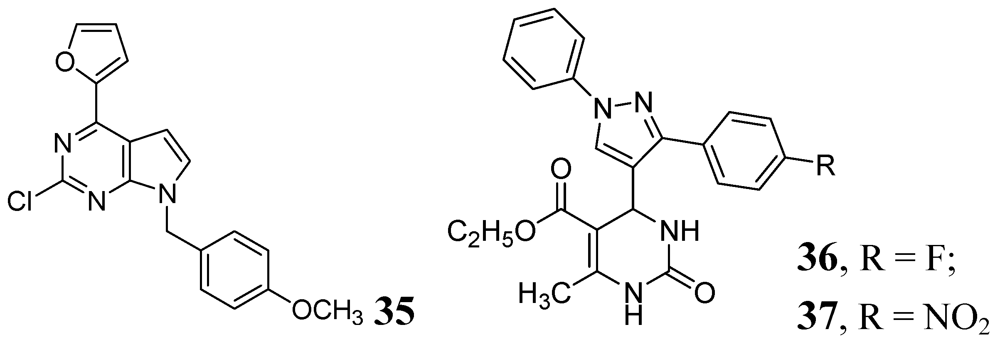
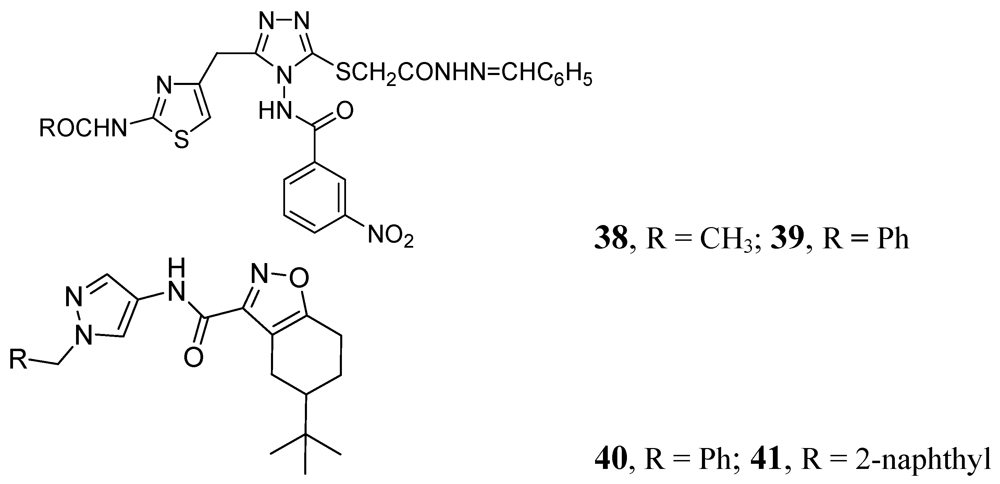
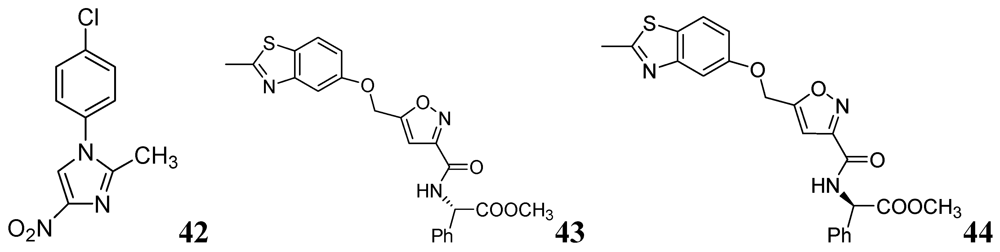
3.1.3.6. Miscellaneous
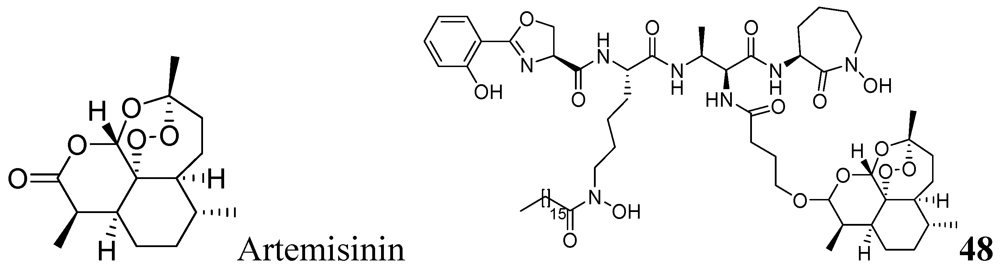
3.1.3.6.1. Artemisinin Analog
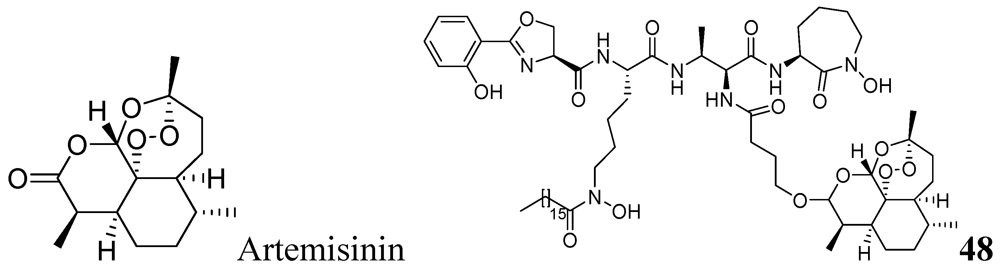
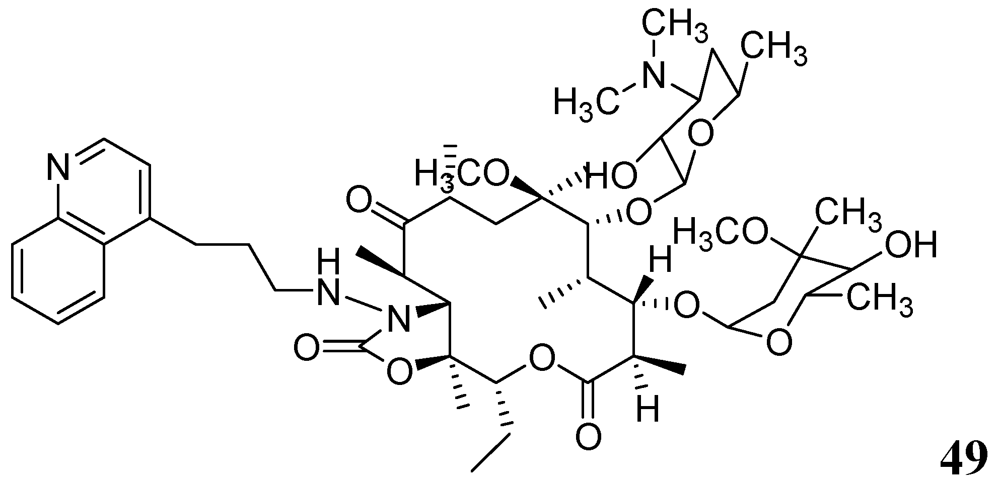
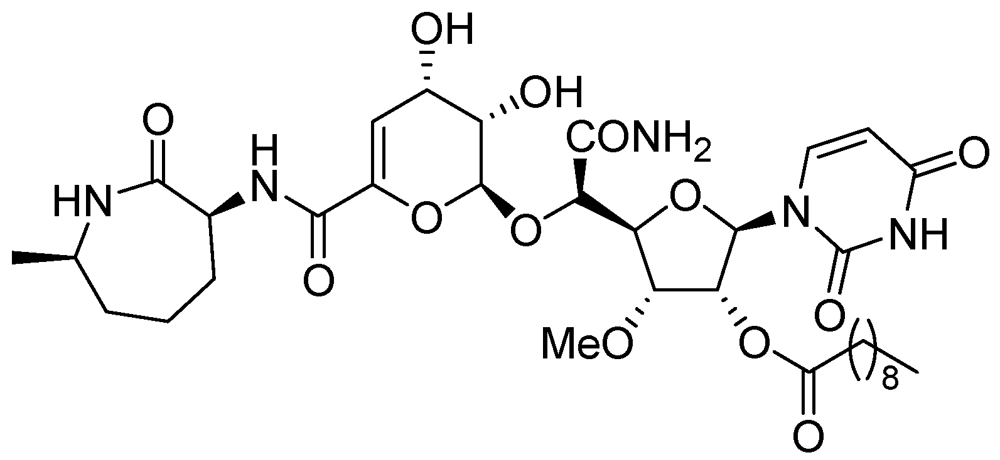
3.1.3.6.2. Macrolides
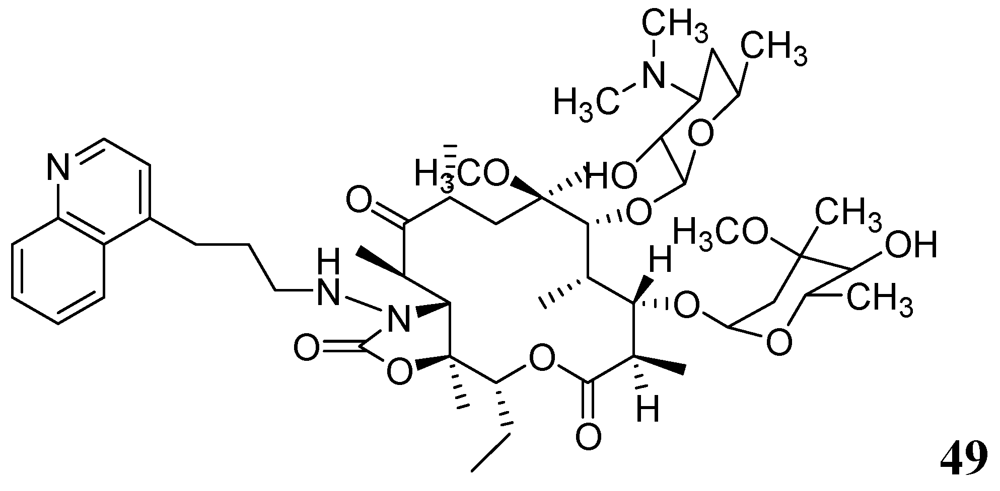
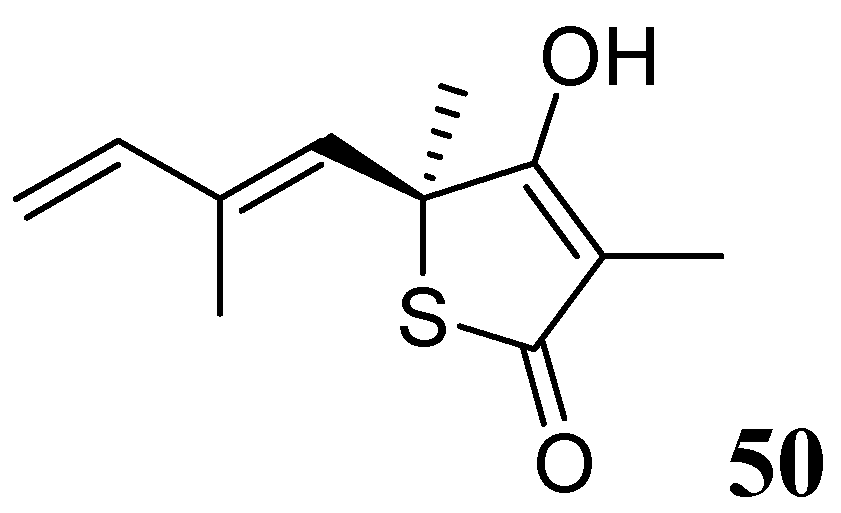
3.1.3.6.3. Thiolactomycin
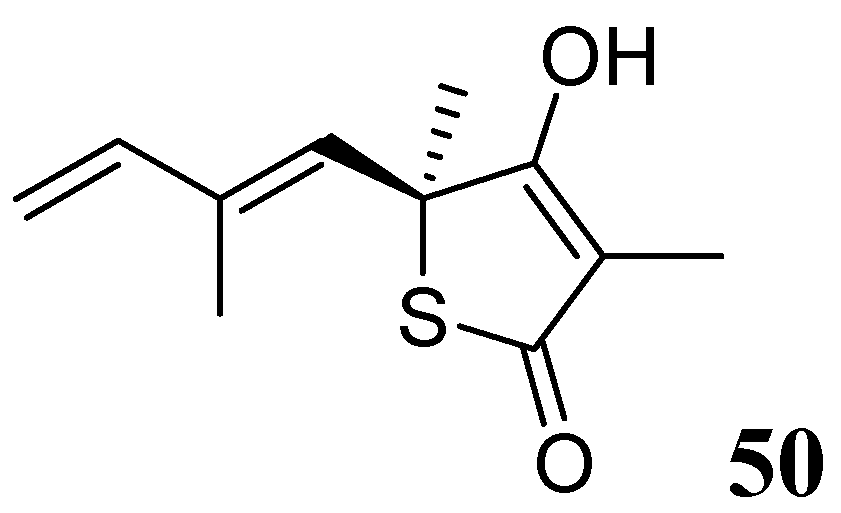
3.2. Molecules in Development
3.2.1. Molecules at Pre-Clinical Stage
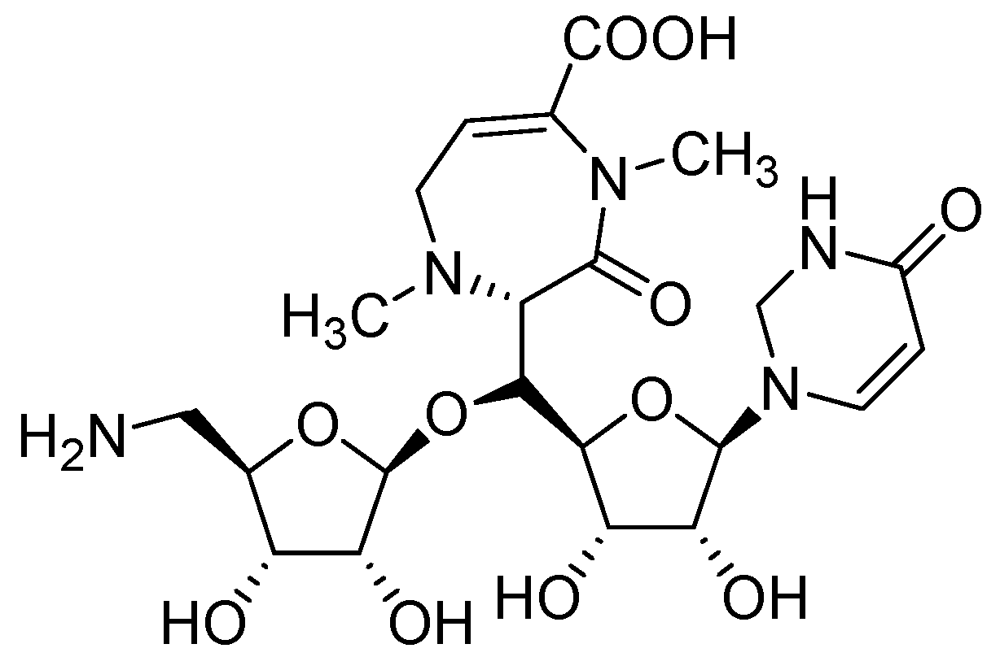
3.2.1.1. CPZEN-45
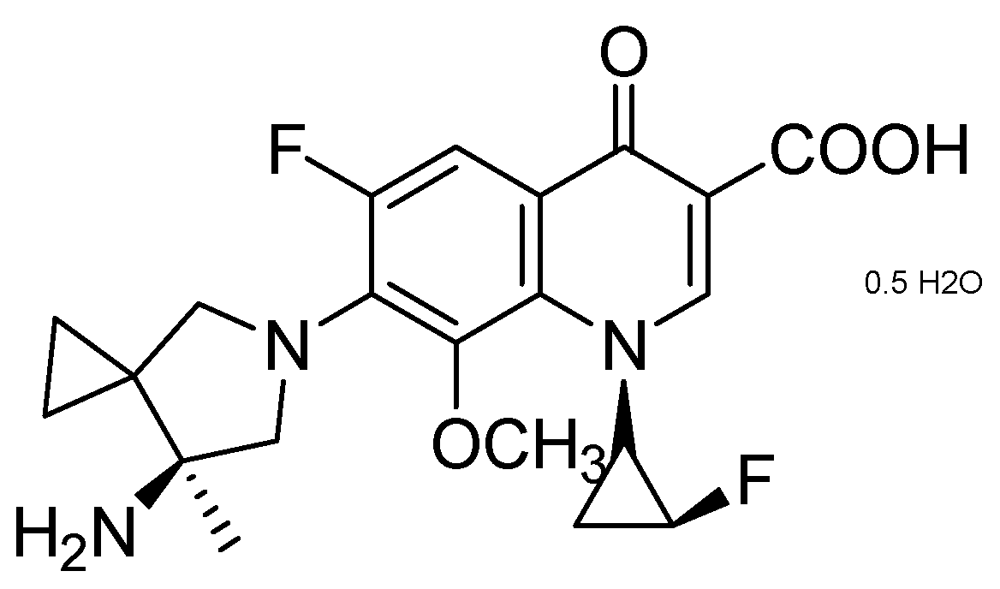
3.2.1.2. Quinolone DC-159a
3.2.1.3. SQ-609
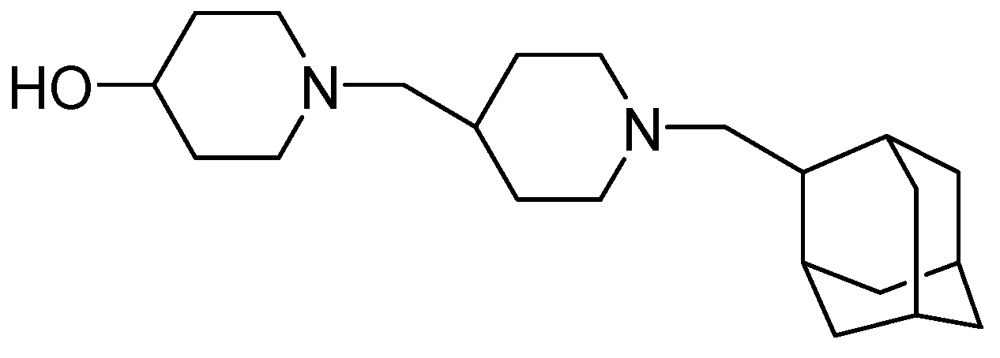
3.2.1.4. SQ-641
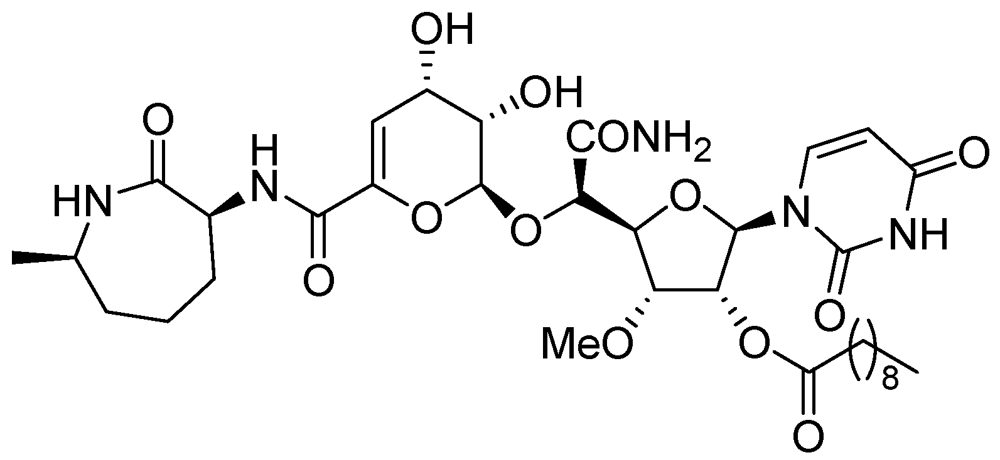
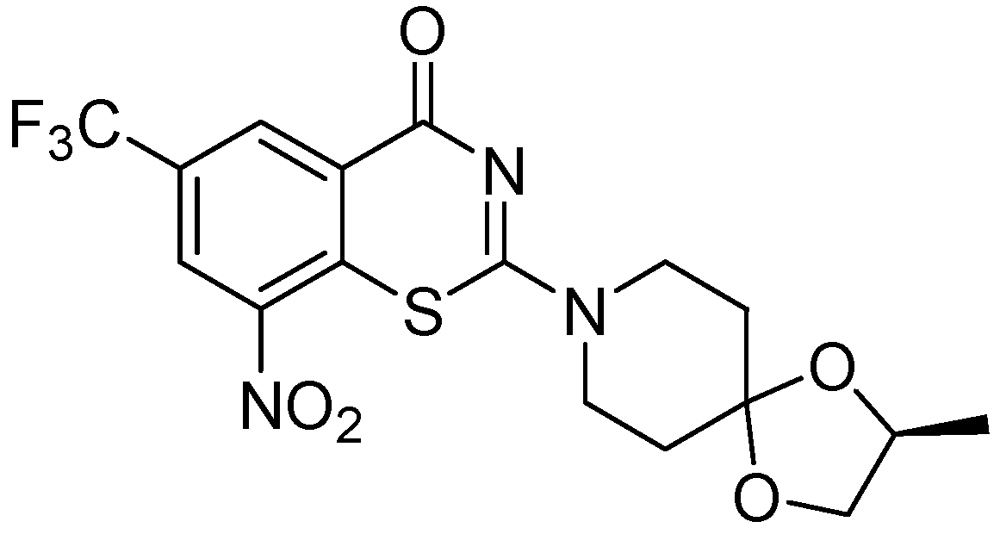
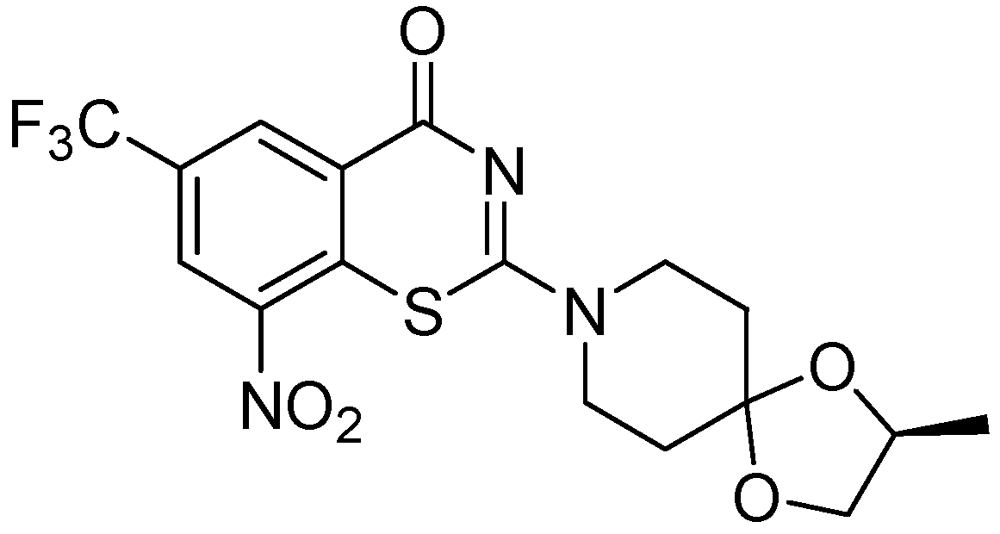
3.2.1.5. Benzothiazinone (BTZ-043)
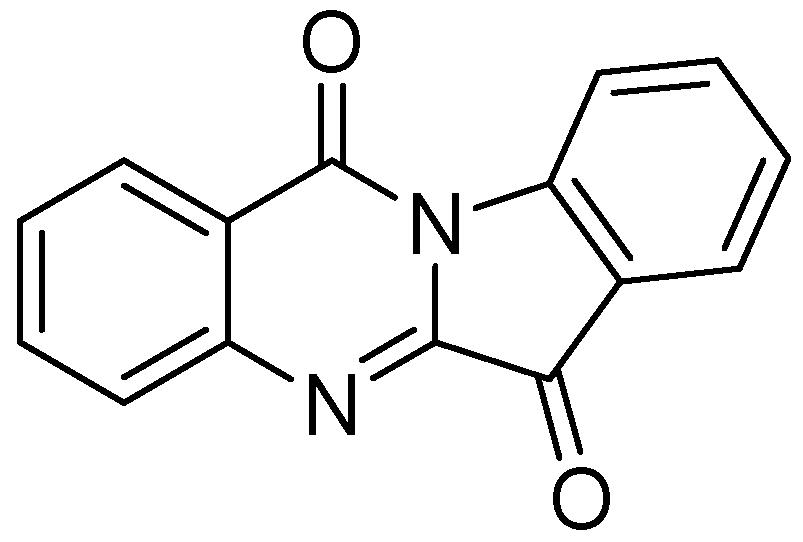
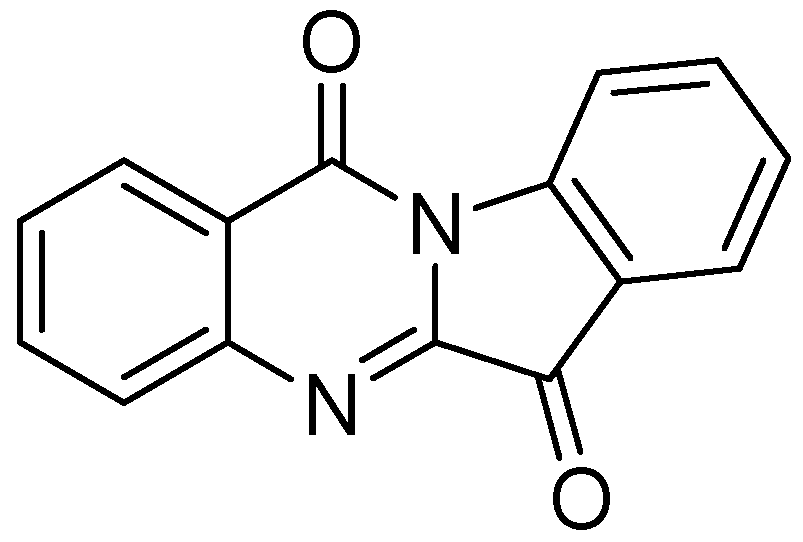
3.2.1.6. Tryptanthrin
3.2.2. Molecules in Phase I Clinical Trials
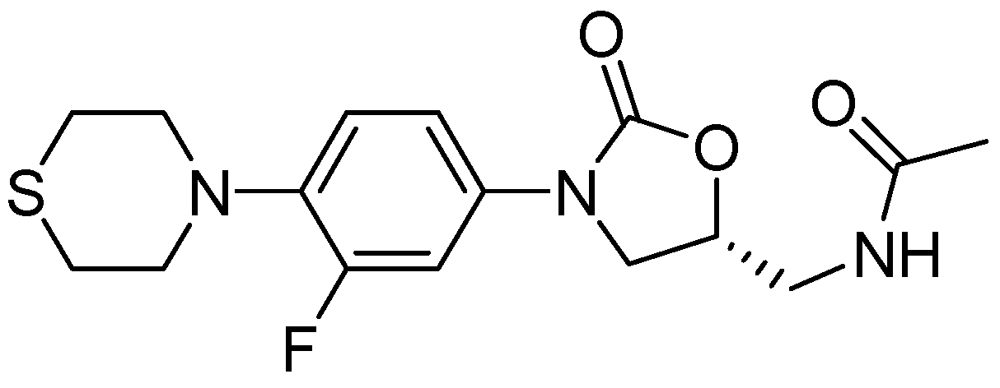
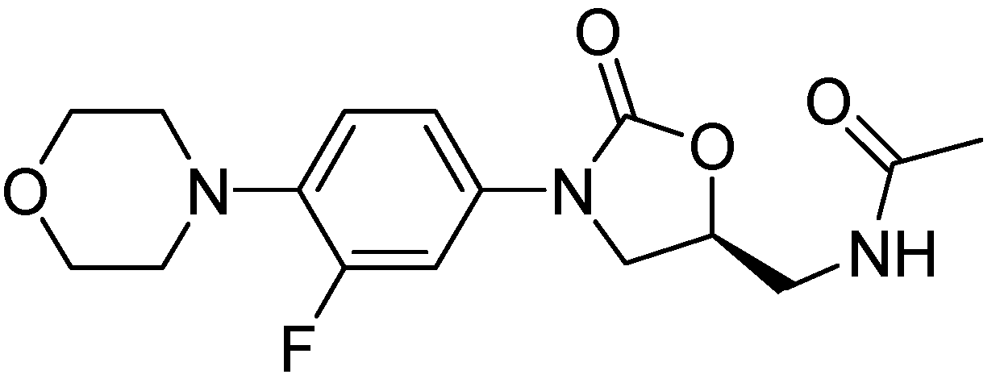
AZD-5847
3.2.3. Molecules in Phase II Clinical Trials
3.2.3.1. PNU-100480
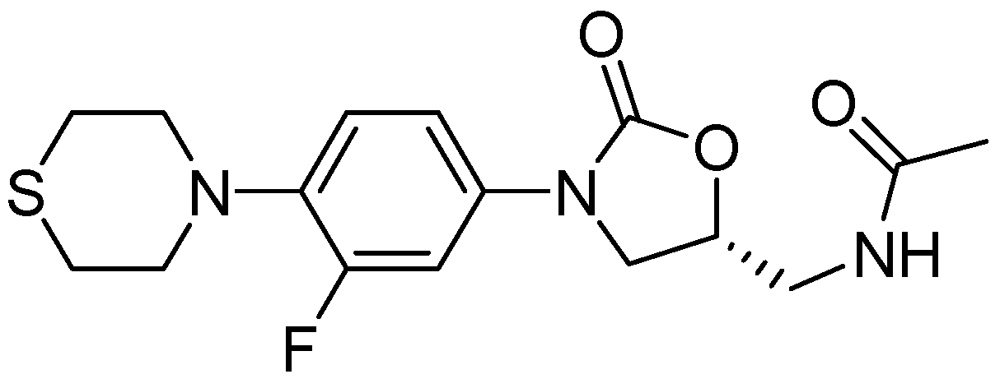
3.2.3.2. LL-3858 or Sudoterb
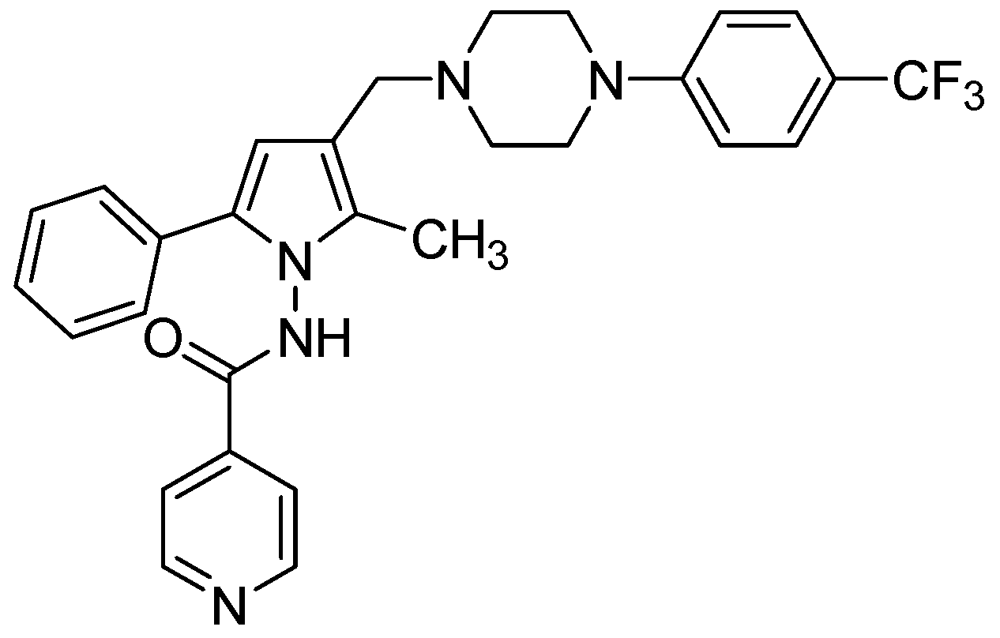
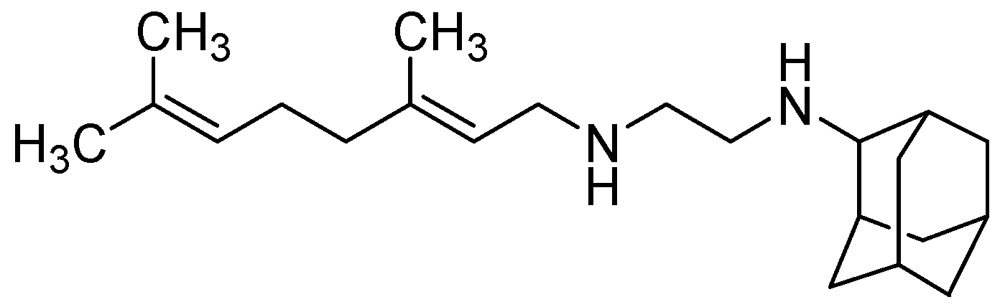
3.2.3.3. SQ-109
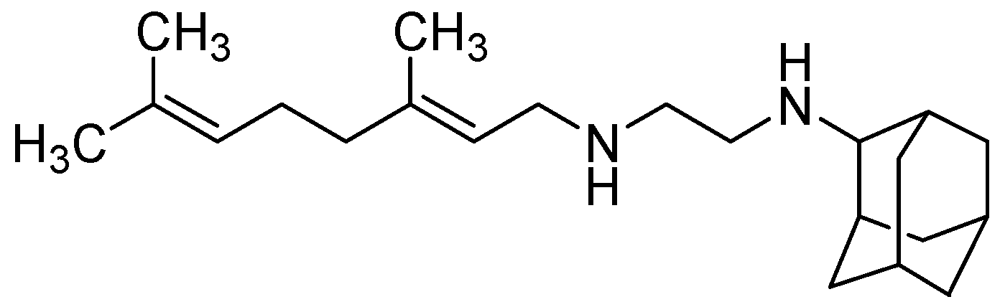
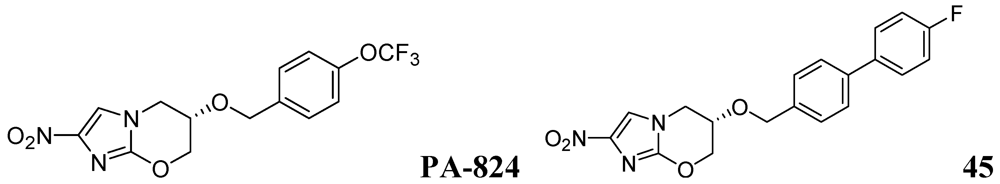
3.2.3.4. Nitroimidazoles
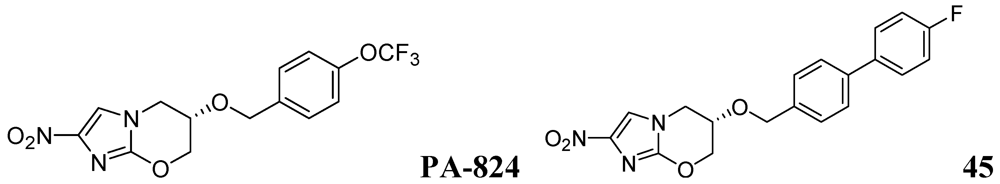
PA-824
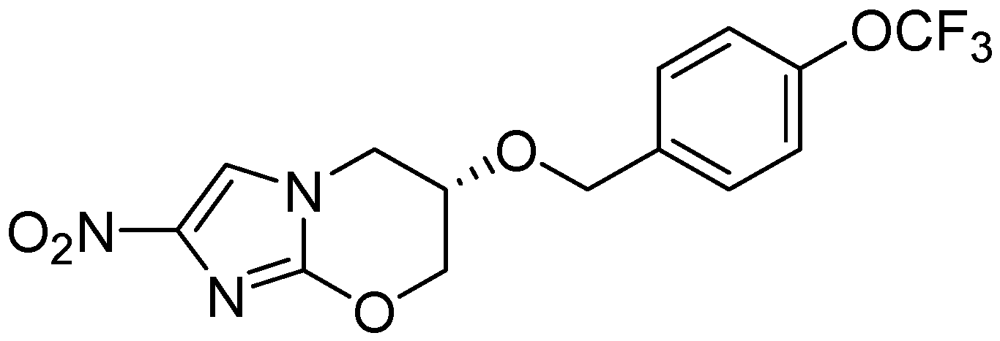
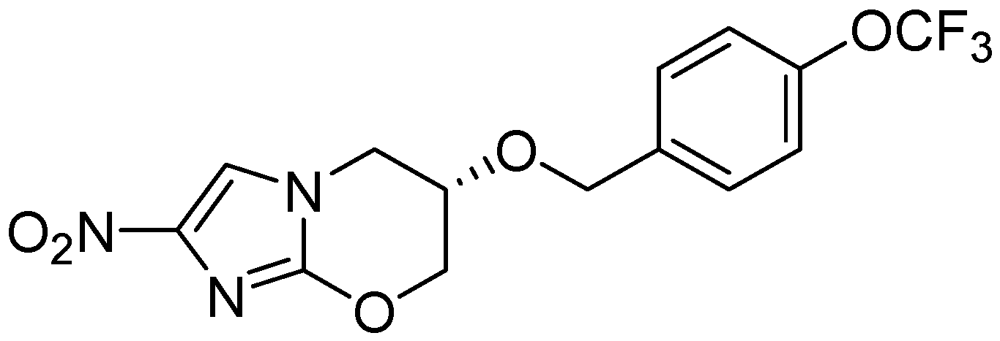
3.2.3.5. OPC-67683 (Delamanid)
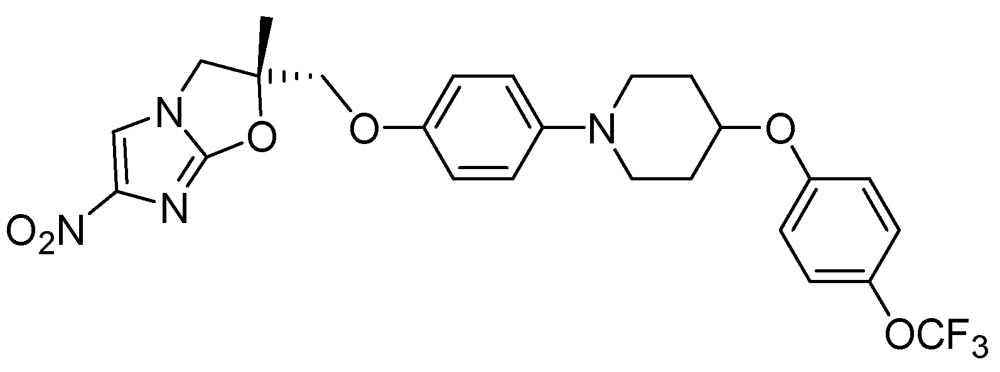
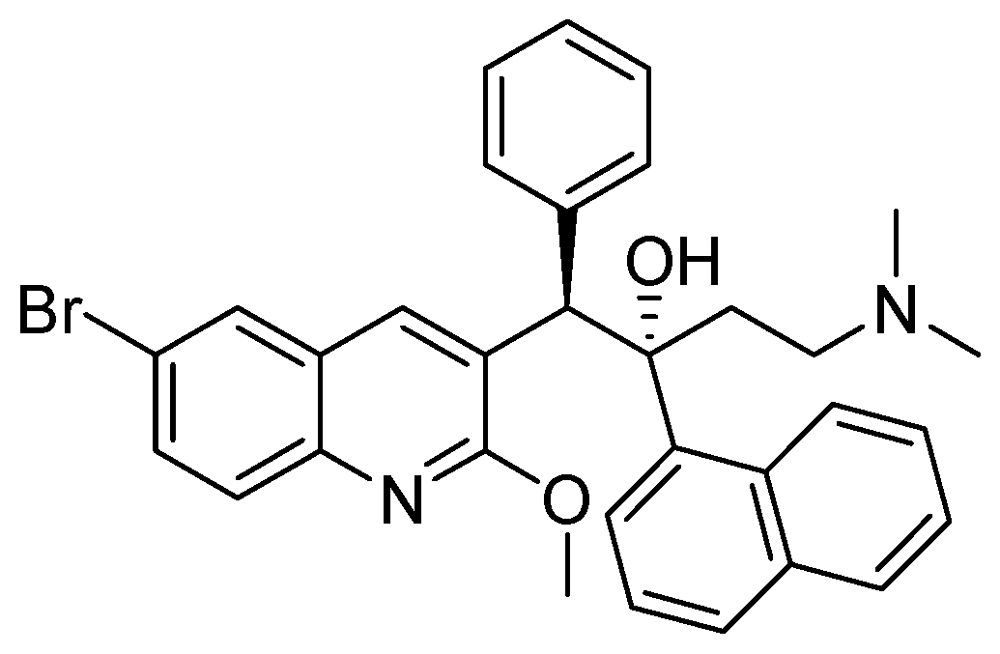
3.2.3.6. TMC-207 or R-207910 or Bedaquiline

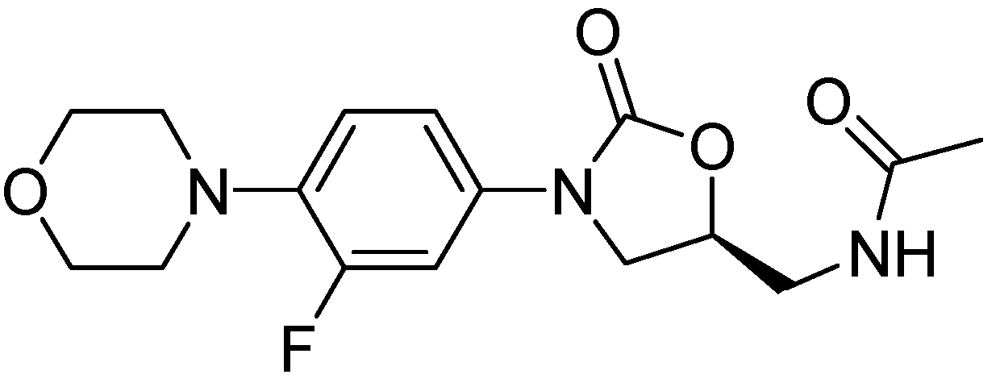
3.2.3.7. Linezolid for the Treatment of MDR-Tuberculosis
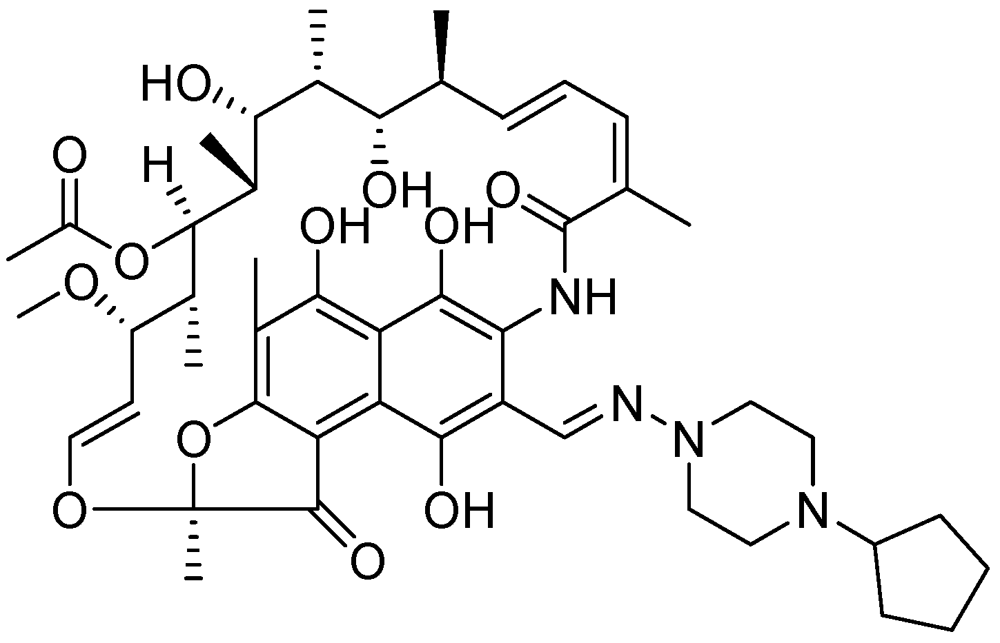
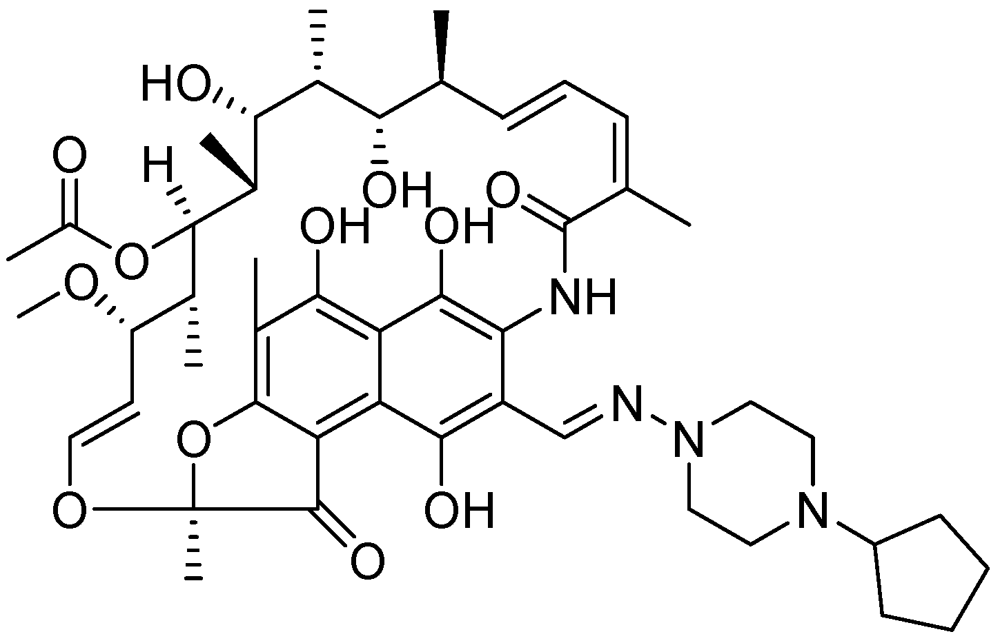
3.2.3.8. Rifapentine (TBTC Study)
3.2.4. Molecules in Phase III Clincal Trials
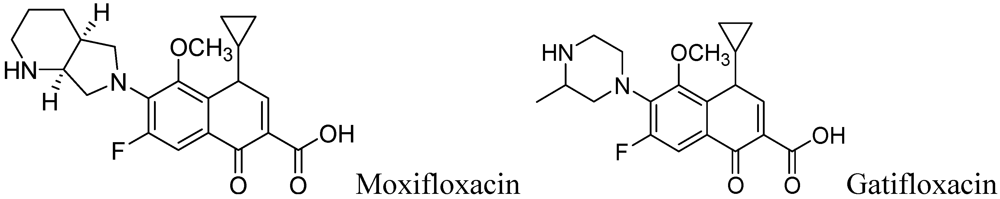
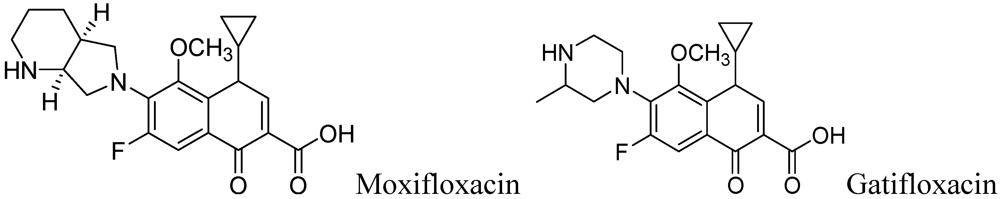
Fluoroquinolones (Moxifloxacin and Gatifloxacin)
4. Conclusions
References
- World Health Organisation (WHO). WHO Global Task Force outlines measures to combat XDR-TB worldwide (17 October 2006). Available online: http://www.who.int/mediacentre/news/notes/2006/np29/en/index.html/ (accessed on 4 April 2012).
- Rowland, K. Totally drug-resistant TB emerges in India, Nature News. Available online: http://www.nature.com/news/totally-drug-resistant-tb-emerges-in-india-1.9797/ (accessed on 9 June 2012).
- Kaufmann, S.H.E. Tuberculosis vaccines—A new kid on the block. Nat. Med. 2011, 17, 159–160. [Google Scholar] [CrossRef]
- Nau, R.; Prange, H.W.; Menck, S.; Kolenda, H.; Visser, K.; Seydel, J.K. Penetration of rifampicin into the cerebrospinal fluid of adults with uninflamed meninges. J. Antimicrob. Chemother. 1992, 29, 719–724. [Google Scholar] [CrossRef]
- Tomioka, H. Current status of some antituberculosis drugs and the development of new antituberculous agents with special reference to their in vitro and in vivo antimicrobial activities. Curr. Pharm. Des. 2006, 12, 4047–4070. [Google Scholar] [CrossRef]
- Aristoff, P.A.; Garcia, G.A.; Kirchhoff, P.D.; Hollis Showalter, H.D. Rifamycins-obstacles and opportunities. Tuberculosis 2010, 90, 94–118. [Google Scholar] [CrossRef]
- The American Thoracic Society [ATS]. Available online: http://www.thoracic.org/assemblies/mtpi/resources/istc-report.pdf (accessed on 4 April 2012).
- Telenti, A.; Imboden, P.; Marchesi, F.; Schmidheini, T.; Bodmer, T. Direct, automated detection of rifampin-resistant mycobacterium tuberculosis by polymerase chain reaction and single-strand conformation polymorphism analysis. Antimicrob. Agents Chemother. 1993, 37, 2054–2058. [Google Scholar] [CrossRef]
- International Programme on Chemical Safety [INCHEM]. Available online: http://www.inchem.org/documents/pims/pharm/rifam.htm#SectionTitle:2.1%20Main%20—risks%20and%20target%20organs/ and reference therein/ (accessed on 4 April 2012).
- Drobac, P.C.; del Castillo, H.; Sweetland, A.; Anca, G.; Joseph, J.K.; Furin, J.; Shin, S. Treatment of multidrug-resistant tuberculosis during pregnancy: Long-term follow-up of 6 children with intrauterine exposure to second-line agents. Clin. Infect. Dis. 2005, 40, 1689–1692. [Google Scholar] [CrossRef]
- Peters, C.; Nienhaus, A. Case report-tuberculosis in a health care worker during pregnancy. Pneumologie 2008, 62, 695–698. [Google Scholar] [CrossRef]
- Baciewicz, A.M.; Chrisman, C.R.; Finch, C.K.; Self, T.H. Update on rifampin and rifabutin drug interactions. Am. J. Med. Sci. 2008, 335, 126–136. [Google Scholar] [CrossRef]
- Singh, B.; Mitchison, D.A. Bactericidal activity of streptomycin and isoniazid against tubercle bacilli. Br. Med. J. 1954, 130, 130–132. [Google Scholar]
- Timmins, G.S.; Deretic, V. Mechanisms of action of isoniazid. Mol. Microbiol. 2006, 62, 1220–1227. [Google Scholar] [CrossRef]
- Suarez, J.; Ranguelova, K.; Jarzecki, A.A.; Manzerova, J.; Krymov, V.; Zhao, X.; Yu, S.; Metlitsky, L.; Gerfen, G.J.; Magliozzo, R.S. An oxyferrous heme/protein-based radical intermediate is catalytically competent in the catalase reaction of Mycobacterium tuberculosis catalase-peroxidase (KatG). J. Biol. Chem. 2009, 284, 7017–7029. [Google Scholar]
- McIlleron, H.; Willemse, M.; Werely, C.J.; Hussey, G.D.; Schaaf, H.S.; Smith, P.J.; Donald, P.R. Isoniazid plasma concentrations in a cohort of South African children with tuberculosis: Implications for international pediatric dosing guidelines. Clin. Infect. Dis. 2009, 48, 1547–1553. [Google Scholar] [CrossRef]
- Ellard, G.A.; Gammon, P.T. Pharmacokinetics of Isoniazid Metabolism in Man. J. Pharmacokinet. Biopharm. 1976, 4, 83–113. [Google Scholar] [CrossRef]
- International Programme on Chemical Safety [INCHEM]. Available online: http://www.inchem.org/documents/pims/pharm/pim288.htm#SectionTitle:2.1%20%20Main%20risks%20and%20target%20organs/ (accessed on 4 April 2012).
- Heifets, L. Antimycobacterial agents: Pyrazinamide. In Antimicrobial Therapy and Vaccines; Yu, V.L., Merigan, T.C., Barriere, S.L., Eds.; Williams and Wilkins: Baltimore, MD, USA, 1999. [Google Scholar]
- Hong Kong Chest Service, Medical Research Council. Controlled trial of four thrice weekly regimens and a daily regimen given for 6 months for pulmonary tuberculosis. Lancet 1981, 1, 171–174.
- Donald, P.R.; Seifart, H. Cerebrospinal fluid pyrazinamide concentrations in children with tuberculous meningitis. Pediatr. Infect. Dis. J. 1988, 7, 469–471. [Google Scholar] [CrossRef]
- Zhang, Y.; Mitchison, D. The Curious characteristics of pyrazinamide: A review. Int. J. Tuberc. Lung Dis. 2003, 7, 6–21. [Google Scholar]
- Zimhony, O.; Vilcheze, C.; Arai, M.; Welch, J.; Jacobs, W.R. Pyrazinoic acid and its n’Propyl Ester Inhibit Fatty Acid Synthase I in Replicating Tubercle Bacilli. Antimicrob. Agents Chemother. 2007, 51, 752–754. [Google Scholar] [CrossRef]
- Mitchison, D.; Davies, G. The chemotherapy of tuberculosis: Past, present and future. Int. J. Tuberc. Lung Dis. 2012, 16, 724–732. [Google Scholar] [CrossRef]
- Scorpio, A.; Zhang, Y. Mutations In pnca, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat. Med. 1996, 2, 662–667. [Google Scholar] [CrossRef]
- Scorpio, A.; Lindholm-Levy, P.; Heifets, L.; Gilman, R.; Siddiqi, S.; Cynamon, M.; Zhang, Y. Characterization of pncA Mutations in pyrazinamide-resistant Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1997, 41, 540–543. [Google Scholar]
- Jureen, P.; Werngren, J.; Toro, J.C.; Hoffner, S. Pyrazinamide resistance and PNCA gene mutations in Mycobacterium tuberculosis. Antimicrob. Agents. Chemother. 2008, 52, 1852–1854. [Google Scholar]
- Lacroix, C.; Phan Hoang, T.; Nouveau, J.; Guyonnaud, C.; Laine, G.; Duwoos, H.; Lafont, O. Pharmacokinetics of pyrazinamide and its metabolites in healthy subjects. Eur. J. Clin. Pharmacol. 1989, 36, 395–400. [Google Scholar] [CrossRef]
- Forget, E.J.; Menzies, D. Adverse reactions to first-line antituberculosis drugs. Expert Opin. Drug Saf. 2006, 5, 231–249. [Google Scholar] [CrossRef]
- Telenti, A.; Philipp, W.J.; Sreevatsan, S.; Bernasconi, C.; Stockbauer, K.E.; Wieles, B.; Musser, J.M.; Jacobs W.R., Jr. The Emb operon, a gene cluster of Mycobacterium tuberculosis involved in resistance to ethambutol. Nat. Med. 1997, 3, 567–570. [Google Scholar] [CrossRef]
- Belanger, A.E.; Besra, G.S.; Ford, M.E.; Mikusova, K.; Belisle, J.T.; Brennan, P.J.; Inamine, J.M. The Embab genes of Mycobacterium avium encode an arabinosyl transferase involved in cell wall arabinan biosynthesis that is the target for the antimycobacterial drug ethambutol. Proc. Natl. Acad. Sci. USA 1996, 93, 11919–11924. [Google Scholar]
- Peloquin, C.A.; Bulpitt, A.E.; Jaresko, G.S.; Jelliffe, R.W.; Childs, J.M.; Nix, D.E. Pharmacokinetics of ethambutol under fasting conditions, with food, and with antacids. Antimicrob. Agents Chemother. 1999, 43, 568–572. [Google Scholar]
- Ethambutol (Ethambutol Hydrochloride)-Indications and Dosage. Available online: http://www.druglib.com/druginfo/ethambutol/indications_doses/ (accessed on 4 April 2012).
- Lim, S.A. Ethambutol-associated optic neuropathy. Ann. Acad. Med. Singapore 2006, 35, 274–278. [Google Scholar]
- Source partly from: North Dakota Department of Health. 2011. Available online: http://www.ndhealth.gov/disease/tb/Documents/Second line TB drugs.pdf (accessed on 4 April 2012).
- Heifets, L. MIC as a quantitative measurement of susceptibility of M. Avium to seven antituberculosis drugs. Antimicrob. Agents Chemother. 1988, 32, 1131–1136. [Google Scholar] [CrossRef]
- Heifets, L.; Lindholm-Levy, P.A. Comparison of bactericidal activities of streptomycin, amikacin, kanamycin, and capreomycin against M. Avium and M. tuberculosis. Antimicrob. Agents Chemother. 1989, 33, 1298–1303. [Google Scholar] [CrossRef]
- Johansen, S.K.; Maus, C.E.; Plikaytis, B.B.; Douthwaite, S. Douthwaite S. Capreomycin binds across the ribosomal subunit interface using Tlya-encoded 2'-O-methylations in 16S and 23S rRNAs. Mol. Cell 2006, 23, 173–182. [Google Scholar] [CrossRef]
- Centre for Disease Control and Prevention [CDC] (1994). Available online: http://wonder.cdc.gov/wonder/prevguid/p0000413/p0000413.asp#head006001000000000/ (accessed on 4 April 2012).
- Lu, Y.; Zheng, M.Q.; Wang, B.; Zhao, W.J.; Li, P.; Chu, N.H.; Liang, B.W. Activities of clofazimine against Mycobacterium tuberculosis in vitro and in vivo. Zhonghua Jie He He Hu Xi Za Zhi 2008, 31, 752–755. [Google Scholar]
- Rastogi, N.; Goh, K.S.; Bryskier, A.; Devallois, A. In vitro activities of levofloxacin used alone and in combination with first- and second-line antituberculous drugs against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1996, 40, 1610–1616. [Google Scholar]
- Vacher, S.; Pellegrin, J.L.; Leblanc, F.; Fourche, J.; Maugein, J. Comparative antimycobacterial activities of ofloxacin, ciprofloxacin and grepafloxacin. J. Antimicrob. Chemother. 1999, 44, 647–652. [Google Scholar] [CrossRef]
- Trimble, K.A.; Clark, R.B.; Sanders, W.E., Jr.; Frankel, J.W.; Cacciatore, R.; Valdez, H. Activity of ciprofloxacin against Mycobacteria in vitro: Comparison of BACTEC and macrobroth dilution methods. J. Antimicrob. Chemother. 1987, 19, 617–622. [Google Scholar] [CrossRef]
- Inderlied, C.B.; Salfinger, M. Antimycobacterial agents and susceptibility tests. In Manual of Clinical Microbiology, III; Murray, P.R., Baron, E.J., Pfaller, M.A., Tenover, F.C., Yolken, R.H., Eds.; ASM Press: Washington, DC, USA, 1999; pp. 1601–1623. [Google Scholar]
- Vanheusden, V.; Munier-Lehmann Froeyen, M.; Busson, R.; Rozenski, J.; Herdewijn, P.; van Calenbergh, S. Discovery of bicyclic thymidine analogues as selective and high-affinity inhibitors of Mycobacterium tuberculosis thymidine monophosphate kinase. J. Med. Chem. 2004, 47, 6187–6194. [Google Scholar] [CrossRef]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E., 3rd; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar]
- Johar, M.; Manning, T.; Kunimoto, D.Y.; Kumar, R. Synthesis and in vitro anti-mycobacterial activity of 5-substituted pyrimidine nucleosides. Bioorg. Med. Chem. 2005, 13, 6663–6671. [Google Scholar] [CrossRef]
- Franzblau, S.G.; Witzig, R.S.; McLaughlin, J.C.; Torres, P.; Madico, G.; Hernandez, A.; Degnan, M.T.; Cook, M.B.; Quenzer, V.K.; Ferguson, R.M.; et al. Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis Isolates by using the microplate alamar blue assay. J. Clin. Microbiol. 1998, 36, 362–366. [Google Scholar]
- Vanheusden, V.; Munier-Lehmann, H.; Pochet, S.; Herdewijn, P.; van Calenbergh, S. Synthesis and evaluation of thymidine-5O-monophosphate analogues as inhibitors of Mycobacterium tuberculosis thymidylate kinase. Bioorg. Med. Chem. Lett. 2002, 12, 2695–2698. [Google Scholar] [CrossRef]
- Vanheusden, V.; van Rompaey, P.; Munier-Lehmann, H.; Pochet, S.; Herdewijn, P.; van Calenbergh, S. Thymidine and thymidine-5'O-monophosphate analogues as inhibitors of Mycobacterium tuberculosis thymidylate kinase. Bioorg. Med. Chem. Lett. 2003, 13, 3045–3048. [Google Scholar]
- Pochet, S.; Dugue, L.; Labesse, G.; Delepierre, M.; Munier-Lehmann, H. Comparative study of purine and pyrimidine nucleoside analogues acting on the thymidylate kinases of Mycobacterium tuberculosis and of humans. ChemBioChem 2003, 4, 742–747. [Google Scholar] [CrossRef]
- Rai, D.; Johar, M.; Manning, T.; Agrawal, B.; Kunimoto, D.Y.; Kumar, R. Design and studies of novel 5-substituted alkynylpyrimidine nucleosides as potent inhibitors of mycobacteria. J. Med. Chem. 2005, 48, 7012–7017. [Google Scholar]
- Johar, M.; Manning, T.; Tse, C.; Desroches, N.; Agrawal, B.; Kunimoto, Y.; Kumar, R. Growth inhibition of Mycobacterium bovis, Mycobacterium tuberculosis and Mycobacterium avium in vitro: Effect of 1-b-D-2'-b-D-arabinofuranosyl and 1-(2-deoxy-2-fluoro-b-D-2'-ribofuranosyl) pyrimidine nucleoside. J. Med. Chem. 2007, 50, 3696–3705. [Google Scholar] [CrossRef]
- Rai, D.; Johar, M.; Srivastav, N.C.; Manning, T.; Agrawal, B.; Kunimoto, D.Y.; Kumar, R. Inhibition of Mycobacterium tuberculosis, Mycobacterium bovis, and Mycobacterium avium by novel dideoxy nucleosides. J. Med. Chem. 2007, 50, 4766–4774. [Google Scholar]
- Srivastav, N.C.; Manning, T.; Kunimoto, D.Y.; Kumar, R. Studies on acyclic pyrimidines as inhibitors of mycobacteria. Bioorg. Med. Chem. 2007, 15, 2045–2053. [Google Scholar] [CrossRef]
- Shakya, N.; Srivastav, N.C.; Desroches, N.; Agrawal, B.; Kunimoto, D.Y.; Kumar, R. 3-Bromo analogues of pyrimidine nucleosides as a new class of potent inhibitors of Mycobacterium tuberculosis. J. Med. Chem. 2010, 53, 4130–4140. [Google Scholar]
- Bermudez, L.E.; Inderlied, C.B.; Kolonoski, P.; Wu, M.; Aralar, P.; Young, L.S. Telithromycin is active against Mycobacterium avium in mice despite lacking significant activity in standard in vitro and macrophage assays and is associated with low frequency of resistance during treatment. Antimicrob. Agents Chemother. 2001, 45, 2210–2214. [Google Scholar] [CrossRef]
- Srivastav, N.C.; Rai, D.; Tse, C.; Agrawal, B.; Kunimoto, D.Y.; Kumar, R. Inhibition of mycobacterial replication by pyrimidines possessing various c-5 functionalities and related 20-deoxynucleoside analogues using in vitro and in vivo models. J. Med. Chem. 2010, 53, 6180–6187. [Google Scholar] [CrossRef]
- Kogler, M.; Vanderhoydonck, B.; de Jonghe, S.; Rozenski, J.; van Belle, K.; Herman, J.; Louat, T.; Parchina, A.; Sibley, C.; Lescrinier, E.; et al. Synthesis and evaluation of 5-substituted 2'-deoxyuridine monophosphate analogues as inhibitors of flavin-dependent thymidylate synthase in mycobacterium tuberculosis. J. Med. Chem. 2011, 54, 4847–4862. [Google Scholar]
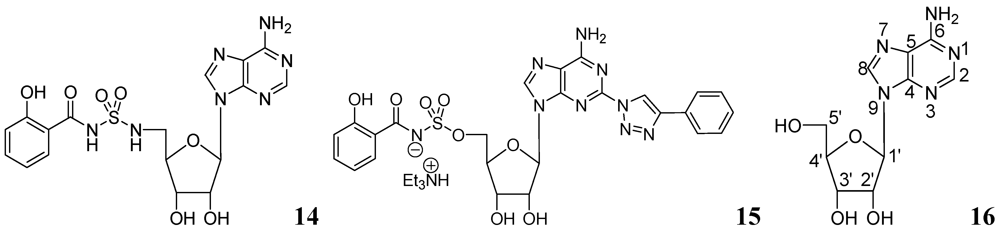
- Somu, R.V.; Boshoff, H.; Qiao, C.; Bennett, E.; Barry, C.E.; Aldrich, C.C. Rationally designed nucleoside antibiotics that inhibit siderophore biosynthesis of Mycobacterium tuberculosis. J. Med. Chem. 2006, 49, 31–34. [Google Scholar]
- Gupte, A.; Boshoff, H.I.; Wilson, D.J.; Neres, J.; Labello, N.P.; Somu, R.; Xing, C.; Barry, C.E.; Aldrich, C.C. Inhibition of siderophore biosynthesis by 2-triazole substituted analogues of 5'-O-[N-(Salicyl)sulfamoyl]adenosine: Antibacterial nucleosides effective against Mycobacterium tuberculosis. J. Med. Chem. 2008, 51, 7495–7507. [Google Scholar]
- Long, M.C.; Parker, W.B. Structure-activity relationship for nucleoside analogs as inhibitors or substrates of adenosine kinase from Mycobacterium tuberculosis I. Modifications to the adenine moiety. Biochem. Pharmacol. 2006, 71, 1671–1682. [Google Scholar]
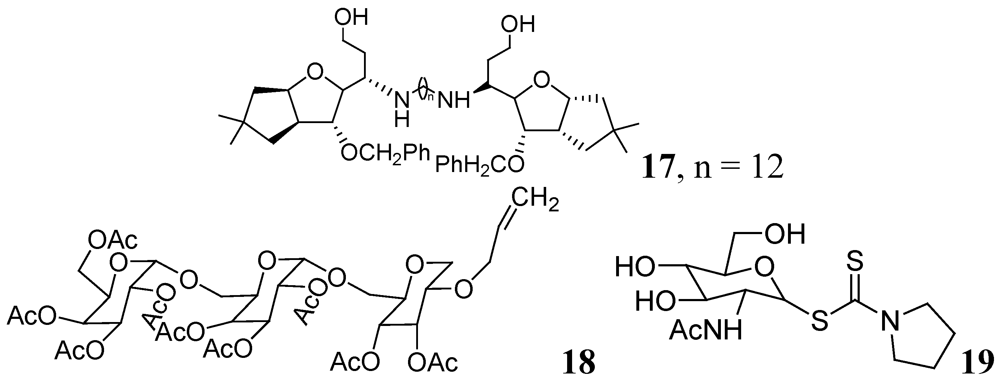
- Tripathi, R.P.; Tiwari, V.K.; Tewari, N.; Katiyar, D.; Saxena, N.; Sinha, S.; Gaikwad, A.; Srivastava, A.; Chaturvedi, V.; Manju, Y.K.; et al. Synthesis and antitubercular activities of Bis-glycosylated diamino alcohols. Bioorg. Med. Chem. 2005, 13, 5668–5679. [Google Scholar]
- Chiba, T.; Takii, T.; Nishimura, K.; Yamamoto, Y.; Morikawa, H.; Abec, C.; Onozaki, K. Synthesis of new sugar derivatives from stachys sieboldi miq and antibacterial evaluation against Mycobacterium tuberculosis, Mycobacterium avium, and Staphylococcus aureus. Bioorg. Med. Chem. Lett. 2007, 17, 2487–2491. [Google Scholar]
- Horita, Y.; Takii, T.; Kuroishi, R.; Chiba, T.; Ogawa, K.; Kremer, L.; Sato, Y.; Lee, Y.; Hasegawa, T.; Onozaki, K. Synthesis and evaluation of anti-tubercular activity of new dithiocarbamate sugar derivatives. Bioorg. Med. Chem. Lett. 2011, 21, 899–903. [Google Scholar]
- Sriram, D.; Aubry, A.; Yogeeswari, P.; Fisher, L.M. Gatifloxacin derivatives: Synthesis, antimycobacterial activities, and inhibition of Mycobacterium tuberculosis DNA gyrase. Bioorg. Med. Chem. Lett. 2006, 16, 2982–2985. [Google Scholar]
- Dinakaran, M.; Senthilkumar, P.; Yogeeswari, P.; China, A.; Nagaraja, V.; Sriram, D. Novel ofloxacin derivatives: Synthesis, antimycobacterial and toxicological evaluation. Bioorg. Med. Chem. Lett. 2008, 18, 1229–1236. [Google Scholar]
- Dinakaran, M.; Senthilkumar, P.; Yogeeswari, P.; China, A.; Nagaraja, V.; Sriram, D. Antimycobacterial activities of novel 2-(sub)-3-fluoro/nitro-5, 12-dihydro-5-oxobenzothiazolo[3,2-A]quinoline-6-carboxylic acid. Bioorg. Med. Chem. 2008, 16, 3408–3418. [Google Scholar]
- Senthilkumar, P.; Dinakaran, M.; Yogeeswari, P.; Sriram, D.; China, A.; Nagaraja, V. Synthesis and antimycobacterial activities of novel 6-nitroquinolone-3-carboxylic acids. Eur. J. Med. Chem. 2009, 44, 345–358. [Google Scholar]
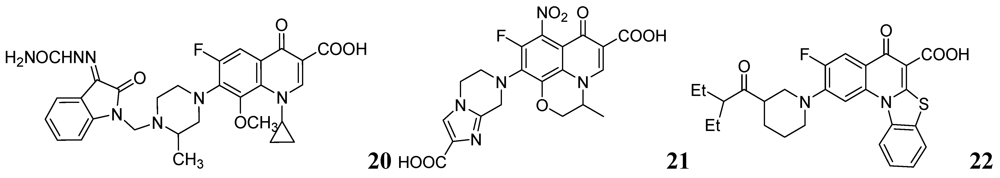
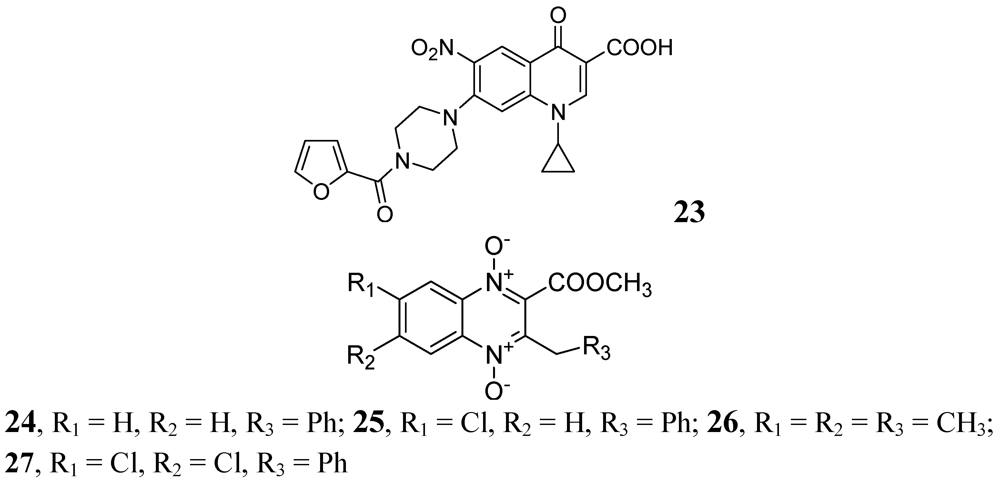
- Jaso, A.; Zarranz, B.; Aldana, I.; Monge, A. Synthesis of new quinoxaline-2-carboxylate 1,4-dioxide derivatives as anti-Mycobacterium tuberculosis agents. J. Med. Chem. 2005, 48, 2019–2025. [Google Scholar] [CrossRef]
- Vicente, E.; Pérez-Silanes, S.; Lima, L.M.; Ancizu, S.; Burguete, A.; Solano, B.; Villar, R.; Aldana, I.; Monge, A. Selective activity against Mycobacterium tuberculosis of new quinoxaline 1,4-di-N-oxides. Bioorg. Med. Chem. 2009, 17, 385–389. [Google Scholar]
- Ancizu, S.; Moreno, E.; Solano, B.; Burguete, A.; Torres, E.; Pérez-Silanes, S.; Aldana, I.; Monge, A. New 3-methylquinoxaline-2-carboxamide 1,4-di-N-oxide derivatives as anti-Mycobacterium tuberculosis agents. Bioorg. Med. Chem. 2010, 18, 2713–2719. [Google Scholar]
- Carta, A.; Palomba, M.; Paglietti, G.; Molicotti, P.; Paglietti, B.; Cannas, S.; Zanetti, S. [1,2,3]Triazolo[4,5-h]quinolones. A new class of potent antitubercular agents against multidrug resistantnt Mycobacterium tuberculosis strains. Bioorg. Med. Chem. Lett. 2007, 17, 4791–4794. [Google Scholar] [CrossRef]
- Upadhayaya, R.S.; Shinde, P.; Kadam, S.A.; Bawane, A.N.; Sayyed, A.Y.; Kardile, R.A.; Gitay, P.N.; Lahore, S.V.; Dixit, S.S.; Földesi, A.; et al. Synthesis and antimycobacterial activity of prodrugs of indeno[2,1-c]quinoline derivatives. Eur. J. Med. Chem. 2011, 46, 1306–1324. [Google Scholar]
- Lilienkampf, A.; Mao, J.; Wan, B.; Wang, Y.; Franzblau, S.G.; Kozikowski, A. Structure-activity relationships for a series of quinoline-based compounds active against replicating and nonreplicating Mycobacterium tuberculosis. J. Med. Chem. 2009, 52, 2109–2118. [Google Scholar]
- Khoje, A.D.; Kulendrn, A.; Charnock, C.; Wan, B.; Franzblau, S.; Gundersen, L.L. Synthesis of non-purine analogs of 6-aryl-9-benzylpurines, and their antimycobacterial activities. Compounds modified in the imidazole ring. Bioorg. Med. Chem. 2010, 18, 7274–7282. [Google Scholar]
- Trivedi, A.R.; Bhuva, V.R.; Dholariya, B.P.; Dodiya, D.K.; Kataria, V.B.; Shah, V.H. Novel dihydropyrimidines as a potential new class of antitubercular agents. Bioorg. Med. Chem. Lett. 2010, 20, 6100–6102. [Google Scholar]
- Shiradkar, M.R.; Murahari, K.K.; Gangadasu, H.R.; Suresh, T.; Kalyan, C.A.; Panchal, D.; Kaur, R.; Burange, P.; Ghogare, J.; Mokale, V.; et al. Synthesis of new S-derivatives of clubbed triazolyl thiazole as anti-Mycobacterium tuberculosis agents. Bioorg. Med. Chem. 2007, 15, 3997–4008. [Google Scholar]
- Velaparthi, S.; Brunsteiner, R.; Petukhov, P.A. 5-tert-Butyl-N-pyrazol-4-yl-4,5,6,7-tetrahydrobenzo[d]isoxazole-3-carboxamide derivatives as novel potent inhibitors of Mycobacterium tuberculosis pantothenate synthetase: Initiating a quest for new antitubercular drugs. J. Med. Chem. 2008, 51, 1999–2002. [Google Scholar] [CrossRef]
- Walczak, K.; Gondela, A.; Suwinski, J. Synthesis and anti-tuberculosis activity of N-Aryl-C-nitroazoles. Eur. J. Med. Chem. 2004, 39, 849–853. [Google Scholar] [CrossRef]
- Huang, Q.; Mao, J.; Wan, B.; Wang, Y.; Brun, R.; Franzblau, S.G.; Kozikowski, A.P. Searching for new cures for tuberculosis: design, synthesis, and biological evaluation of 2-methylbenzothiazoles. J. Med. Chem. 2009, 52, 6757–6767. [Google Scholar] [CrossRef]
- Palmer, B.D.; Thompson, A.M.; Sutherland, H.S.; Blaser, A.; Kmentova, I.; Franzblau, S.G.; Wan, B.; Wang, Y.; Ma, Z.; Denny, W.A. Synthesis and structure-activity studies of biphenyl analogues of the tuberculosis drug (6S)-2-nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824). J. Med. Chem. 2010, 53, 282–294. [Google Scholar]
- Hearn, M.J.; Cynamon, M.H.; Chen, M.F.; Coppins, R.; Davis, J.; Joo-On Kang, H.; Noble, A.; Tu-Sekine, B.; Terrot, M.S.; Trombino, D.; et al. Preparation and antitubercular activities in vitro and in vivo of novel schiff bases of isoniazid. Eur. J. Med. Chem. 2009, 44, 4169–4178. [Google Scholar]
- Lourenço, M.C.; de Lima Ferreira, M.; de Souza, M.V.N.; Peralta, M.A.; Vasconcelos, T.R.A.; Henriques, M.G.M.O. Synthesis and anti-mycobacterial activity of (E)-N'-(monosubstituted-benzylidene) isonicotinohydrazide derivatives. Eur. J. Med. Chem. 2008, 43, 1344–1347. [Google Scholar]
- Miller, M.J.; Walz, A.J.; Zhu, H.; Wu, C.; Moraski, G.; Mcollmann, U.; Tristani, E.M.; Crumbliss, A.L.; Ferdig, M.T.; Checkley, L.; et al. Design, synthesis, and study of a mycobactin-artemisinin conjugat that has selective and potent activity against tuberculosis and malaria. J. Am. Chem. Soc. 2011, 133, 2076–2079. [Google Scholar]
- Falzari, K.; Zhu, Z.; Pan, D.; Liu, H.; Hongmanee, P.; Franzblau, S.G. In vitro and in vivo activities of macrolide derivatives against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2005, 49, 1447–1454. [Google Scholar] [CrossRef]
- Kim, P.; Zhang, Y.M.; Shenoy, G.; Nguyen, Q.A.; Boshoff, H.I.; Manjunatha, U.H.; Goodwin, M.B.; Lonsdale, J.; Price, A.C.; Miller, D.J.; et al. Structure-activity relationships at the 5-position of thiolactomycin: An intact (5R)-isoprene unit is required for activity against the condensing enzymes from Mycobacterium tuberculosis and Escherichia coli. J. Med. Chem. 2006, 49, 159–171. [Google Scholar]
- Slayden, R.A.; Lee, R.E.; Armour, J.W.; Cooper, A.M.; Orme, I.M.; Brennan, P.J.; Besra, G.S. Antimycobacterial action of thiolactomycin: An inhibitor of fatty acid and mycolic acid synthesis. Antimicrob. Agents Chemother. 1996, 40, 2813–2819. [Google Scholar]
- McFadden, J.M.; Medghalchi, S.M.; Thupari, J.N.; Pinn, M.L.; Vadlamudi, A.; Miller, K.I.; Kuhajda, F.P.; Townsend, C.A. Application of a flexible synthesis of (5R)-thiolactomycin to develop new inhibitors of type I fatty acid synthase. J. Med.Chem. 2005, 48, 946–961. [Google Scholar]
- Miyakawa, S.; Suzuki, K.; Noto, T.; Harada, Y.; Okazaki, H. Thiolactomycin, a new antibiotic. IV. Biological properties and chemotherapeutic activity in mice. J. Antibiot. 1982, 35, 411–419. [Google Scholar]
- Working Group on New Drugs. (WGND) (8 December 2011). Available online: http://www.newtbdrugs.org/ (accessed on 4 April 2012).
- TB Alliance, and Tuberculosis Trial Consortium [TBTC]). Available online: http://www.cdc.gov/tb/topic/research/tbtc/introduction.htm/ (accessed on 26 June 2012).
- Hirano, S.; Ichikawa, S.; Matsuda, A. Structure-activity relationship of truncated analogs of caprazamycins as potential anti-tuberculosis agents. Bioorg. Med. Chem. 2008, 16, 5123–5133. [Google Scholar] [CrossRef]
- Bogatcheva, E.; Hanrahan, C.; Nikonenko, B.; de los Santos, G.; Reddy, V.; Chen, C.; Barbosa, F.; Einck, L.; Nacy, C.; Protopopova, M. Identification of SQ609 as a lead compound from a library of dipiperidines. Bioorg. Med. Chem. Lett. 2011, 21, 5353–5357. [Google Scholar]
- Mitscher, L.A.; Baker, W. Tuberculosis: A search for novel therapy starting with natural products. Med. Res. Rev. 1998, 18, 363–374. [Google Scholar] [CrossRef]
- Global Alliance for TB Drug Development. “Background”. Available online: http://www.tballiance.org/new/portfolio/html-portfolio-item.php?id=29/ (accessed on 9 June 2012).
- Balasubramanian, V.; Gaonkar, S.; Solapure, S.; Sambandamurthy, V.; Shandil, R.; Mahesh, K.N.; Sharma, S.; Kaur, P.; Deepthi, R.; Subbulakshmi, V.; et al. (Scheduled Presentation on Monday, 19 September 2011) AZD5847, an Oxazolidinone for the Treatment of Tuberculosis: Pre-clinical Studies. [Presentation no. F1-1364]. American Society for Microbiology, 1752 N Street NW Washington, DC. USA. Available online: http://www.abstractsonline.com/plan/ViewAbstract.aspx?mID=2789&sKey=5f3fa01a-9c86-4ebd-8cab-50756d6faa6f&cKey=c393491b-4fdf-42a8-8857-3756dce2a517&mKey={0C918954-D607-46A7-8073-44F4B537A439}/ (accessed on 4 April 2012).
- Alffenaar, J.W.C.; van der Laan, T.; Simons, S.; van der Werf, T.S.; van de Kasteele, P.J.; de Neeling, H.; van Soolingen, D. Susceptibility of Clinical Mycobacterium tuberculosis isolates to a potentially less toxic derivate of linezolid, PNU-100480. Antimicrob. Agents Chemother. 2011, 55, 1287–1289. [Google Scholar]
- Sinha, R.K.; Arora, S.K.; Sinha, N.; Modak, V.M. In vivo activity of LL4858 against Mycobacterium tuberculosis [abstract F-1116]. In Program and Abstracts of the 44th Interscience Conference on Antimicrobial Agents and Chemotherapy (Washington, DC); American Society for Microbiology: Washington, DC, USA, 2004; p. 212. [Google Scholar]
- Lee, R.; Protopopova, M.; Crooks, E.; Slayden, R.A.; Terrot, M.; Barry, C.E. Combinatorial lead optimization of [1,2]-diamines based on ethambutol as potential antituberculosis preclinical candidates. J. Comb. Chem. 2003, 5, 172–187. [Google Scholar] [CrossRef]
- Jia, L.; Tomaszewski, J.E.; Hanrahan, C.; Coward, L.; Noker, P.; Gorman, G.; Nikonenko, B.; Protopopova, M. Pharmacodynamics and pharmacokinetics of SQ109, a new diamine-based antitubercular drug. Br. J. Pharmacol. 2005, 144, 80–87. [Google Scholar]
- Stover, C.K.; Warrener, P.; van Devanter, D.R.; Sherman, D.R.; Arain, T.M.; Langhorne, M.H.; Anderson, S.W.; Towell, J.A.; Yuan, Y.; McMurray, D.N.; et al. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 2000, 405, 962–966. [Google Scholar]
- Nuermberger, E.; Tyagi, S.; Williams, K.N.; Rosenthal, I.; Bishai, W.R.; Grosset, J.H. Rifapentine, moxifloxacin, or dna vaccine improves treatment of latent tuberculosis in a mouse model. Am. J. Respir. Crit. Care Med. 2005, 172, 1452–1456. [Google Scholar] [CrossRef]
- Lenaerts, A.J.; Gruppo, V.; Marietta, K.S.; Johnson, C.M.; Driscoll, D.K.; Tompkins, N.M.; Rose, J.D.; Reynolds, R.C.; Orme, I.M. Preclinical testing of the nitroimidazopyran PA-824 for activity against Mycobacterium tuberculosis in a series of in vitro and in vivo models. Antimicrob. Agents Chemother. 2005, 49, 2294–2301. [Google Scholar]
- Tyagi, S.; Nuermberger, E.; Yoshimatsu, T.; Williams, K.; Rosenthal, I.; Bishai, W.; Grosset, J. Bactericidal activity of the nitroimidazopyran PA-824 in the murine model of tuberculosis. Antimicrob. Agents Chemother. 2005, 49, 2289–2293. [Google Scholar]
- Matsumoto, M.; Hshizume, H.; Tomishige, T.; Kawasaki, M. In vitro and in vivo efficacy of novel antituberculous candidate OPC-67683 [abstract F-1462]. In Program and abstracts of the 45th Interscience Conference on Antimicrobial Agents and Chemotherapy (Washington, DC); American Society for Microbiology: Washington, DC, USA, 2005. [Google Scholar]
- Kawasaki, M.; Yamamoto, K.; Matusmoto, M. Mechanism of action of OPC-67683 against Mycobacterium tuberculosis [abstract F-1463]. In Program and abstracts of the 45th Interscience Conference on Antimicrobial Agents and Chemotherapy (Washington, DC); American Society for Microbiology: Washington, DC, USA, 2005. [Google Scholar]
- Andries, K.; Verhasselt, P.; Guillemont, J.; Göhlmann, H.W.H.; Neefs, J.M.; Winkler, H.; Gestel, J.V.; Timmerman, P.; Zhu, M.; Lee, E.; et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 2005, 307, 223–227. [Google Scholar]
- Huitric, E.; Verhasselt, P.; Andries, K.; Hoffner, S.E. In vitro antimycobacterial spectrum of a diarylquinoline atp-synthase inhibitor. Antimicrob. Agents Chemother. 2007, 51, 4202–4204. [Google Scholar] [CrossRef]
- Alcalá, L.; Ruiz-Serrano, M.J.; Turégano, C.P.F.; de Viedma, D.G.; Díaz-Infantes, M.; Marín-Arriaza, M.; Bouza, E. In vitro activities of linezolid against clinical isolates of Mycobacterium tuberculosis that are susceptible or resistant to first-line antituberculous drugs. Antimicrob. Agents Chemother. 2003, 47, 416–417. [Google Scholar]
- Migliori, G.B.; Eker, B.; Richardson, M.D.; Sotgiu, G.; Zellweger, J.P.; Skrahina, A.; Ortmann, J.; Girardi, E.; Hosffmann, H.; Besozzi, G.; Bevilacqua, N.; et al. A retrospective TBNET assessment of linezolid, safety, tolerability and efficacy in multidrug resistant tuberculosis. Eur. Respir. J. 2009, 34, 387–393. [Google Scholar]
- Gerson, S.L.; Kaplan, S.L.; Bruss, J.B.; Le, V.; Arellano, F.M.; Hafkin, B.; Kuter, D.J. Hematologic effects of linezolid: Summary of clinical experience. Antimicrob. Agents Chemother. 2002, 46, 2723–2726. [Google Scholar]
- Heifets, L.; Sanchez, T.; Vanderkolk, J.; Pham, V. Development of rifapentine susceptibility tests for Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1999, 43, 25–28. [Google Scholar]
- Williams, D.L.; Spring, L.; Collins, L.; Miller, L.P.; Heifets, L.B.; Gangadharam, P.R.J.; Gillis, T.P. Contribution of rpoB mutations to development of rifamycin cross-resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1998, 42, 1853–1857. [Google Scholar]
- Gosling, R.D.; Uiso, L.O.; Sam, N.E.; Bongard, E.; Kanduma, E.G.; Nyindo, M.; Morris, R.W.; Gillespie, S.H. The bactericidal activity of moxifloxacin in patients with pulmonary tuberculosis. Am. J. Respir. Crit. Care Med. 2003, 168, 1342–1345. [Google Scholar] [CrossRef]
- Pletz, M.W.R.; de Roux, A.; Roth, A.; Neumann, K.H.; Mauch, H.; Lode, H. Early bactericidal activity of moxifloxacin in treatment of pulmonary tuberculosis: A prospective, randomized study. Antimicrob. Agents Chemother. 2004, 48, 780–782. [Google Scholar]
- Tortoli, E.; Dionisio, D.; Fabbri, C. Evaluation of moxifloxacin activity in vitro against Mycobacterium tuberculosis, including resistant and multidrug-resistant strains. J. Chemother. 2004, 16, 334–336. [Google Scholar]
- Bayer. Avelox® (moxifloxacin hydrochloride) Product Safety. Available online: http://www.avelox.com/en/physician/product_information/avelox_safety/index.php (accessed on 4 April 2012).
- Nuermberger, E.L.; Yoshimatsu, T.; Tyagi, S.; Williams, K.; Rosenthal, I.; O’brien, R.J.; Vernon, A.A.; Chaisson, R.E.; Bishai, W.R.; Grosset, J.H. Moxifloxacin-containing regimens of reduced duration produce a stable cure in murine tuberculosis. Am. J. Respir. Crit. Care Med. 2004, 170, 1131–1134. [Google Scholar] [CrossRef]
- Paramasivan, C.N.; Sulochana, S.; Kubendiran, G.; Venkatesan, P.; Mitchison, D.A. Bactericidal action of gatifloxacin, rifampin, and isoniazid on logarithmic- and stationary-phase cultures of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2005, 49, 627–631. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shakya, N.; Garg, G.; Agrawal, B.; Kumar, R. Chemotherapeutic Interventions Against Tuberculosis. Pharmaceuticals 2012, 5, 690-718. https://doi.org/10.3390/ph5070690
Shakya N, Garg G, Agrawal B, Kumar R. Chemotherapeutic Interventions Against Tuberculosis. Pharmaceuticals. 2012; 5(7):690-718. https://doi.org/10.3390/ph5070690
Chicago/Turabian StyleShakya, Neeraj, Gaurav Garg, Babita Agrawal, and Rakesh Kumar. 2012. "Chemotherapeutic Interventions Against Tuberculosis" Pharmaceuticals 5, no. 7: 690-718. https://doi.org/10.3390/ph5070690
APA StyleShakya, N., Garg, G., Agrawal, B., & Kumar, R. (2012). Chemotherapeutic Interventions Against Tuberculosis. Pharmaceuticals, 5(7), 690-718. https://doi.org/10.3390/ph5070690