Pathogenesis and Antifungal Drug Resistance of the Human Fungal Pathogen Candida glabrata

Abstract
: Candida glabrata is a major opportunistic human fungal pathogen causing superficial as well as systemic infections in immunocompromised individuals and several other patient cohorts. C. glabrata represents the second most prevalent cause of candidemia and a better understanding of its virulence and drug resistance mechanisms is thus of high medical relevance. In contrast to the diploid dimorphic pathogen C. albicans, whose ability to undergo filamentation is considered a major virulence trait, C. glabrata has a haploid genome and lacks the ability to switch to filamentous growth. A major impediment for the clinical therapy of C. glabrata infections is its high intrinsic resistance to several antifungal drugs, especially azoles. Further, the development of antifungal resistance, particularly during prolonged and prophylactic therapies is diminishing efficacies of therapeutic interventions. In addition, C. glabrata harbors a large repertoire of adhesins involved in the adherence to host epithelia. Interestingly, genome plasticity, phenotypic switching or the remarkable ability to persist and survive inside host immune cells further contribute to the pathogenicity of C. glabrata. In this comprehensive review, we want to emphasize and discuss the mechanisms underlying virulence and drug resistance of C. glabrata, and discuss its ability to escape from the host immune surveillance or persist inside host cells.1. Introduction
Candida species are currently the fourth-leading cause of hospital-acquired bloodstream infections, reaching a mortality rate of up to ∼35–40% for systemic or disseminated infections [1,2]. Systemic mycoses can occur in patients with severely impaired immune systems (AIDS), people with organ or bone marrow transplants, cancer patients undergoing chemotherapy or in intensive care unit (ICU) patients, as well as both neonates and the elderly. The high mortality observed with systemic candidemia can be explained at least in part by a lack of fast and accurate diagnostic tools and in some cases by inefficient antifungal therapies. Therefore, there is a need for basic as well as clinical research to understand the molecular mechanisms of pathogenicity, to define the pathways and genetic networks driving the transition from commensalism (i.e. colonization) to host dissemination, and to develop novel antifungal drugs and diagnostic tools in order to improve treatment of fungal infections, especially those caused by C. glabrata.
Among all Candida species C albicans is still the most frequently isolated species, followed by C. glabrata accounting for ∼15–20% in Europe and ∼20% in North America of all clinical Candida spp isolates [1,3,4]. When compared to C. albicans, relatively little is known about the molecular mechanisms enabling C. glabrata to become a successful human pathogen. The genome organization indicates a synteny relationship to the well-known model non-pathogenic baker's yeast Saccharomyces cerevisiae. However, although haploid, C. glabrata lacks a sexual cycle and mating has never been observed. Moreover, prominent important virulence factors operating in C. albicans such as the formation of true hyphae, are absent in C. glabrata yet it managed to become a successful human pathogen. In this review, we want to summarize recent progress in the identification and characterization of different virulence factors and drug resistance mechanisms of C. glabrata (Table 1). For space constraints, we will limit this review to C. glabrata, but would like to refer to numerous excellent recent and comprehensive reviews addressing the pathobiology of C. albicans [5-10].
2. Adherence
Adherence to host cells and tissues is considered as a key virulence factor of many human fungal pathogens. Members of the ALS gene family encoding adhesins play a crucial role for interactions of C. albicans with host tissues [31,32]. In C. glabrata, the genome harbors a large group of putative GPI-anchored cell wall proteins [33], many of which are potential covalently-bound adhesins. The epithelial adhesin (EPA) gene family represents the largest group in C. glabrata, comprising at least 23 related genes, most of them located in subtelomeric regions [11,34]. The absolute number of EPA genes varies in different strain backgrounds and clinical isolates. For example, the BG2 strain contains 23, whereas the standard laboratory strain ATCC2001 (CBS138) strain carries only 17 EPA genes, lacking, for example, EPA4 and EPA5 [34,35]. The major epithelial adhesins, Epa1, Epa6 and Epa7, display different binding specificities concerning decoration of host cell ligands containing a terminal galactose residue [36]. Morover, the C. glabrata genome harbors a variety of additional putative adhesin families (Awp, Pwp), covalently surface-bound enzymatically active (Gas) or protein families of unknown function (Cwp, Pir). The presence of adhesin-like proteins (Awp1-4) in the cell surface strongly depends on the strain background and the growth phase [33,37].
In vitro, C. glabrata adherence to epithelial tissue is largely mediated by the major lectin Epa1, whereas other EPA genes are expressed at rather low level [11,12]. The adhesins EPA6 and EPA7 have been implicated in C. glabrata biofilm formation [13]. Epa6 seems to be a major player in biofilm formation, since it is highly induced during this phenomenon, and its absence reduces biofilms in vitro. Biofilms often typically display a higher resistance to several antifungal drugs. This is of special relevance, since C. glabrata naturally displays an inherent high azole resistance. Furthermore, EPA6 expression is also induced by exposure to sorbic acid and parabens, which are used as preservatives in food and health products. The transcription factors Flo8 and Mss11 control weak organic acid stress induction of EPA6, leading to an increased adherence to vaginal epithelium due to the low pH in this environment [14].
The subtelomeric localization of most EPA genes places their expression under the control of the Sir-dependent chromatin silencing machinery [12]. In C. glabrata, this machinery depends on orthologoues of the S. cerevisiae silencing machinery, including Rap1, Sir2, Sir3, Sir4 and Rif1 [11,38]. For instance, expression of EPA1, EPA6 and EPA7 is induced in cells lacking the silencing genes SIR3 and RIF1. In a murine model of disseminated candidiasis, C. glabrata silencing mutants are hyper-adherent to epithelial cells and more efficient in colonizing the kidney [12].
The transcriptional regulation of EPA gene expression is also controlled by host environmental signals such as limited nicotinic acid levels as present in the human urinary tract [15]. Interestingly, C. glabrata is an auxotroph for nicotinic acid (NA) and thus often causes urinary tract infections, since the low NA levels are sufficient to support C. glabrata growth. At the same time, the lack of NA, a precursor of NAD+ which is also a cofactor for the histone modifier Sir2, decreases Sir2 activity, resulting in the derepression of EPA6. In consequence, this leads to an increased adherence of C. glabrata to host tissues. Consistently, a triple epa1Δ epa6Δ epa7Δ mutant fails to colonize the bladder [15]. Notably, the NA auxotrophy of C. glabrata may actually reflect its close adaptation to the human host or even indicate adaptive co-evolution with the host, and enables C. glabrata to efficiently colonize a specific host niche.
Moreover, C. glabrata lacks most of the Biosynthesis of Nicotinic Acid (BNA) genes, and therefore must aquire any and all NAD+ precursors from its host environment. The NA uptake requires the membrane transporters Tna1, Tnr1 and Tnr2 [39]. In addition to NA, C. glabrata can utilize several different NAD+ precursors, including nicotinamide and nicotinamide riboside. During infections, nicotinamide riboside appears as the prime source of NAD+ [40]. Expression of the dedicated transporters in response to NA limitation is regulated by another histone modifier, the histone deacetylase Hst1. Interestingly, a lack of transporters again results in enhanced EPA6 expression, implying a function in growth and adhesion during the infection process [39].
The C. glabrata YPS family comprising 11 cell wall genes is involved in interaction with host cells. The corresponding proteins share significant similarities with the S. cerevisiae yapsins (YPS). These GPI-anchored aspartyl proteases comprise five distinct proteins implicated in cell wall remodeling [41]. In C. albicans, secreted aspartyl proteases have been intimately associated with virulence [42]. The C. glabrata genes YPS3 - YPS11 are located in a specific gene cluster; expression of six cluster genes is induced after internalization by macrophages. Furthermore, YPS1 and YPS7 are implicated in cell wall integrity and cellular survival in stationary phase. Strains lacking the yapsins YPS1 and YPS7 or those lacking all eleven YPS genes, show attenuated virulence, implicating the YPS gene cluster in infectious processes [16]. Interestingly, the major adhesin Epa1 is stabilized in CgypsΔ mutants, implying a direct or indirect role of the Yps proteases in Epa1 processing and/or proteolytic turnover. Consequently, yps gene deletion strains display increased adherence to epithelial cells [16].
3. Hypervirulence Factor ACE2
Exploiting a library of insertional signature-tagged mutants, the C. glabrata Ace2 transcription factor of the RAM (Regulation of Ace2 transcription factor and polarized Morphogenesis) network [43], has been identified as a hypervirulence factor [17]. The orthologous baker's yeast transcription factor Ace2 localizes only to the daughter cell nucleus, activates expression of early G1-phase genes, and mediates the separation of mother and daughter cells. Ace2 controls expression of a set of distinct cell wall target genes, including the chitinase CTS1, the putative glucanase SCW11 and DSE genes implicated in the actual cell separation process [44,45]. Deletion of ACE2 causes cell separation defects, resulting in pseudohyphal growth, clumping cells and detectable agar invasion [46].
Interestingly, the lack of the C. glabrata transcription factor Ace2 also causes cell separation defects, leading to the formation of large cell aggregates [17]. Strikingly, ace2Δ cells are hypervirulent in a neutropenic mouse infection model, causing 100% lethality after four days. The hypervirulent phenotype may, at least in part, be caused by drastically elevated proinflammatory cytokines due to abnormal exposure of fungal surface components in ace2Δ cells [17]. A proteomic analysis hints some 123 protein changes in the ace2Δ mutant. Consistent with expression data, morphogenesis and cell wall remodeling genes are down-regulated [18]. Notably, abundant cytoplasmic proteins are also detectable in the ace2Δ secretome, which are otherwise not found in the wild type supernatants [19]. These cytoplasmic proteins may increase immunogenicity, triggering an exacerbated immune response [18]. Noteworthy, the lack of ACE2 changes the murine immune response only to C. glabrata but not to C. albicans mutants, which are rather attenuated in virulence [20]. In addition, the hypervirulent effect of ace2Δ cells is only observed in immunosuppressed mice [20]. Strikingly, while being highly pathogenic to humans, C. glabrata is very efficiently cleared when injected into immunocompetent mice. However, a number of inconsistencies exist in the literature concerning the use of mouse models for studying C. glabrata virulence. Since killing of wild type mice by systemic C. glabrata infections is at least a controversial issue, mice may better serve as in vivo systems to monitor growth, dissemination and colonization of organs and tissues [47].
4. Model Systems to Study Virulence of C. glabrata
Infection of mice with C. glabrata does not lead to the development of systemic candidiasis and subsequent death. Therefore, immunosuppressed mice are obtained by using 200 mg/kg cyclophosphamide administered three days before infecting with C. glabrata. This model system yields mortality rates of up to 100% after five days of infection without any evidence for necrosis or inflammation. However, high fungal burdens such as 2 × 108 C. glabrata cells injected into the lateral tail vein are required, while lower burdens increase mouse survival [48]. This mouse model suggests the hypervirulence of C. glabrata ace2Δ cells [17,20], as well as the increased virulence of Pdr1 gain-of-function mutants [24]. As indicated above, survival of infected mice is not the best readout for C. glabrata virulence, since severe immunosuppression is required to obtain fungal killing. Nevertheless, immunocompetent mice have been successfully used to study the virulence or fitness phenotypes in vivo [16]. For example, for YPS deletion strains, the level of tissue colonization was used as a measure of fungal virulence [16]. Hence, quantifying organ colonization of C. glabrata strains in normal mice is a useful assay for fitness in vivo and thus directly relates to virulence [47].
In addition, C. glabrata virulence has been also investigated in a Drosophila melanogaster infection model. Whereas wild type flies survive the injection of 7,500 C. glabrata cells, MyD88 mutant flies show strongly increased mortality [49]. Hence, this fly model could be used in the future to screen a larger number of C. glabrata strains for virulence phenotypes.
Another invertebrate model system used to study virulence of C. albicans and Cryptococcus neoformans is the Greater Wax Moth Galleria mellonella [50,51]. For survival assays, C. albicans cells are injected into the haemocoel of G. mellonella larvae. Notably, good correlations between virulence phenotypes in the invertebrate model and in murine models of systemic candidiasis exist [50,52]. Therefore, this convenient and inexpensive model system may be suitable to study virulence of C. glabrata on a large scale.
A Caenorhabditis elegans infection model has also been used to screen a library of 83 C. albicans transcription factor mutants for alterations in virulence. Five mutants were identified, two of which were previously shown to have defects in virulence in a murine model of candidiasis [53]. This model system may be suitable for high-throughput screening of putative antifungal compounds [54]. Whether this model system is also suitable to study C. glabrata virulence remains unclear.
5. Pigmentation as Virulence Factor
Many pathogenic fungi can produce pigments some of which are implicated in virulence [55,56,57]. Such pigments have diverse biological functions, including antioxidative effects [58,59], which counteract reactive oxygen species (ROS) produced by the host immune system to kill and eliminate invading microbial pathogens [60].
While C. glabrata was hitherto believed to be an unpigmented yeast species, recent work demonstrates the production of indole-derived pigments [61]. Pigment production requires the presence of tryptophan as the sole nitrogen source in the medium. Furthermore, the chemical composition was similar to the pigments produced by the lipophilic yeast Malassezia furfur [61], which are distinct from pigments produced by other pathogenic fungi via the melanin synthesis pathway [57]. Interestingly, pigment production by C. glabrata proceeds via the Ehrlich pathway [21], which mediates degradation of aromatic amino acids in S. cerevisiae [62]. Deletion of ARO8 encoding an aromatic aminotransferase catalyzing a transamination reaction in the Ehrlich pathway, reduces pigmentation. In addition, aro8Δ mutants show increased susceptibility to H2O2 treatment. A similar phenotype was observed in wild type cells growing in non-pigment-inducing media. Furthermore, pigments may protect fungal cells against neutrophil attack, since a lack of pigments leads to killing hypersensitivity [21], suggesting a possible role for pigments in the survival of C. glabrata within the host. Similarly, pigmentation may also protect filamentous fungi like Aspergillus fumigatus from host killing [56].
6. Genome Plasticity and Tandem Repeats
Like the random chromosome alterations frequently observed in C. albicans, the C. glabrata genome of clinical isolates, although as yet not much appreciated, also appears to undergo chromosomal alterations, including chromosome loss, translocations and aneuploidy. The analysis of 40 clinical isolates shows drastic differences in their genome organization, suggesting a highly dynamic genome [63]. Chromosomal rearrangements, translocations, chromosome fusions and inter-chromosomal duplications lead to distinct karyotypes. Strikingly, C. glabrata is even able to perform de novo chromosome generation [63]. The authors speculate that the lack of a sexual cycle may cause tolerance to frequent chromosomal rearrangements, providing an explanation for the remarkable clonal population diversity. Interestingly, chromosomal rearrangements often occur at the same loci, as several isolates showed duplicated segments carrying genes associated with drug resistance (CDR1, CDR2) or survival in macrophages (YPS gene cluster [16]) [63]. This genomic plasticity of C. glabrata may therefore serve as a compensatory mechanism to allow for rapid adaptation to changing host conditions / environments and maybe compensate the absence of a functional sexual cycle.
These genome dynamics is also reflected in the high number of minisatellite sequences found in C. glabrata genomes [35]. Tandem repeats and selective domain amplifications are commonly found in both pathogenic and non-pathogenic yeast species, often occurring in adhesin or flocculation genes [35,64,65]. Notably, the majority of minisatellites are not conserved between baker's yeast and C. glabrata, although genes carrying minisatellites appear conserved. Remarkably, C. glabrata also harbors unusual types of minisatellites, so-called compound minisatellites with intermingled repeats and megasatellites containing long repeated motifs [35]. The evolutionary mechanism and origin of these minisatellites remains unclear, but a large number of EPA adhesion genes carry such repeats. A plausible hypothesis is that unusual minisatellites relate to high-frequency chromosomal rearrangements. This would further diversify expression as well as function of adhesion genes, which are considered important pathogenicity genes.
7. Phenotypic Switching of C. glabrata
Two distinct morphologies, core and irregular wrinkled, which result from high-frequency phenotypic switching mechanisms, were recently discovered in C. glabrata. The core system is composed of four phenotypes identified on the basis of their colony color on plates containing CuSO4. They are called white (Wh), light brown (LB), dark brown (DB) and very dark brown (vDB) [66,67]. In addition, cells of each of the core phenotypes can switch to the irregular wrinkled (IWr) phenotype and reverse back to core phenotypes. Most clinical isolates may undergo phenotypic switching, with DB being the most frequently observed species [67]. In addition, there are differences in the frequency of switching phenotypes depending on the sites of host colonization. For instance, vaginal isolates prefer DB, whereas genetically identical cells from the oral cavity were predominantly displaying the Wh phenotype [68]. These results strongly suggest roles for phenotypic switching in the adaptation to different host niches. Indeed, after injecting a mixture of DB and Wh or DB and IWr in a 50:50 ratio into a mouse model of systemic infection, mainly DB species appear in the spleen, liver and kidney after plating organ homogenates [69]. The outcome is similar for all organs, suggesting an advantage of DB cells over other switching phenotypes within the host. Importantly, the observed advantage of DB cells over Wh cells is not caused by increases in switching towards the DB phenotype, but rather arises from preferred organ colonization, as demonstrated by GFP-tagging of either the DB or the Wh cells in the injection mixture [69]. Different host niches may favor other switching phenotypes than DB. Although the molecular mechanisms underlying phenotypic switching in C. glabrata are enigmatic, switching might be important in C. glabrata infections and host colonization.
8. Resistance to Oxidative Stress and Survival Inside the Phagolysosome
The first encounters of C. glabrata with the host innate immune cells include phagocytic cells [70]. Remarkably, many pathogens have developed different strategies to escape from the phagosome following internalization. For example, C. albicans destroys macrophages by switching to the hyphal growth while C. neoformans can lyse macrophages or escape via phagosomal extrusion [71,72]. To date, little is known how C. glabrata responds to host cell phagocytosis and how it can survive and persist inside the phagolysosome. The lack of morphogenesis does not allow physical killing of host cells by C. glabrata. Moreover, phagolysosome maturation brings a hostile environment for pathogens, including hydrolytic enzymes as well as a lower pH due to acidification [73,74]. In addition, pathogen adhesion triggers extracellular host-derived ROS to kill pathogens [60]. Therefore, antioxidant activities seem plausible virulence factors in different pathogenic fungi [75-77]. For example, A. fumigatus lacking catalases normally degrading H2O2, shows attenuated virulence in a rat model of invasive aspergillosis [78]. For C. glabrata, a lack of CTA1, the gene encoding the only catalase, results in hypersensitivity to H2O2. Interestingly, C. glabrata strains show higher peroxide resistance than S. cerevisiae or C. albicans, suggesting a high intrinsic resistance to oxidative stress [23]. Indeed, Cta1 expression is induced after phagocytosis and both the number of peroxisomes and Cta1 localization to peroxisomes is enhanced [22]. Notably, peroxisome numbers decrease after prolonged residence of C. glabrata in the phagolysosome, perhaps via autophagy, to help C. glabrata surviving in the nutrient-limited environment. Deletion of ATG11 or ATG17 results in defects in the reduction of peroxisomes and reduced survival upon phagocytosis. Thus, in addition to surviving ROS attacks, recycling of potential internal nutrients or the use of host-derived nutrients may be beneficial for the persistence and survival of C. glabrata in the host after phagocytosis.
9. Mechanisms of Antifungal Resistance in C. glabrata & Modulation of Drug Susceptibility
For space constraints, we shall limit the discussion on antifungal drug resistance, but would like to refer to numerous excellent recent reviews on the use of antifungal drugs and the mechanisms of antifungal resistance in fungal pathogens [7,79-81]. It has been widely recognized that C. glabrata displays inherently high resistance to several antifungal drugs, limiting the efficacy of some antifungal drugs used in clinical therapy [7,79-81].
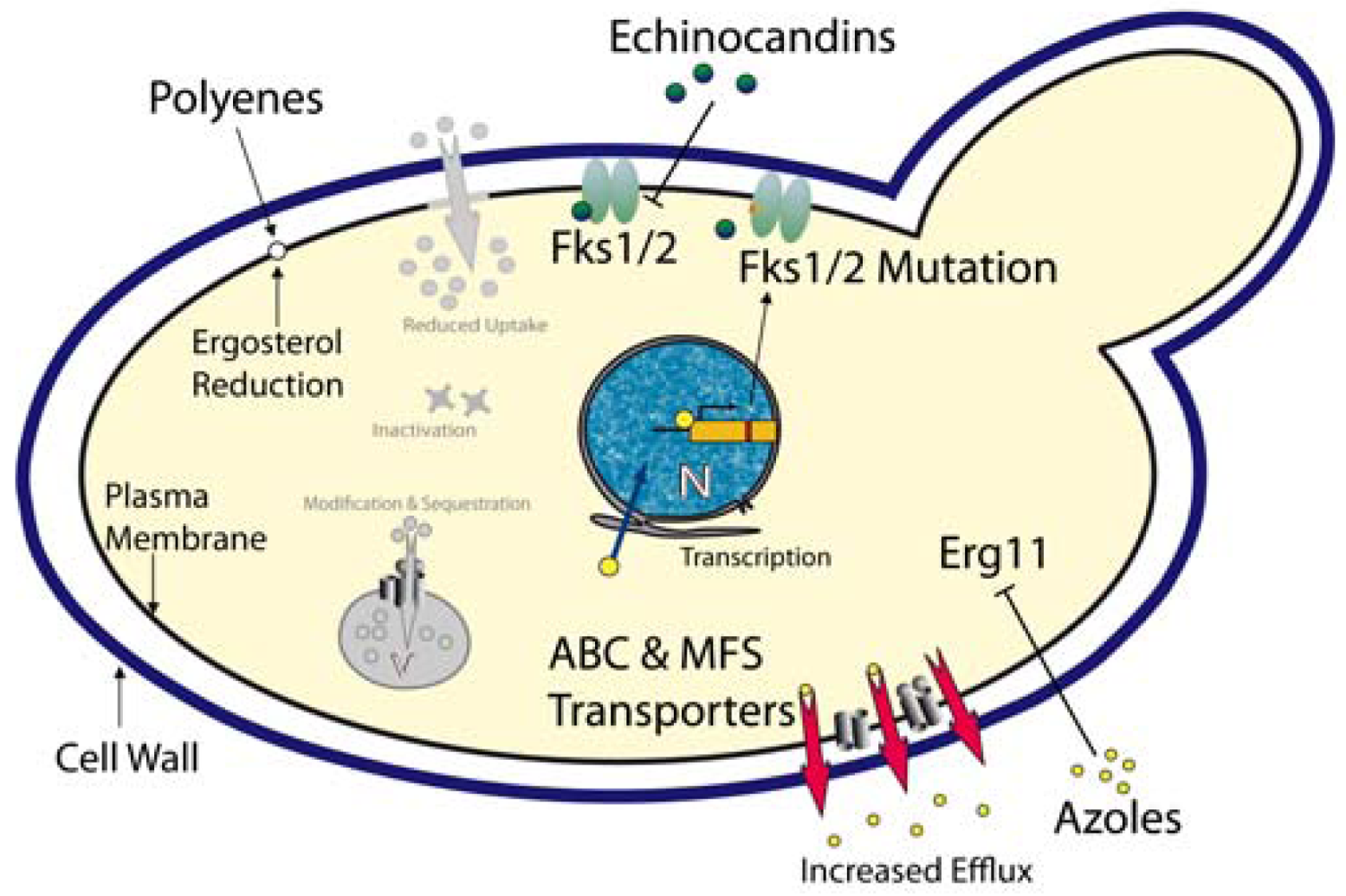
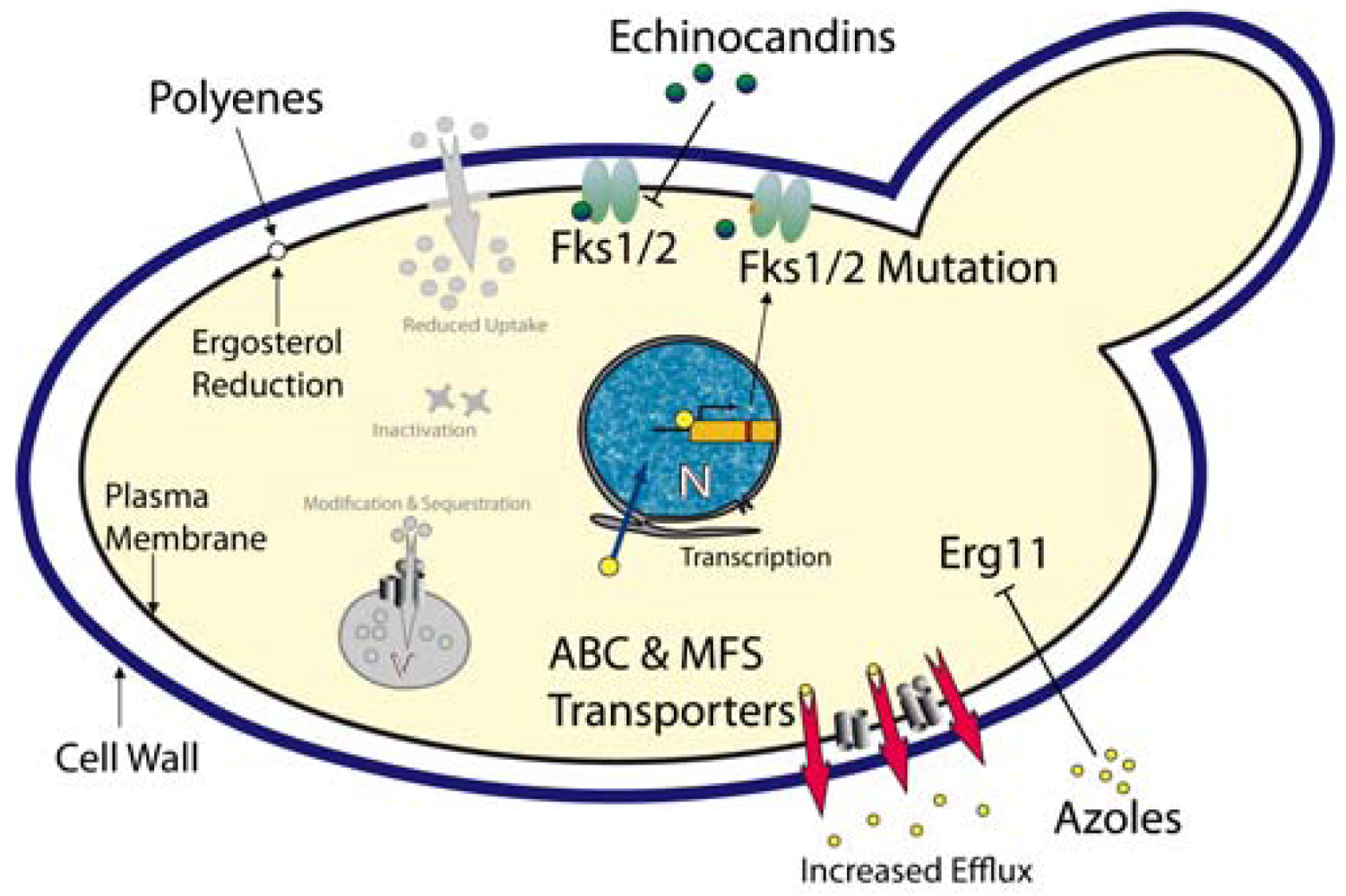
Azoles – These compounds represent most widely used class of antifungal drugs and they have been used to treat fungal infections for several decades. The cellular target of the azoles is the lanosterol 14-α-demethylase, encoded by the ERG11 gene [82]. Inhibition of this enzyme efficiently blocks ergosterol biosynthesis, an essential fungal membrane component (Figure 1). When compared with other Candida spp, C. glabrata shows an inherently reduced azole susceptibility. In addition, prolonged and prophylactic treatment with azoles often results in the emergence of clinically resistant C. glabrata strains. For C. albicans, one azole resistance mechanism is the overexpression or mutation of the azole target Erg11 [83,84]. However, in C. glabrata azole-resistant clinical isolates, neither overexpression nor ERG11 mutations seem to mediate resistance [85-87]. However, the transcriptional induction and massive up-regulation of drug efflux pumps, especially members of the ABC (ATP-binding cassette) transporter family and the major facilitator family efficiently prevent intracellular azole accumulation [88-91]. Three ABC transporters are involved in C. glabrata azole resistance: Cdr1, Cdr2 (Pdh1) and Snq2 [25,92,93].
Aus1, another fungal ABC transporter implicated in sterol uptake in yeast [94], may be somehow involved in a low intrinsic susceptibility of C. glabrata to azoles but the mechanisms behind remain unclear. The growth-inhibition by fluconazole is suppressed by the addition of serum to the medium, perhaps because C. glabrata can take up cholesterol from the serum under these conditions [95]. In addition, this effect was dependent on the presence of Aus1, implying that Aus1 might also be involved in the uptake of sterols in C. glabrata [96]. The authors propose that Aus1-mediated uptake of cholesterol from the medium can rescue the lack of membrane ergosterol as caused by Erg11 inhibition. This might contribute to the low susceptibility of C. glabrata to azoles.
Polyenes - Polyenes are fungicidal antifungals and have been used for more than 50 years. These substances intercalate into ergosterol-containing membranes (i.e. mostly plasma membrane), thereby forming pores which result in leakage of cellular components, collapse of ion and electrical gradients and ultimately lead to cell death (Figure 1). Unfortunately, adverse side effects such as severe nephrotoxicity to the host rather than resistance make a long-term use of this class of antifungals difficult [97]. Resistance or decreased susceptibility to amphotericin B, the most prominent polyene, was reported in clinical isolates of different Candida spp including C. glabrata [98]. A reduction of the ergosterol content in the plasma membrane appears to correlate with reduced susceptibilities to amphotericin B [99].
Echinocandins - The echinocandin antifungals are inhibitors of the Fks1/Fks2 1,3-β-D-glucan synthases, the enzymes responsible for synthesis of 1,3-β-d-glucan, a major and essential cell wall component of all fungi [100]. The approved candin drugs (caspofungin, anidulafungin and micafungin) are non-competitive Fks1/Fks2 inhibitors, disrupting the integrity and structural organization of the cell wall, thereby exerting fungicidal action [101]. As expected, mutations in FKS1 and FKS2 encoding the catalytical subunits of the 1,3-β-d-glucan synthases mediate echinocandin resistance in C. glabrata [27-29] (Figure 1). Some 11 new mutations detected in FKS1 or FKS2 of C. glabrata clinical isolates cause reduced susceptibility to echinocandins. However, reduced enzymatic activities of Fks1/2 mutant variants might affect fitness in the host and therefore promote low frequency of echinocandin resistance [30]. Notably, ectopic overexpression of the Cdr2 ABC transporter causes efflux-mediated tolerance to caspofungin in C. albicans laboratory strains, as well as in clinical isolates [102].
Taken together, a composite multidrug resistance phenotype is often caused by the parallel or consecutive activation of a number of distinct mechanisms operating in all living cells from bacteria to cancer cells [103-106]. Thus, while mechanisms such as reduced drug uptake, intracellular catabolism, target gene mutations, overexpression and gene amplification, signaling and stress response pathways, membrane lipid changes, vacuolar sequestration operate in most infectious microbes, clinical resistance in C. glabrata patient isolates may result mainly from transporter-mediated drug efflux.
Finally, several mechanisms are operating in C. glabrata to modulate antifungal resistance phenotypes. The Zn(2)-Cys(6) transcription factor Pdr1 controls the expression of at least three ABC transporters involved in azole resistance in C. glabrata [25,86]. Pdr1 may directly bind antifungal drugs or other xenobiotics to become transcriptionally active and trigger transcription of drug efflux pumps leading to multidrug resistance [107]. As in the non-pathogenic baker's yeast, gain-of-function mutations in PDR1 result in a constitutively active transcription factor, and these are prevalent in the majority of azole-resistant clinical isolates. In addition, strains harboring constitutively active PDR1 alleles also show increased virulence in mice, even in the absence of azole treatment [24]. These results strongly suggest that Pdr1 may account for most cases of clinical azole resistance in C. glabrata. Notably, similar to yeast, Pdr1 may also mediate azole resistance in response to mitochondrial dysfunction in C. glabrata [108-110].
Interestingly, chromatin modification may account for novel mechanisms mediating drug resistance in certain fungi. For example, in S. cerevisiae, deletion of RPD3 encoding a histone deacetylase also results in enhanced susceptibility to azoles. Furthermore, Rpd3 is required for up-regulation of an ABC transporter in response to cycloheximide [111]. In addition, treatment of C. albicans with histone deacetylase inhibitors also strongly increases the sensitivity of fungal pathogens to different classes of antifungal agents [112,113]. Notably, induction of the ABC transporters CDR1 and CDR2 upon fluconazole treatment is significantly reduced in the presence of the histone deacetylase inhibitor trichostatin A [112]. Hence, histone-modifying enzymes might also be involved in the regulation of drug resistance in C. glabrata and should be considered as potential future drug targets.
10. Conclusions and Perspectives
C. glabrata is a very successful human pathogen, accounting for up to 25% of all clinical Candida infections. Although new insights concerning the molecular mechanisms mediating virulence are surfacing, many open questions concerning host adaptability and pathogenicity remain. Hence, the field needs as much as possible genome-wide and global approaches, whereby researchers can make use of tools such as a deletion collection, an overexpression collection or an epitope-tagged ORFome just to name a few tools that have been revolutionizing research in the non-pathogenic baker's yeast [114-116]. Importantly, we need to have a catalogue of potential virulence genes, similar to attempts initiated for C. albicans or Cryptococcus neoformans [117,118]. However, to exploit such a tool, the community needs to develop appropriate mammalian models of virulence, enabling studies on commensalism, immune evasion, colonization, host dissemination and pathogenic conversion. Furthermore, there is a need for a quantitative understanding of the dynamic interplay of mammalian hosts with fungal pathogens. Deep-sequencing [119,120] as well as single cell imaging will be highly beneficial to understand invasion and dissemination in the host. Comparative functional genomics is taking advantage of the comparison with the non-pathogenic and well-studied relative S. cerevisiae, hoping for new insights. The genome organization show rather limited differences, some of which may suffice to explain the striking differences in virulence. However, the remarkable and nearly endless combinatorial complexity of genetic interactions [121] will make it difficult to come up with meaningful cause-consequence conclusions based on small genomic or genetic differences identified by comparing S. cerevisiae and C. glabrata.
Hence, the future calls for integration of different disciplines, including mathematics and molecular approaches delivering quantitative data. The use of systems biology approaches such as predictive mathematical modeling of quantitative biological data will facilitate a better understanding of the complexity underlying fungal pathogenicity. Furthermore, diagnostic tools exploiting molecular methods will have to improve concerning both speed and reliability, since this will facilitate clinical therapy. Of course, the clinical day-to-day reality is always in need for new and more efficacious antifungal drugs once the persistent difficulties concerning the rapid and accurate diagnosis of fungal species causing diseases have been overcome. In addition to classical small molecule drugs, novel antibody-based approaches may aid both diagnostic and therapeutic approaches, and even vaccines are now entering stage as feasible approaches to cure or combat systemic fungal disease.

{kind=link}
| Gene | Deletion phenotype | References |
|---|---|---|
| EPA gene family | Reduced adherence, organ colonization and biofilm formation | [11,12,13,14,15] |
| SIR3, RIF1 | Increased adherence and kidney colonization | [12] |
| YPS gene family | Reduced organ colonization, increased adherence | [16] |
| ACE2 | Hypervirulence, cell separation defect | [17,18,19,20] |
| ARO8 | Reduced pigmentation, increased susceptibility to oxidative stress | [21] |
| CTA1 | Increased susceptibility to oxidative stress | [22,23] |
| ATG11, ATG17 | Reduced survival upon phagocytosis | [22] |
| PDR1 | Increased azole susceptibility; GOF mutations: increased virulence and organ colonization, azole resistance | [24] |
| CDR1, CDR2, SNQ2 | Reduced azole resistance | [25,26] |
| FKS1, FKS2 | Mutations lead to echinocandin resistance | [27,28,29,30] |
Acknowledgments
We thank all our laboratory members for critical and helpful discussions. Research on fungal pathogens in our group has been supported by grants from the ERA-Net Pathogenomics project FunPath (FWF-AP-I0125-B09), the Christian Doppler Society, and the Austrian FFG (ETB-CanVac Project). MT is a fellow of the Vienna Biocenter PhD Programme.
References
- Pfaller, M.A.; Diekema, D.J. Epidemiology of invasive candidiasis: a persistent public health problem. Clin. Microbiol. Rev. 2007, 20, 133–163. [Google Scholar]
- Nace, H.L.; Horn, D.; Neofytos, D. Epidemiology and outcome of multiple-species candidemia at a tertiary care center between 2004 and 2007. Diagn. Microbiol. Infect. Dis. 2009, 64, 289–294. [Google Scholar]
- Pfaller, M.A.; Boyken, L.; Hollis, R.J.; Kroeger, J.; Messer, S.A.; Tendolkar, S.; Diekema, D.J. In vitro susceptibility of invasive isolates of Candida spp. to anidulafungin, caspofungin, and micafungin: six years of global surveillance. J. Clin. Microbiol. 2008, 46, 150–156. [Google Scholar]
- Tortorano, A.M.; Kibbler, C.; Peman, J.; Bernhardt, H.; Klingspor, L.; Grillot, R. Candidaemia in Europe: epidemiology and resistance. Int. J. Antimicrob. Agents 2006, 27, 359–366. [Google Scholar]
- Wilson, D.; Thewes, S.; Zakikhany, K.; Fradin, C.; Albrecht, A.; Almeida, R.; Brunke, S.; Grosse, K.; Martin, R.; Mayer, F.; Leonhardt, I.; Schild, L.; Seider, K.; Skibbe, M.; Slesiona, S.; Waechtler, B.; Jacobsen, I.; Hube, B. Identifying infection-associated genes of Candida albicans in the postgenomic era. FEMS Yeast Res. 2009, 9, 688–700. [Google Scholar]
- Brown, A.J.; Odds, F.C.; Gow, N.A. Infection-related gene expression in Candida albicans. Curr. Opin. Microbiol. 2007, 10, 307–313. [Google Scholar]
- Cannon, R.D.; Lamping, E.; Holmes, A.R.; Niimi, K.; Tanabe, K.; Niimi, M.; Monk, B.C. Candida albicans drug resistance another way to cope with stress. Microbiology 2007, 153, 3211–3217. [Google Scholar]
- Biswas, S.; Van Dijck, P.; Datta, A. Environmental sensing and signal transduction pathways regulating morphopathogenic determinants of Candida albicans. Microbiol. Mol. Biol. Rev. 2007, 71, 348–376. [Google Scholar]
- Kumamoto, C.A.; Vinces, M.D. Contributions of hyphae and hypha-co-regulated genes to Candida albicans virulence. Cell. Microbiol. 2005, 7, 1546–1554. [Google Scholar]
- Schaller, M.; Borelli, C.; Korting, H.C.; Hube, B. Hydrolytic enzymes as virulence factors of Candida albicans. Mycoses 2005, 48, 365–377. [Google Scholar]
- De Las Penas, A.; Pan, S.J.; Castano, I.; Alder, J.; Cregg, R.; Cormack, B.P. Virulence-related surface glycoproteins in the yeast pathogen Candida glabrata are encoded in subtelomeric clusters and subject to RAP1- and SIR-independent transcriptional silencing. Genes Dev. 2003, 17, 2245–2258. [Google Scholar]
- Castano, I.; Pan, S.J.; Zupancic, M.; Hennequin, C.; Dujon, B.; Cormack, B.P. Telomere length control and transcriptional regulation of subtelomeric adhesins in Candida glabrata. Mol. Microbiol. 2005, 55, 1246–1258. [Google Scholar]
- Iraqui, I.; Garcia-Sanchez, S.; Aubert, S.; Dromer, F.; Ghigo, J.M.; d'Enfert, C.; Janbon, G. The Yak1p kinase controls expression of adhesins and biofilm formation in Candida glabrata in a Sir4p-dependent pathway. Mol. Microbiol. 2005, 55, 1259–1271. [Google Scholar]
- Mundy, R.D.; Cormack, B. Expression of Candida glabrata Adhesins after Exposure to Chemical Preservatives. J. Infect. Dis. 2009, 199, 1891–1898. [Google Scholar]
- Domergue, R.; Castano, I.; De Las Penas, A.; Zupancic, M.; Lockatell, V.; Hebel, J.R.; Johnson, D.; Cormack, B.P. Nicotinic acid limitation regulates silencing of Candida adhesins during UTI. Science 2005, 308, 866–870. [Google Scholar]
- Kaur, R.; Ma, B.; Cormack, B.P. A family of glycosylphosphatidylinositol-linked aspartyl proteases is required for virulence of Candida glabrata. Proc. Natl. Acad. Sci. USA 2007, 104, 7628–7633. [Google Scholar]
- Kamran, M.; Calcagno, A.M.; Findon, H.; Bignell, E.; Jones, M.D.; Warn, P.; Hopkins, P.; Denning, D.W.; Butler, G.; Rogers, T.; Muhlschlegel, F.A.; Haynes, K. Inactivation of transcription factor gene ACE2 in the fungal pathogen Candida glabrata results in hypervirulence. Eukaryot. Cell 2004, 3, 546–552. [Google Scholar]
- Stead, D.; Findon, H.; Yin, Z.; Walker, J.; Selway, L.; Cash, P.; Dujon, B.A.; Hennequin, C.; Brown, A.J.; Haynes, K. Proteomic changes associated with inactivation of the C. glabrata ACE2 virulence-moderating gene. Proteomics 2005, 5, 1838–1848. [Google Scholar]
- Stead, D.A.; Walker, J.; Holcombe, L.; Gibbs, S.R.; Yin, Z.; Selway, L.; Butler, G.; Brown, A.J.; Haynes, K. Impact of the transcriptional regulator, Ace2, on the Candida glabrata secretome. Proteomics 2009, 10, 212–223. [Google Scholar]
- MacCallum, D.M.; Findon, H.; Kenny, C.C.; Butler, G.; Haynes, K.; Odds, F.C. Different consequences of ACE2 and SWI5 gene disruptions for virulence of pathogenic and nonpathogenic yeasts. Infect. Immun. 2006, 74, 5244–5248. [Google Scholar]
- Brunke, S.; Seider, K.; Almeida, R.S.; Heyken, A.; Fleck, C.B.; Brock, M.; Barz, D.; Rupp, S.; Hube, B. Candida glabrata tryptophan-based pigment production via the Ehrlich pathway. Mol. Microbiol. 2010, 76, 25–47. [Google Scholar]
- Roetzer, A.; Gratz, N.; Kovarik, P.; Schüller, C. Autophagy supports Candida glabrata survival during phagocytosis. Cell. Microbiol. 2010, 12, 199–216. [Google Scholar]
- Cuellar-Cruz, M.; Briones-Martin-del-Campo, M.; Canas-Villamar, I.; Montalvo-Arredondo, J.; Riego-Ruiz, L.; Castano, I.; De Las Penas, A. High resistance to oxidative stress in the fungal pathogen Candida glabrata is mediated by a single catalase, Cta1p, and is controlled by the transcription factors Yap1p, Skn7p, Msn2p, and Msn4p. Eukaryot. Cell 2008, 7, 814–825. [Google Scholar]
- Ferrari, S.; Ischer, F.; Calabrese, D.; Posteraro, B.; Sanguinetti, M.; Fadda, G.; Rohde, B.; Bauser, C.; Bader, O.; Sanglard, D. Gain of function mutations in CgPDR1 of Candida glabrata not only mediate antifungal resistance but also enhance virulence. PLoS Pathog. 2009, 5, e1000268. [Google Scholar]
- Torelli, R.; Posteraro, B.; Ferrari, S.; La Sorda, M.; Fadda, G.; Sanglard, D.; Sanguinetti, M. The ATP-binding cassette transporter-encoding gene CgSNQ2 is contributing to the CgPDR1-dependent azole resistance of Candida glabrata. Mol. Microbiol. 2008, 68, 186–201. [Google Scholar]
- Sanglard, D.; Ischer, F.; Bille, J. Role of ATP-binding-cassette transporter genes in high-frequency acquisition of resistance to azole antifungals in Candida glabrata. Antimicrob. Agents Chemother. 2001, 45, 1174–1183. [Google Scholar]
- Katiyar, S.; Pfaller, M.; Edlind, T. Candida albicans and Candida glabrata clinical isolates exhibiting reduced echinocandin susceptibility. Antimicrob. Agents Chemother. 2006, 50, 2892–2894. [Google Scholar]
- Thompson, G.R., 3rd; Wiederhold, N.P.; Vallor, A.C.; Villareal, N.C.; Lewis, J.S., 2nd; Patterson, T.F. Development of caspofungin resistance following prolonged therapy for invasive candidiasis secondary to Candida glabrata infection. Antimicrob. Agents Chemother. 2008, 52, 3783–3785. [Google Scholar]
- Cleary, J.D.; Garcia-Effron, G.; Chapman, S.W.; Perlin, D.S. Reduced Candida glabrata susceptibility secondary to an FKS1 mutation developed during candidemia treatment. Antimicrob. Agents Chemother. 2008, 52, 2263–2265. [Google Scholar]
- Garcia-Effron, G.; Lee, S.; Park, S.; Cleary, J.D.; Perlin, D.S. Effect of Candida glabrata FKS1 and FKS2 mutations on echinocandin sensitivity and kinetics of 1,3-β-d-glucan synthase: implication for the existing susceptibility breakpoint. Antimicrob. Agents Chemother. 2009, 53, 3690–3699. [Google Scholar]
- Sundstrom, P. Adhesins in Candida albicans. Curr. Opin. Microbiol. 1999, 2, 353–357. [Google Scholar]
- Hoyer, L.L. The ALS gene family of C. albicans. Trends Microbiol. 2001, 9, 176–180. [Google Scholar]
- Weig, M.; Jansch, L.; Gross, U.; De Koster, C.G.; Klis, F.M.; De Groot, P.W. Systematic identification in silico of covalently bound cell wall proteins and analysis of protein-polysaccharide linkages of the human pathogen Candida glabrata. Microbiology 2004, 150, 3129–3144. [Google Scholar]
- Kaur, R.; Domergue, R.; Zupancic, M.L.; Cormack, B.P. A yeast by any other name: C. glabrata and its interaction with the host. Curr. Opin. Microbiol. 2005, 8, 378–384. [Google Scholar]
- Thierry, A.; Bouchier, C.; Dujon, B.; Richard, G.F. Megasatellites: a peculiar class of giant minisatellites in genes involved in cell adhesion and pathogenicity in Candida glabrata. Nucleic. Acids Res. 2008, 36, 5970–5982. [Google Scholar]
- Zupancic, M.L.; Frieman, M.; Smith, D.; Alvarez, R.A.; Cummings, R.D.; Cormack, B.P. Glycan microarray analysis of Candida glabrata adhesin ligand specificity. Mol. Microbiol. 2008, 68, 547–559. [Google Scholar]
- de Groot, P.W.; Kraneveld, E.A.; Yin, Q.Y.; Dekker, H.L.; Gross, U.; Crielaard, W.; de Koster, C.G.; Bader, O.; Klis, F.M.; Weig, M. The cell wall of the human pathogen Candida glabrata: differential incorporation of novel adhesin-like wall proteins. Eukaryot. Cell 2008, 7, 1951–1964. [Google Scholar]
- Rusche, L.N.; Kirchmaier, A.L.; Rine, J. The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annu. Rev. Biochem. 2003, 72, 481–516. [Google Scholar]
- Ma, B.; Pan, S.J.; Domergue, R.; Rigby, T.; Whiteway, M.; Johnson, D.; Cormack, B.P. High-affinity transporters for NAD+ precursors in C. glabrata are regulated by Hst1 and induced in response to niacin limitation. Mol. Cell. Biol. 2009, 29, 4067–4079. [Google Scholar]
- Ma, B.; Pan, S.J.; Zupancic, M.L.; Cormack, B.P. Assimilation of NAD(+) precursors in Candida glabrata. Mol. Microbiol. 2007, 66, 14–25. [Google Scholar]
- Krysan, D.J.; Ting, E.L.; Abeijon, C.; Kroos, L.; Fuller, R.S. Yapsins are a family of aspartyl proteases required for cell wall integrity in Saccharomyces cerevisiae. Eukaryot. Cell 2005, 4, 1364–1374. [Google Scholar]
- Albrecht, A.; Felk, A.; Pichova, I.; Naglik, J.R.; Schaller, M.; de Groot, P.; Maccallum, D.; Odds, F.C.; Schafer, W.; Klis, F.; Monod, M.; Hube, B. Glycosylphosphatidylinositol-anchored proteases of C. albicans target proteins necessary for both cellular processes and host-pathogen interactions. J. Biol. Chem. 2006, 281, 688–694. [Google Scholar]
- Nelson, B.; Kurischko, C.; Horecka, J.; Mody, M.; Nair, P.; Pratt, L.; Zougman, A.; McBroom, L.D.; Hughes, T.R.; Boone, C.; Luca, F.C. RAM: a conserved signaling network that regulates Ace2p transcriptional activity and polarized morphogenesis. Mol. Biol. Cell 2003, 14, 3782–3803. [Google Scholar]
- Colman-Lerner, A.; Chin, T.E.; Brent, R. Yeast Cbk1 and Mob2 activate daughter-specific genetic programs to induce asymmetric cell fates. Cell 2001, 107, 739–750. [Google Scholar]
- O'Conallain, C.; Doolin, M.T.; Taggart, C.; Thornton, F.; Butler, G. Regulated nuclear localisation of the yeast transcription factor Ace2p controls expression of chitinase (CTS1) in Saccharomyces cerevisiae. Mol. Gen. Genet. 1999, 262, 275–282. [Google Scholar]
- King, L.; Butler, G. Ace2p, a regulator of CTS1 (chitinase) expression, affects pseudohyphal production in S. cerevisiae. Curr. Genet. 1998, 34, 183–191. [Google Scholar]
- Jacobsen, I.D.; Brunke, S.; Seider, K.; Schwarzmuller, T.; Firon, A.; d'Enfert, C.; Kuchler, K.; Hube, B. C. glabrata persistence in mice does not depend on host immunosuppression and is unaffected by fungal amino acid auxotrophy. Infect. Immun. 2010, 78, 1066–1077. [Google Scholar]
- Calcagno, A.M.; Bignell, E.; Warn, P.; Jones, M.D.; Denning, D.W.; Muhlschlegel, F.A.; Rogers, T.R.; Haynes, K. C. glabrata STE12 is required for wild-type levels of virulence and nitrogen starvation induced filamentation. Mol. Microbiol. 2003, 50, 1309–1318. [Google Scholar]
- Roetzer, A.; Gregori, C.; Jennings, A.M.; Quintin, J.; Ferrandon, D.; Butler, G.; Kuchler, K.; Ammerer, G.; Schüller, C. C. glabrata environmental stress response involves S. cerevisiae Msn2/4 orthologous transcription factors. Mol. Microbiol. 2008, 69, 603–620. [Google Scholar]
- Brennan, M.; Thomas, D.Y.; Whiteway, M.; Kavanagh, K. Correlation between virulence of Candida albicans mutants in mice and Galleria mellonella larvae. FEMS Immunol. Med. Microbiol. 2002, 34, 153–157. [Google Scholar]
- Mylonakis, E.; Moreno, R.; El Khoury, J.B.; Idnurm, A.; Heitman, J.; Calderwood, S.B.; Ausubel, F.M.; Diener, A. Galleria mellonella as a model system to study Cryptococcus neoformans pathogenesis. Infect. Immun. 2005, 73, 3842–3850. [Google Scholar]
- Fuchs, B.B.; Eby, J.; Nobile, C.J.; El Khoury, J.B.; Mitchell, A.P.; Mylonakis, E. Role of filamentation in Galleria mellonella killing by C. albicans. Microbes Infect. 2010, 12, 488–496. [Google Scholar]
- Pukkila-Worley, R.; Peleg, A.Y.; Tampakakis, E.; Mylonakis, E. Candida albicans hyphal formation and virulence assessed using a Caenorhabditis elegans infection model. Eukaryot. Cell 2009, 8, 1750–1758. [Google Scholar]
- Okoli, I.; Coleman, J.J.; Tempakakis, E.; An, W.F.; Holson, E.; Wagner, F.; Conery, A.L.; Larkins-Ford, J.; Wu, G.; Stern, A.; Ausubel, F.M.; Mylonakis, E. Identification of antifungal compounds active against Candida albicans using an improved high-throughput Caenorhabditis elegans assay. PLoS One 2009, 4, e7025. [Google Scholar]
- Nosanchuk, J.D.; Gomez, B.L.; Youngchim, S.; Diez, S.; Aisen, P.; Zancope-Oliveira, R.M.; Restrepo, A.; Casadevall, A.; Hamilton, A.J. Histoplasma capsulatum synthesizes melanin-like pigments in vitro and during mammalian infection. Infect. Immun. 2002, 70, 5124–5131. [Google Scholar]
- Jahn, B.; Koch, A.; Schmidt, A.; Wanner, G.; Gehringer, H.; Bhakdi, S.; Brakhage, A.A. Isolation and characterization of a pigmentless-conidium mutant of Aspergillus fumigatus with altered conidial surface and reduced virulence. Infect. Immun. 1997, 65, 5110–5117. [Google Scholar]
- Kwon-Chung, K.J.; Tom, W.K.; Costa, J.L. Utilization of indole compounds by C. neoformans to produce a melanin-like pigment. J. Clin. Microbiol. 1983, 18, 1419–1421. [Google Scholar]
- Jacobson, E.S.; Tinnell, S.B. Antioxidant function of fungal melanin. J. Bacteriol. 1993, 175, 7102–7104. [Google Scholar]
- Jacobson, E.S. Pathogenic roles for fungal melanins. Clin. Microbiol. Rev. 2000, 13, 708–717. [Google Scholar]
- Morgenstern, D.E.; Gifford, M.A.; Li, L.L.; Doerschuk, C.M.; Dinauer, M.C. Absence of respiratory burst in X-linked chronic granulomatous disease mice leads to abnormalities in both host defense and inflammatory response to Aspergillus fumigatus. J. Exp. Med. 1997, 185, 207–218. [Google Scholar]
- Mayser, P.; Wenzel, M.; Kramer, H.J.; Kindler, B.L.; Spiteller, P.; Haase, G. Production of indole pigments by Candida glabrata. Med. Mycol. 2007, 45, 519–524. [Google Scholar]
- Hazelwood, L.A.; Tai, S.L.; Boer, V.M.; de Winde, J.H.; Pronk, J.T.; Daran, J.M. A new physiological role for Pdr12p in S. cerevisiae: export of aromatic and branched-chain organic acids produced in amino acid catabolism. FEMS Yeast Res. 2006, 6, 937–945. [Google Scholar]
- Polakova, S.; Blume, C.; Zarate, J.A.; Mentel, M.; Jorck-Ramberg, D.; Stenderup, J.; Piskur, J. Formation of new chromosomes as a virulence mechanism in yeast Candida glabrata. Proc. Natl. Acad. Sci. USA 2009, 106, 2688–2693. [Google Scholar]
- Verstrepen, K.J.; Fink, G.R. Genetic and epigenetic mechanisms underlying cell-surface variability in protozoa and fungi. Annu. Rev. Genet. 2009, 43, 1–24. [Google Scholar]
- Levdansky, E.; Romano, J.; Shadkchan, Y.; Sharon, H.; Verstrepen, K.J.; Fink, G.R.; Osherov, N. Coding tandem repeats generate diversity in Aspergillus fumigatus genes. Eukaryot. Cell 2007, 6, 1380–1391. [Google Scholar]
- Lachke, S.A.; Srikantha, T.; Tsai, L.K.; Daniels, K.; Soll, D.R. Phenotypic switching in Candida glabrata involves phase-specific regulation of the metallothionein gene MT-II and the newly discovered hemolysin gene HLP. Infect. Immun. 2000, 68, 884–895. [Google Scholar]
- Lachke, S.A.; Joly, S.; Daniels, K.; Soll, D.R. Phenotypic switching and filamentation in Candida glabrata. Microbiology 2002, 148, 2661–2674. [Google Scholar]
- Brockert, P.J.; Lachke, S.A.; Srikantha, T.; Pujol, C.; Galask, R.; Soll, D.R. Phenotypic switching and mating type switching of Candida glabrata at sites of colonization. Infect. Immun. 2003, 71, 7109–7118. [Google Scholar]
- Srikantha, T.; Daniels, K.J.; Wu, W.; Lockhart, S.R.; Yi, S.; Sahni, N.; Ma, N.; Soll, D.R. Dark brown is the more virulent of the switch phenotypes of Candida glabrata. Microbiology 2008, 154, 3309–3318. [Google Scholar]
- Nicola, A.M.; Casadevall, A.; Goldman, D.L. Fungal killing by mammalian phagocytic cells. Curr. Opin. Microbiol. 2008, 11, 313–317. [Google Scholar]
- Alvarez, M.; Casadevall, A. Phagosome extrusion and host-cell survival after C. neoformans phagocytosis by macrophages. Curr. Biol. 2006, 16, 2161–2165. [Google Scholar]
- Ma, H.; Croudace, J.E.; Lammas, D.A.; May, R.C. Expulsion of live pathogenic yeast by macrophages. Curr. Biol. 2006, 16, 2156–2160. [Google Scholar]
- Geisow, M.J.; D'Arcy Hart, P.; Young, M.R. Temporal changes of lysosome and phagosome pH during phagolysosome formation in macrophages: studies by fluorescence spectroscopy. J. Cell. Biol. 1981, 89, 645–652. [Google Scholar]
- Levitz, S.M.; Nong, S.H.; Seetoo, K.F.; Harrison, T.S.; Speizer, R.A.; Simons, E.R. Cryptococcus neoformans resides in an acidic phagolysosome of human macrophages. Infect. Immun. 1999, 67, 885–890. [Google Scholar]
- Chaves, G.M.; Bates, S.; Maccallum, D.M.; Odds, F.C. C. albicans GRX2, encoding a putative glutaredoxin, is required for virulence in a murine model. Genet. Mol. Res. 2007, 6, 1051–1063. [Google Scholar]
- Missall, T.A.; Pusateri, M.E.; Lodge, J.K. Thiol peroxidase is critical for virulence and resistance to nitric oxide and peroxide in the fungal pathogen, Cryptococcus neoformans. Mol. Microbiol. 2004, 51, 1447–1458. [Google Scholar]
- Frohner, I.E.; Bourgeois, C.; Yatsyk, K.; Majer, O.; Kuchler, K. Candida albicans cell surface superoxide dismutases degrade host-derived reactive oxygen species to escape innate immune surveillance. Mol. Microbiol. 2009, 71, 240–252. [Google Scholar]
- Paris, S.; Wysong, D.; Debeaupuis, J.P.; Shibuya, K.; Philippe, B.; Diamond, R.D.; Latge, J.P. Catalases of Aspergillus fumigatus. Infect. Immun. 2003, 71, 3551–3562. [Google Scholar]
- Goffeau, A. Drug resistance: the fight against fungi. Nature 2008, 452, 541–542. [Google Scholar]
- Cowen, L.E.; Steinbach, W.J. Stress, drugs, and evolution: the role of cellular signaling in fungal drug resistance. Eukaryot. Cell 2008, 7, 747–764. [Google Scholar]
- Odds, F.C.; Brown, A.J.; Gow, N.A. Antifungal agents: mechanisms of action. Trends Microbiol. 2003, 11, 272–279. [Google Scholar]
- Vanden Bossche, H.; Koymans, L.; Moereels, H. P450 inhibitors of use in medical treatment: focus on mechanisms of action. Pharmacol. Ther. 1995, 67, 79–100. [Google Scholar]
- Lopez-Ribot, J.L.; McAtee, R.K.; Lee, L.N.; Kirkpatrick, W.R.; White, T.C.; Sanglard, D.; Patterson, T.F. Distinct patterns of gene expression associated with development of fluconazole resistance in serial C. albicans isolates from human immunodeficiency virus-infected patients with oropharyngeal candidiasis. Antimicrob. Agents Chemother. 1998, 42, 2932–2937. [Google Scholar]
- Perea, S.; Lopez-Ribot, J.L.; Kirkpatrick, W.R.; McAtee, R.K.; Santillan, R.A.; Martinez, M.; Calabrese, D.; Sanglard, D.; Patterson, T.F. Prevalence of molecular mechanisms of resistance to azole antifungal agents in Candida albicans strains displaying high-level fluconazole resistance isolated from human immunodeficiency virus-infected patients. Antimicrob. Agents Chemother. 2001, 45, 2676–2684. [Google Scholar]
- Henry, K.W.; Nickels, J.T.; Edlind, T.D. Upregulation of ERG genes in Candida species by azoles and other sterol biosynthesis inhibitors. Antimicrob. Agents Chemother. 2000, 44, 2693–2700. [Google Scholar]
- Vermitsky, J.P.; Edlind, T.D. Azole resistance in Candida glabrata: coordinate upregulation of multidrug transporters and evidence for a Pdr1-like transcription factor. Antimicrob. Agents Chemother. 2004, 48, 3773–3781. [Google Scholar]
- Sanguinetti, M.; Posteraro, B.; Fiori, B.; Ranno, S.; Torelli, R.; Fadda, G. Mechanisms of azole resistance in clinical isolates of Candida glabrata collected during a hospital survey of antifungal resistance. Antimicrob. Agents Chemother. 2005, 49, 668–679. [Google Scholar]
- Sanglard, D.; Kuchler, K.; Ischer, F.; Pagani, J.L.; Monod, M.; Bille, J. Mechanisms of resistance to azole antifungal agents in C. albicans isolates from AIDS patients involve specific multidrug transporters. Antimicrob. Agents Chemother. 1995, 39, 2378–2386. [Google Scholar]
- Sanglard, D.; Ischer, F.; Calabrese, D.; Majcherczyk, P.A.; Bille, J. The ATP binding cassette transporter gene CgCDR1 from C. glabrata is involved in the resistance of clinical isolates to azole antifungal agents. Antimicrob. Agents Chemother. 1999, 43, 2753–2765. [Google Scholar]
- Sanglard, D.; Ischer, F.; Monod, M.; Bille, J. Cloning of Candida albicans genes conferring resistance to azole antifungal agents: characterization of CDR2, a new multidrug ABC transporter gene. Microbiology 1997, 143, 405–416. [Google Scholar]
- White, T.C. Increased mRNA levels of ERG16, CDR, and MDR1 correlate with increases in azole resistance in Candida albicans isolates from a patient infected with human immunodeficiency virus. Antimicrob. Agents Chemother. 1997, 41, 1482–1487. [Google Scholar]
- Bennett, J.E.; Izumikawa, K.; Marr, K.A. Mechanism of increased fluconazole resistance in Candida glabrata during prophylaxis. Antimicrob. Agents Chemother. 2004, 48, 1773–1777. [Google Scholar]
- Miyazaki, H.; Miyazaki, Y.; Geber, A.; Parkinson, T.; Hitchcock, C.; Falconer, D.J.; Ward, D.J.; Marsden, K.; Bennett, J.E. Fluconazole resistance associated with drug efflux and increased transcription of a drug transporter gene, PDH1, in Candida glabrata. Antimicrob. Agents Chemother. 1998, 42, 1695–1701. [Google Scholar]
- Sipos, G.; Kuchler, K. Fungal ATP-binding cassette (ABC) transporters in drug resistance & detoxification. Curr. Drug Targets 2006, 7, 471–481. [Google Scholar]
- Nakayama, H.; Izuta, M.; Nakayama, N.; Arisawa, M.; Aoki, Y. Depletion of the squalene synthase (ERG9) gene does not impair growth of Candida glabrata in mice. Antimicrob. Agents Chemother. 2000, 44, 2411–2418. [Google Scholar]
- Nakayama, H.; Tanabe, K.; Bard, M.; Hodgson, W.; Wu, S.; Takemori, D.; Aoyama, T.; Kumaraswami, N.S.; Metzler, L.; Takano, Y.; Chibana, H.; Niimi, M. The Candida glabrata putative sterol transporter gene CgAUS1 protects cells against azoles in the presence of serum. J. Antimicrob. Chemother. 2007, 60, 1264–1272. [Google Scholar]
- Ellis, D. Amphotericin B: spectrum and resistance. J. Antimicrob. Chemother. 2002, 49 Suppl 1, 7–10. [Google Scholar]
- Sterling, T.R.; Merz, W.G. Resistance to amphotericin B: emerging clinical and microbiological patterns. Drug Resist. Updat. 1998, 1, 161–165. [Google Scholar]
- Nolte, F.S.; Parkinson, T.; Falconer, D.J.; Dix, S.; Williams, J.; Gilmore, C.; Geller, R.; Wingard, J.R. Isolation and characterization of fluconazole- and amphotericin B-resistant Candida albicans from blood of two patients with leukemia. Antimicrob. Agents Chemother. 1997, 41, 196–199. [Google Scholar]
- Douglas, C.M. Fungal beta(1,3)-D-glucan synthesis. Med. Mycol. 2001, 39 Suppl 1, 55–66. [Google Scholar]
- Perlin, D.S. Resistance to echinocandin-class antifungal drugs. Drug Resist. Updat. 2007, 10, 121–130. [Google Scholar]
- Schuetzer-Muehlbauer, M.; Willinger, B.; Krapf, G.; Enzinger, S.; Presterl, E.; Kuchler, K. The Candida albicans Cdr2p ATP-binding cassette (ABC) transporter confers resistance to caspofungin. Mol. Microbiol. 2003, 48, 225–235. [Google Scholar]
- Gulshan, K.; Moye-Rowley, W.S. Multidrug resistance in fungi. Eukaryot. Cell 2007, 6, 1933–1942. [Google Scholar]
- Li, X.Z.; Nikaido, H. Efflux-mediated drug resistance in bacteria. Drugs 2004, 64, 159–204. [Google Scholar]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar]
- Jones, P.M.; George, A.M. Multidrug resistance in parasites: ABC transporters, P-glycoproteins and molecular modelling. Int. J. Parasitol. 2005, 35, 555–566. [Google Scholar]
- Thakur, J.K.; Arthanari, H.; Yang, F.; Pan, S.J.; Fan, X.; Breger, J.; Frueh, D.P.; Gulshan, K.; Li, D.K.; Mylonakis, E.; Struhl, K.; Moye-Rowley, W.S.; Cormack, B.P.; Wagner, G.; Naar, A.M. A nuclear receptor-like pathway regulating multidrug resistance in fungi. Nature 2008, 452, 604–609. [Google Scholar]
- Bouchara, J.P.; Zouhair, R.; Le Boudouil, S.; Renier, G.; Filmon, R.; Chabasse, D.; Hallet, J.N.; Defontaine, A. In vivo selection of an azole-resistant petite mutant of Candida glabrata. J. Med. Microbiol. 2000, 49, 977–984. [Google Scholar]
- Tsai, H.F.; Krol, A.A.; Sarti, K.E.; Bennett, J.E. Candida glabrata PDR1, a transcriptional regulator of a pleiotropic drug resistance network, mediates azole resistance in clinical isolates and petite mutants. Antimicrob. Agents Chemother. 2006, 50, 1384–1392. [Google Scholar]
- Hallstrom, T.C.; Moye-Rowley, W.S. Multiple signals from dysfunctional mitochondria activate the pleiotropic drug resistance pathway in Saccharomyces cerevisiae. J. Biol. Chem. 2000, 275, 37347–37356. [Google Scholar]
- Borecka-Melkusova, S.; Kozovska, Z.; Hikkel, I.; Dzugasova, V.; Subik, J. RPD3 and ROM2 are required for multidrug resistance in Saccharomyces cerevisiae. FEMS Yeast Res. 2008, 8, 414–424. [Google Scholar]
- Smith, W.L.; Edlind, T.D. Histone deacetylase inhibitors enhance Candida albicans sensitivity to azoles and related antifungals: correlation with reduction in CDR and ERG upregulation. Antimicrob. Agents Chemother. 2002, 46, 3532–3539. [Google Scholar]
- Wurtele, H.; Tsao, S.; Lepine, G.; Mullick, A.; Tremblay, J.; Drogaris, P.; Lee, E.H.; Thibault, P.; Verreault, A.; Raymond, M. Modulation of histone H3 lysine 56 acetylation as an antifungal therapeutic strategy. Nat. Med. 2010, 16, 774–780. [Google Scholar]
- Scherens, B.; Goffeau, A. The uses of genome-wide yeast mutant collections. Genome Biol. 2004, 5, 229. [Google Scholar]
- Jones, G.M.; Stalker, J.; Humphray, S.; West, A.; Cox, T.; Rogers, J.; Dunham, I.; Prelich, G. A systematic library for comprehensive overexpression screens in Saccharomyces cerevisiae. Nat. Methods 2008, 5, 239–241. [Google Scholar]
- Kumar, A.; Cheung, K.H.; Ross-Macdonald, P.; Coelho, P.S.; Miller, P.; Snyder, M. TRIPLES: a database of gene function in Saccharomyces cerevisiae. Nucleic Acids Res 2000, 28, 81–84. [Google Scholar]
- Liu, O.W.; Chun, C.D.; Chow, E.D.; Chen, C.; Madhani, H.D.; Noble, S.M. Systematic genetic analysis of virulence in the human fungal pathogen Cryptococcus neoformans. Cell 2008, 135, 174–188. [Google Scholar]
- Homann, O.R.; Dea, J.; Noble, S.M.; Johnson, A.D. A phenotypic profile of the Candida albicans regulatory network. PLoS Genet. 2009, 5, e1000783. [Google Scholar]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008, 320, 1344–1349. [Google Scholar]
- Hegedus, Z.; Zakrzewska, A.; Agoston, V.C.; Ordas, A.; Racz, P.; Mink, M.; Spaink, H.P.; Meijer, A.H. Deep sequencing of the zebrafish transcriptome response to mycobacterium infection. Mol. Immunol. 2009, 46, 2918–2930. [Google Scholar]
- Costanzo, M.; Baryshnikova, A.; Bellay, J.; Kim, Y.; Spear, E.D.; Sevier, C.S.; Ding, H.; Koh, J.L.; Toufighi, K.; Mostafavi, S.; et al. The genetic landscape of a cell. Science 2010, 327, 425–431. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tscherner, M.; Schwarzmüller, T.; Kuchler, K. Pathogenesis and Antifungal Drug Resistance of the Human Fungal Pathogen Candida glabrata. Pharmaceuticals 2011, 4, 169-186. https://doi.org/10.3390/ph4010169
Tscherner M, Schwarzmüller T, Kuchler K. Pathogenesis and Antifungal Drug Resistance of the Human Fungal Pathogen Candida glabrata. Pharmaceuticals. 2011; 4(1):169-186. https://doi.org/10.3390/ph4010169
Chicago/Turabian StyleTscherner, Michael, Tobias Schwarzmüller, and Karl Kuchler. 2011. "Pathogenesis and Antifungal Drug Resistance of the Human Fungal Pathogen Candida glabrata" Pharmaceuticals 4, no. 1: 169-186. https://doi.org/10.3390/ph4010169
APA StyleTscherner, M., Schwarzmüller, T., & Kuchler, K. (2011). Pathogenesis and Antifungal Drug Resistance of the Human Fungal Pathogen Candida glabrata. Pharmaceuticals, 4(1), 169-186. https://doi.org/10.3390/ph4010169